
Regulatory Role of Sex Hormones in Cardiovascular Calcification


Abstract
:1. Clinical Consequences of Cardiovascular Calcification
2. Types of Cardiovascular Calcification
2.1. Atherosclerotic Intimal Calcification
2.2. Medial Calcification
2.3. Valvular Calcification
2.4. Pharmaceutical Strategies
3. Current Understanding of Cardiovascular Calcification
3.1. Calcification Is Similar to Physiological Bone Formation
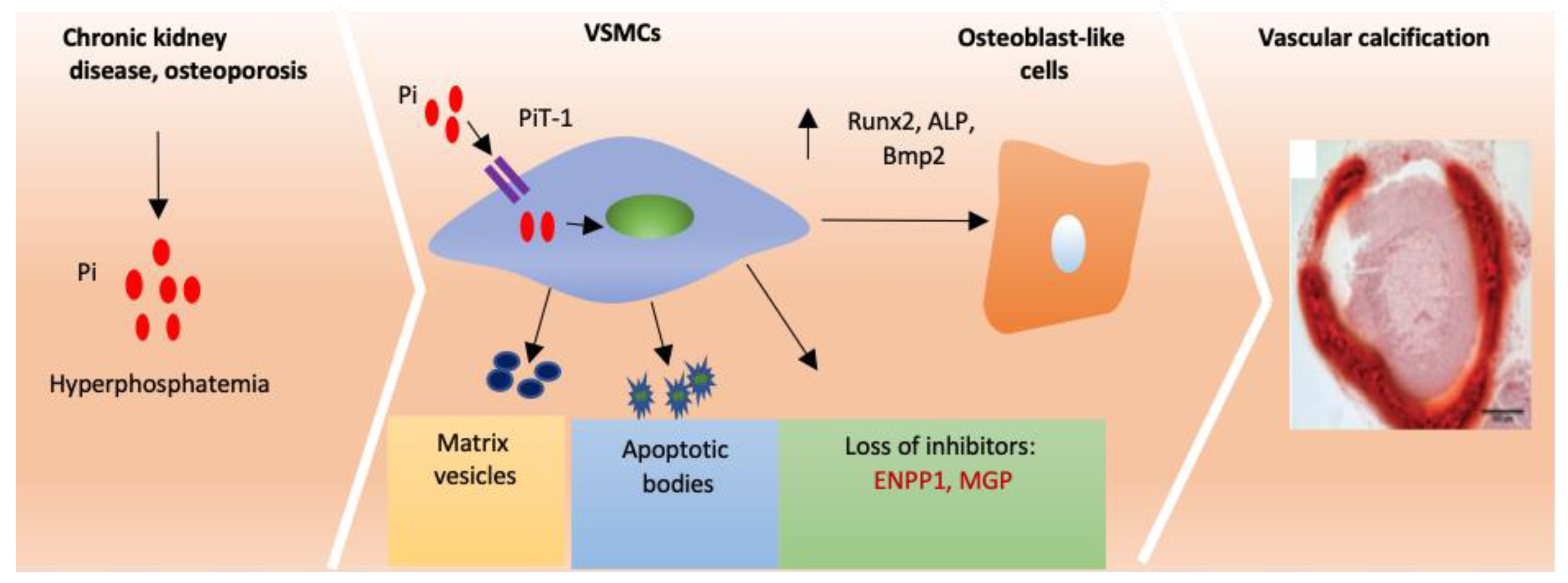
3.2. Loss of Endogenous Inhibitors Induces Vascular Calcification
3.3. Matrix Vesicles and Apoptotic Bodies Promote Cardiovascular Calcification
4. The Role of Sex and Sex Hormones in Cardiovascular Calcification
4.1. Sex difference Exists in Cardiovascular Calcification
4.2. Estrogen and Activation of the Estrogen Receptor Prevents Calcification
4.3. Testosterone Is a Risk Factor for Cardiovascular Calcification
5. Sex Hormones Mediate Cellular Signalling Pathways in the Cardiovascular System
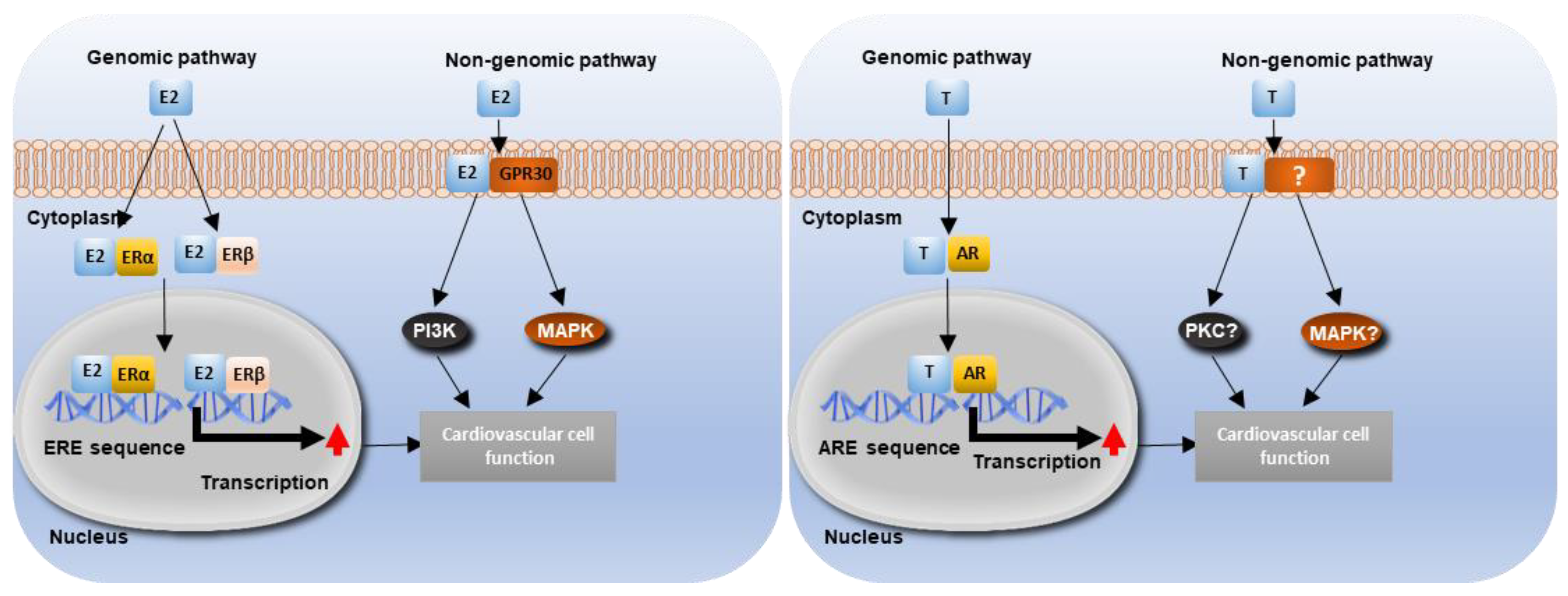
5.1. Estrogen Signalling and Cardiovascular Function
5.2. Testosterone Signalling and Cardiovascular Function
6. Animal Models Offer Insights into Sex Differences in Cardiovascular Calcification
6.1. Rodent Models
6.2. Large Animal Models
7. Future Perspectives
Funding
Conflicts of Interest
References
- Liu, W.; Zhang, Y.; Yu, C.-M.; Ji, Q.-W.; Cai, M.; Zhao, Y.-X.; Zhou, Y.-J. Current understanding of coronary artery calcification. J. Geriatr. Cardiol. 2015, 12, 668–675. [Google Scholar]
- Chen, Q.; Wang, Z.-Y.; Chen, L.-Y.; Hu, H.-Y. Roles of High Mobility Group Box 1 in Cardiovascular Calcification. Cell. Physiol. Biochem. 2017, 42, 427–440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shanahan, C.M.; Cary, N.R.B.; Salisbury, J.R.; Proudfoot, D.; Weissberg, P.L.; Edmonds, M.E. Medial Localization of Mineralization-Regulating Proteins in Association with Mönckeberg’s Sclerosis. Circulation 1999, 100, 2168–2176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Christian, R.C.; Harrington, S.; Edwards, W.D.; Oberg, A.L.; Fitzpatrick, L.A. Estrogen Status Correlates with the Calcium Content of Coronary Atherosclerotic Plaques in Women. J. Clin. Endocrinol. Metab. 2002, 87, 1062–1067. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Miller, V.M.; Miller, J.D. Influences of Sex and Estrogen in Arterial and Valvular Calcification. Front. Endocrinol. 2019, 10, 622. [Google Scholar] [CrossRef]
- Amann, K. Media Calcification and Intima Calcification Are Distinct Entities in Chronic Kidney Disease: Figure 1. Clin. J. Am. Soc. Nephrol. 2008, 3, 1599–1605. [Google Scholar] [CrossRef] [Green Version]
- Steitz, S.A.; Speer, M.Y.; Curinga, G.; Yang, H.-Y.; Haynes, P.A.; Aebersold, R.; Schinke, T.; Karsenty, G.; Giachelli, C.M. Smooth Muscle Cell Phenotypic Transition Associated with Calcification. Circ. Res. 2001, 89, 1147–1154. [Google Scholar] [CrossRef]
- Nadra, I.; Mason, J.C.; Philippidis, P.; Florey, O.; Smythe, C.D.; McCarthy, G.M.; Landis, R.C.; Haskard, D.O. Proinflammatory Activation of Macrophages by Basic Calcium Phosphate Crystals via Protein Kinase C and MAP Kinase Pathways. Circ. Res. 2005, 96, 1248–1256. [Google Scholar] [CrossRef] [Green Version]
- Bentzon, J.F.; Otsuka, F.; Virmani, R.; Falk, E. Mechanisms of Plaque Formation and Rupture. Circ. Res. 2014, 114, 1852–1866. [Google Scholar] [CrossRef]
- Libby, P.; Pasterkamp, G. Requiem for the ‘vulnerable plaque’. Eur. Heart J. 2015, 36, 2984–2987. [Google Scholar] [CrossRef] [Green Version]
- Bardeesi, A.S.A.; Gao, J.; Zhang, K.; Yu, S.; Wei, M.; Liu, P.; Huang, H. A novel role of cellular interactions in vascular calcification. J. Transl. Med. 2017, 15, 1–8. [Google Scholar] [CrossRef]
- Manzoor, S.; Ahmed, S.; Ali, A.; Han, K.H.; Sechopoulos, I.; O’Neill, A.; Fei, B.; O’Neill, W.C. Progression of Medial Arterial Calcification in CKD. Kidney Int. Rep. 2018, 3, 1328–1335. [Google Scholar] [CrossRef] [Green Version]
- Ho, C.Y.; Shanahan, C.M. Medial Arterial Calcification. Arter. Thromb. Vasc. Biol. 2016, 36, 1475–1482. [Google Scholar] [CrossRef] [Green Version]
- Lanzer, P.; Boehm, M.; Sorribas, V.; Thiriet, M.; Janzen, J.; Zeller, T.; Hilaire, C.S.; Shanahan, C. Medial vascular calcification revisited: Review and perspectives. Eur. Heart J. 2014, 35, 1515–1525. [Google Scholar] [CrossRef]
- Wu, M.; Rementer, C.; Giachelli, C.M. Vascular Calcification: An Update on Mechanisms and Challenges in Treatment. Calcif. Tissue Int. 2013, 93, 365–373. [Google Scholar] [CrossRef]
- Mohler, E.R.; Gannon, F.; Reynolds, C.; Zimmerman, R.; Keane, M.G.; Kaplan, F.S. Bone Formation and Inflammation in Cardiac Valves. Circulation 2001, 103, 1522–1528. [Google Scholar] [CrossRef] [PubMed]
- Ooyama, T.; Sakamato, H. Elastase in the Prevention of Arterial Ageing and the Treatment of Atherosclerosis. Novartis Found. Symposia 2007, 192, 307–320. [Google Scholar] [CrossRef]
- Schlotter, F.; Halu, A.; Pham, T.; Rogers, M.A.; Sharma, A.; Seidman, C.E.; Loscalzo, J.; Seidman, J.G.; Aikawa, M.; Singh, S.A.; et al. Spatiotemporal Multi-Omics Mapping Generates a Molecular Atlas of the Aortic Valve and Reveals Networks Driving Disease. Circulation 2018, 138, 377–393. [Google Scholar] [CrossRef] [PubMed]
- Bäck, M.; Aranyi, T.; Cancela, M.L.; Carracedo, M.; Conceição, N.; Leftheriotis, G.; Macrae, V.; Martin, L.; Nitschke, Y.; Pasch, A.; et al. Endogenous Calcification Inhibitors in the Prevention of Vascular Calcification: A Consensus Statement From the COST Action EuroSoftCalcNet. Front. Cardiovasc. Med. 2019, 5, 196. [Google Scholar] [CrossRef]
- Small, A.; Kiss, D.; Giri, J.; Anwaruddin, S.; Siddiqi, H.; Guerraty, M.; Chirinos, J.A.; Ferrari, G.; Rader, D.J. Biomarkers of Calcific Aortic Valve Disease. Arter. Thromb. Vasc. Biol. 2017, 37, 623–632. [Google Scholar] [CrossRef] [Green Version]
- Liu, A.C.; Joag, V.R.; Gotlieb, A.I. The Emerging Role of Valve Interstitial Cell Phenotypes in Regulating Heart Valve Pathobiology. Am. J. Pathol. 2007, 171, 1407–1418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Normand, J.; Loire, R.; Zambartas, C. The anatomical aspects of adult aortic stenosis. Eur. Heart J. 1988, 9, 31–36. [Google Scholar] [CrossRef] [PubMed]
- Lu, K.-C.; Wu, C.-C.; Yen, J.-F.; Liu, W.-C. Vascular Calcification and Renal Bone Disorders. Sci. World J. 2014, 2014, 1–20. [Google Scholar] [CrossRef]
- Tinica, G.; Chistol, R.O.; Enache, M.; Constantin, M.M.L.; Ciocoiu, M.; Furnica, C. Long-term graft patency after coronary artery bypass grafting: Effects of morphological and pathophysiological factors. Anatol. J. Cardiol. 2018, 20, 275–282. [Google Scholar] [CrossRef] [PubMed]
- Badran, A.A.; Vohra, H.A.; Livesey, S.A. Unoperated severe aortic stenosis: Decision making in an adult UK-based population. Ann. R. Coll. Surg. Engl. 2012, 94, 416–421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toker, M.E.; Eren, E.; Guler, M.; Kirali, K.; Yanartas, M.; Balkanay, M.; Yakut, C. Second and third cardiac valve reoperations: Factors influencing death and long-term survival. Tex. Heart Inst. J. 2009, 36, 557–562. [Google Scholar]
- Ranganath, N.K.; Koeckert, M.S.; Smith, D.E.; Hisamoto, K.; Loulmet, D.F.; Galloway, A.C.; Grossi, E.A. Aggressive tissue aortic valve replacement in younger patients and the risk of re-replacement: Implications from microsimulation analysis. J. Thorac. Cardiovasc. Surg. 2019, 158, 39–45.e1. [Google Scholar] [CrossRef]
- Chakravarty, T.; Patel, A.; Kapadia, S.; Raschpichler, M.; Smalling, R.W.; Szeto, W.Y.; Abramowitz, Y.; Cheng, W.; Douglas, P.S.; Hahn, R.T.; et al. Anticoagulation After Surgical or Transcatheter Bioprosthetic Aortic Valve Replacement. J. Am. Coll. Cardiol. 2019, 74, 1190–1200. [Google Scholar] [CrossRef]
- Chen, Y.; Zhao, X.; Wu, H. Arterial Stiffness. Arter. Thromb. Vasc. Biol. 2020, 40, 1078–1093. [Google Scholar] [CrossRef]
- Tyson, K.L.; Reynolds, J.L.; McNair, R.; Zhang, Q.; Weissberg, P.L.; Shanahan, C.M. Osteo/Chondrocytic Transcription Factors and Their Target Genes Exhibit Distinct Patterns of Expression in Human Arterial Calcification. Arter. Thromb. Vasc. Biol. 2003, 23, 489–494. [Google Scholar] [CrossRef] [Green Version]
- Zhu, D.; MacKenzie, N.C.W.; Millán, J.L.; Farquharson, C.; Macrae, V.E. The Appearance and Modulation of Osteocyte Marker Expression during Calcification of Vascular Smooth Muscle Cells. PLoS ONE 2011, 6, e19595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Speer, M.Y.; Yang, H.-Y.; Brabb, T.; Leaf, E.; Look, A.; Lin, W.-L.; Frutkin, A.; Dichek, D.; Giachelli, C.M. Smooth Muscle Cells Give Rise to Osteochondrogenic Precursors and Chondrocytes in Calcifying Arteries. Circ. Res. 2009, 104, 733–741. [Google Scholar] [CrossRef]
- Li, X.; Yang, H.-Y.; Giachelli, C.M. Role of the Sodium-Dependent Phosphate Cotransporter, Pit-1, in Vascular Smooth Muscle Cell Calcification. Circ. Res. 2006, 98, 905–912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dalfino, G.; Simone, S.; Porreca, S.; Cosola, C.; Balestra, C.; Manno, C.; Schena, F.P.; Grandaliano, G.; Pertosa, G. Bone morphogenetic protein-2 may represent the molecular link between oxidative stress and vascular stiffness in chronic kidney disease. Atherosclerosis 2010, 211, 418–423. [Google Scholar] [CrossRef] [PubMed]
- Zhu, D.; MacKenzie, N.C.W.; Shanahan, C.M.; Shroff, R.C.; Farquharson, C.; Macrae, V.E. BMP-9 regulates the osteoblastic differentiation and calcification of vascular smooth muscle cells through an ALK1 mediated pathway. J. Cell. Mol. Med. 2015, 19, 165–174. [Google Scholar] [CrossRef]
- Li, X.; Yang, H.-Y.; Giachelli, C.M. BMP-2 promotes phosphate uptake, phenotypic modulation, and calcification of human vascular smooth muscle cells. Atherosclerosis 2008, 199, 271–277. [Google Scholar] [CrossRef] [Green Version]
- Villa-Bellosta, R.; Sorribas, V. Phosphonoformic Acid Prevents Vascular Smooth Muscle Cell Calcification by Inhibiting Calcium-Phosphate Deposition. Arter. Thromb. Vasc. Biol. 2009, 29, 761–766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, F.; Wang, H.; Ren, W.-Y.; Ma, Y.; Liao, Y.-P.; Zhu, J.-H.; Cui, J.; Deng, Z.-L.; Su, Y.-X.; Gan, H.; et al. BMP9/COX-2 axial mediates high phosphate-induced calcification in vascular smooth muscle cells via Wnt/β-catenin pathway. J. Cell. Biochem. 2018, 119, 2851–2863. [Google Scholar] [CrossRef]
- Derwall, M.S.; Malhotra, R.; Lai, C.S.; Beppu, Y.; Aikawa, E.; Seehra, J.S.; Zapol, W.M.; Bloch, K.D.; Yu, P.B. Inhibition of Bone Morphogenetic Protein Signaling Reduces Vascular Calcification and Atherosclerosis. Arter. Thromb. Vasc. Biol. 2012, 32, 613–622. [Google Scholar] [CrossRef] [Green Version]
- Davies, M.R.; Lund, R.J.; Hruska, K.A. BMP-7 Is an Efficacious Treatment of Vascular Calcification in a Murine Model of Atherosclerosis and Chronic Renal Failure. J. Am. Soc. Nephrol. 2003, 14, 1559–1567. [Google Scholar] [CrossRef] [Green Version]
- Mori, K.; Shioi, A.; Jono, S.; Nishizawa, Y.; Morii, H. Expression of matrix Gla protein (MGP) in an in vitro model of vascular calcification. FEBS Lett. 1998, 433, 19–22. [Google Scholar] [CrossRef] [Green Version]
- Luo, G.; Ducy, P.; McKee, M.D.; Pinero, G.J.; Loyer, E.; Behringer, R.R.; Karsenty, G. Spontaneous calcification of arteries and cartilage in mice lacking matrix GLA protein. Nat. Cell Biol. 1997, 386, 78–81. [Google Scholar] [CrossRef] [PubMed]
- Munroe, P.P.; Olgunturk, R.R.; Fryns, J.-P.J.-P.; Van Maldergem, L.; Ziereisen, F.; Yuksel, B.B.; Gardiner, M.R.; Chung, E.E. Mutations in the gene encoding the human matrix Gla protein cause Keutel syndrome. Nat. Genet. 1999, 21, 142–144. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.; Zebboudj, A.F.; Shao, E.; Perez, M.; Boström, K. Regulation of Bone Morphogenetic Protein-4 by Matrix GLA Protein in Vascular Endothelial Cells Involves Activin-like Kinase Receptor 1. J. Biol. Chem. 2006, 281, 33921–33930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zebboudj, A.F.; Imura, M.; Boström, K. Matrix GLA Protein, a Regulatory Protein for Bone Morphogenetic Protein-2. J. Biol. Chem. 2002, 277, 4388–4394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malhotra, R.; Burke, M.F.; Hoeft, K.; Mayeur, C.; Jiramongkolchai, P.; Kumar, R.; Buys, E.S.; Yu, P.B.; Bloch, K.D.; Bloch, D.B.; et al. Inhibition of Bone Morphogenetic Protein Signal Transduction Prevents the Medial Vascular Calcification Associated with Matrix Gla Protein Deficiency. PLoS ONE 2015, 10, e0117098. [Google Scholar] [CrossRef] [PubMed]
- Cranenburg, E.C.M.; Van Spaendonck-Zwarts, K.Y.; Bonafé, L.; Crettol, L.M.; Rödiger, L.A.; Dikkers, F.G.; Van Essen, A.J.; Superti-Furga, A.; Alexandrakis, E.; Vermeer, C.; et al. Circulating matrix γ-carboxyglutamate protein (MGP) species are refractory to vitamin K treatment in a new case of Keutel syndrome. J. Thromb. Haemost. 2011, 9, 1225–1235. [Google Scholar] [CrossRef] [PubMed]
- Schurgers, L.J.; Spronk, H.M.H.; Soute, B.A.M.; Schiffers, P.M.; Demey, J.G.R.; Vermeer, C. Regression of warfarin-induced medial elastocalcinosis by high intake of vitamin K in rats. Blood 2006, 109, 2823–2831. [Google Scholar] [CrossRef]
- Goding, J.W.; Terkeltaub, R.; Maurice, M.; Deterre, P.; Sali, A.; Belli, S.I. Ecto-phosphodiesterase/pyrophosphatase of lymphocytes and non-lymphoid cells: Structure and function of the PC-1 family. Immunol. Rev. 1998, 161, 11–26. [Google Scholar] [CrossRef] [PubMed]
- Nitschke, Y.; Yan, Y.; Buers, I.; Kintziger, K.; Askew, K.; Rutsch, F. ENPP1-Fc prevents neointima formation in generalized arterial calcification of infancy through the generation of AMP. Exp. Mol. Med. 2018, 50, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Okawa, A.; Nakamura, I.; Goto, S.; Moriya, H.; Nakamura, Y.; Ikegawa, S. Mutation in Npps in a mouse model of ossification of the posterior longitudinal ligament of the spine. Nat. Genet. 1998, 19, 271–273. [Google Scholar] [CrossRef] [PubMed]
- MacKenzie, N.C.W.; Zhu, D.; Milne, E.M.; Hof, R.V.T.; Martin, A.; Quarles, D.L.; Millán, J.L.; Farquharson, C.; Macrae, V.E. Altered Bone Development and an Increase in FGF-23 Expression in Enpp1−/− Mice. PLoS ONE 2012, 7, e32177. [Google Scholar] [CrossRef]
- Rutsch, F.; Böyer, P.; Nitschke, Y.; Ruf, N.; Lorenz-Depierieux, B.; Wittkampf, T.; Weissen-Plenz, G.; Fischer, R.-J.; Mughal, Z.; Gregory, J.W.; et al. Hypophosphatemia, Hyperphosphaturia, and Bisphosphonate Treatment Are Associated With Survival Beyond Infancy in Generalized Arterial Calcification of Infancy. Circ. Cardiovasc. Genet. 2008, 1, 133–140. [Google Scholar] [CrossRef] [PubMed]
- Ruf, N.; Uhlenberg, B.; Terkeltaub, R.; Nürnberg, P.; Rutsch, F. The mutational spectrum ofENPP1as arising after the analysis of 23 unrelated patients with generalized arterial calcification of infancy (GACI). Hum. Mutat. 2004, 25, 98. [Google Scholar] [CrossRef]
- Stella, J.; Buers, I.; Van De Wetering, K.; Höhne, W.; Rutsch, F.; Nitschke, Y. Effects of Different Variants in theENPP1Gene on the Functional Properties of Ectonucleotide Pyrophosphatase/Phosphodiesterase Family Member 1. Hum. Mutat. 2016, 37, 1190–1201. [Google Scholar] [CrossRef] [PubMed]
- Roberts, F.; Zhu, D.; Farquharson, C.; Macrae, V.E. ENPP1 in the Regulation of Mineralization and Beyond. Trends Biochem. Sci. 2019, 44, 616–628. [Google Scholar] [CrossRef]
- Huesa, C.; Zhu, D.; Glover, J.D.; Ferron, M.; Karsenty, G.; Milne, E.M.; Millan, J.L.; Ahmed, S.F.; Farquharson, C.; Morton, N.M.; et al. Deficiency of the bone mineralization inhibitor NPP1 protects mice against obesity and diabetes. Dis. Model. Mech. 2014, 7, 1341–1350. [Google Scholar] [CrossRef] [Green Version]
- Roberts, F.L.; Rashdan, N.A.; Phadwal, K.; Markby, G.R.; Dillon, S.; Zoll, J.; Berger, J.; Milne, E.; Orriss, I.R.; Karsenty, G.; et al. Osteoblast-specific deficiency of ectonucleotide pyrophosphatase or phosphodiesterase-1 engenders insulin resistance in high-fat diet fed mice. J. Cell. Physiol. 2021, 236, 4614–4624. [Google Scholar] [CrossRef]
- Golub, E.E. Role of matrix vesicles in biomineralization. Biochim. Biophys. Acta Gen. Subj. 2009, 1790, 1592–1598. [Google Scholar] [CrossRef] [Green Version]
- Cui, L.; Rashdan, N.A.; Zhu, N.; Milne, E.M.; Ajuh, P.; Milne, G.; Helfrich, M.H.; Lim, K.; Prasad, S.; Lerman, D.A.; et al. End stage renal disease-induced hypercalcemia may promote aortic valve calcification via Annexin VI enrichment of valve interstitial cell derived-matrix vesicles. J. Cell. Physiol. 2017, 232, 2985–2995. [Google Scholar] [CrossRef] [Green Version]
- Hutcheson, J.D.; Goettsch, C.; Bertazzo, S.S.; Maldonado, N.; Ruiz, J.L.; Goh, W.; Yabusaki, K.; Faits, T.; Bouten, C.C.; Franck, G.; et al. Genesis and growth of extracellular-vesicle-derived microcalcification in atherosclerotic plaques. Nat. Mater. 2016, 15, 335–343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, N.X.; O’Neill, K.D.; Chen, X.; Moe, S.M. Annexin-Mediated Matrix Vesicle Calcification in Vascular Smooth Muscle Cells. J. Bone Miner. Res. 2008, 23, 1798–1805. [Google Scholar] [CrossRef] [Green Version]
- New, S.E.P.; Goettsch, C.; Aikawa, M.; Marchini, J.F.; Shibasaki, M.; Yabusaki, K.; Libby, P.; Shanahan, C.M.; Croce, K.; Aikawa, E. Macrophage-Derived Matrix Vesicles. Circ. Res. 2013, 113, 72–77. [Google Scholar] [CrossRef]
- Kapustin, A.N.; Chatrou, M.L.L.; Drozdov, I.; Zheng, Y.; Davidson, S.M.; Soong, D.; Furmanik, M.; Sanchis, P.; De Rosales, R.T.M.; Alvarez-Hernandez, D.; et al. Vascular Smooth Muscle Cell Calcification Is Mediated by Regulated Exosome Secretion. Circ. Res. 2015, 116, 1312–1323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goettsch, C.; Hutcheson, J.D.; Aikawa, E. MicroRNA in Cardiovascular Calcification. Circ. Res. 2013, 112, 1073–1084. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaturvedi, P.; Chen, N.X.; O’Neill, K.; McClintick, J.N.; Moe, S.M.; Janga, S.C. Differential miRNA Expression in Cells and Matrix Vesicles in Vascular Smooth Muscle Cells from Rats with Kidney Disease. PLoS ONE 2015, 10, e0131589. [Google Scholar] [CrossRef] [Green Version]
- Chen, N.X.; O’Neill, K.D.; Moe, S.M. Matrix vesicles induce calcification of recipient vascular smooth muscle cells through multiple signaling pathways. Kidney Int. 2018, 93, 343–354. [Google Scholar] [CrossRef] [Green Version]
- Tsuchiya, K.; Nakajima, S.; Hosojima, S.; Nguyen, D.T.; Hattori, T.; Le, T.M.; Hori, O.; Mahib, M.R.; Yamaguchi, Y.; Miura, M.; et al. Caspase-1 initiates apoptosis in the absence of gasdermin D. Nat. Commun. 2019, 10, 1–19. [Google Scholar] [CrossRef] [Green Version]
- Proudfoot, D.; Skepper, J.N.; Hegyi, L.; Bennett, M.R.; Shanahan, C.M.; Weissberg, P.L. Apoptosis Regulates Human Vascular Calcification In Vitro. Circ. Res. 2000, 87, 1055–1062. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Z.; Gordillo-Martinez, F.; Jiang, L.; He, P.; Hong, W.; Wei, X.; Staines, K.A.; Macrae, V.E.; Zhang, C.; Yu, D.; et al. Zinc ameliorates human aortic valve calcification through GPR39 mediated ERK1/2 signalling pathway. Cardiovasc. Res. 2021, 117, 820–835. [Google Scholar] [CrossRef]
- Kim, H.; Kim, H.-J.; Lee, K.; Kim, J.-M.; Kim, H.S.; Kim, J.-R.; Ha, C.-M.; Choi, Y.-K.; Lee, S.J.; Kim, J.-Y.; et al. α-Lipoic acid attenuates vascular calcification via reversal of mitochondrial function and restoration of Gas6/Axl/Akt survival pathway. J. Cell. Mol. Med. 2011, 16, 273–286. [Google Scholar] [CrossRef] [PubMed]
- El Husseini, D.; Boulanger, M.-C.; Fournier, D.; Mahmut, A.; Bossé, Y.; Pibarot, P.; Mathieu, P. High Expression of the Pi-Transporter SLC20A1/Pit1 in Calcific Aortic Valve Disease Promotes Mineralization through Regulation of Akt-1. PLoS ONE 2013, 8, e53393. [Google Scholar] [CrossRef]
- Chavkin, N.W.; Chia, J.J.; Crouthamel, M.H.; Giachelli, C.M. Phosphate uptake-independent signaling functions of the type III sodium-dependent phosphate transporter, PiT-1, in vascular smooth muscle cells. Exp. Cell Res. 2015, 333, 39–48. [Google Scholar] [CrossRef] [Green Version]
- Carracedo, M.; Artiach, G.; Arnardottir, H.; Bäck, M. The resolution of inflammation through omega-3 fatty acids in atherosclerosis, intimal hyperplasia, and vascular calcification. Semin. Immunopathol. 2019, 41, 757–766. [Google Scholar] [CrossRef] [Green Version]
- Gomel, M.A.; Lee, R.; Grande-Allen, K.J. Comparing the Role of Mechanical Forces in Vascular and Valvular Calcification Progression. Front. Cardiovasc. Med. 2019, 5, 197. [Google Scholar] [CrossRef]
- Simard, L.; Côté, N.; Dagenais, F.; Mathieu, P.; Couture, C.; Trahan, S.; Bossé, Y.; Mohammadi, S.; Pagé, S.; Joubert, P.; et al. Sex-Related Discordance Between Aortic Valve Calcification and Hemodynamic Severity of Aortic Stenosis. Circ. Res. 2017, 120, 681–691. [Google Scholar] [CrossRef]
- Nitsche, C.; Koschutnik, M.; Kammerlander, A.; Hengstenberg, C.; Mascherbauer, J. Gender-specific differences in valvular heart disease. Wien. Klin. Wochenschr. 2020, 132, 61–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stout, R.W. Atherosclerosis in males and females. Horm. Atheroscler. 1982, 97–111. [Google Scholar] [CrossRef]
- Pérez-López, F.R.; Larrad-Mur, L.; Kallen, A.; Chedraui, P.; Taylor, H.S. Review: Gender Differences in Cardiovascular Disease: Hormonal and Biochemical Influences. Reprod. Sci. 2010, 17, 511–531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lai, J.; Ge, Y.; Shao, Y.; Xuan, T.; Xia, S.; Li, M. Low serum testosterone level was associated with extensive coronary artery calcification in elderly male patients with stable coronary artery disease. Coron. Artery Dis. 2015, 26, 437–441. [Google Scholar] [CrossRef]
- Calderon-Margalit, R.; Siscovick, D.; Merkin, S.S.; Wang, E.; Daviglus, M.L.; Schreiner, P.J.; Sternfeld, B.; Williams, O.D.; Lewis, C.E.; Azziz, R.; et al. Prospective Association of Polycystic Ovary Syndrome With Coronary Artery Calcification and Carotid-Intima-Media Thickness. Arter. Thromb. Vasc. Biol. 2014, 34, 2688–2694. [Google Scholar] [CrossRef] [Green Version]
- Zhu, D.; Li, X.; Macrae, V.E.; Simoncini, T.; Fu, X. Extragonadal Effects of Follicle-Stimulating Hormone on Osteoporosis and Cardiovascular Disease in Women during Menopausal Transition. Trends Endocrinol. Metab. 2018, 29, 571–580. [Google Scholar] [CrossRef] [Green Version]
- Dalal, P.K.; Agarwal, M. Postmenopausal syndrome. Indian J. Psychiatry 2015, 57, 222–232. [Google Scholar] [CrossRef] [PubMed]
- Nakao, J.; Orimo, H.; Ooyama, T.; Shiraki, M. Low serum estradiol levels in subjects with arterial calcification. Atherosclerosis 1979, 34, 469–474. [Google Scholar] [CrossRef]
- Lopez-Pier, M.A.; Lipovka, Y.; Koppinger, M.P.; Harris, P.R.; Konhilas, J.P. The clinical impact of estrogen loss on cardio-vascular disease in menopausal females. Med. Res. Arch. 2018, 6, 1663. [Google Scholar] [PubMed]
- Hodis, H.N.; Mack, W.J.; Shoupe, D.; Azen, S.P.; Stanczyk, F.Z.; Hwang-Levine, J.; Budoff, M.J.; Henderson, V.W. Methods and baseline cardiovascular data from the Early versus Late Intervention Trial with Estradiol testing the menopausal hormone timing hypothesis. Menopause 2015, 22, 391–401. [Google Scholar] [CrossRef] [Green Version]
- Manson, J.E.; Allison, M.A.; Rossouw, J.E.; Carr, J.J.; Langer, R.D.; Hsia, J.; Kuller, L.H.; Cochrane, B.B.; Hunt, J.R.; Ludlam, S.E.; et al. Estrogen Therapy and Coronary-Artery Calcification. N. Engl. J. Med. 2007, 356, 2591–2602. [Google Scholar] [CrossRef]
- Osako, M.K.; Nakagami, H.; Koibuchi, N.; Shimizu, H.; Nakagami, F.; Koriyama, H.; Shimamura, M.; Miyake, T.; Rakugi, H.; Morishita, R. Estrogen Inhibits Vascular Calcification via Vascular RANKL System. Circ. Res. 2010, 107, 466–475. [Google Scholar] [CrossRef] [Green Version]
- Wu, X.; Zhao, Q.; Chen, Z.; Geng, Y.-J.; Zhang, W.; Zhou, Q.; Yang, W.; Liu, Q.; Liu, H. Estrogen inhibits vascular calcification in rats via hypoxia-induced factor-1α signaling. Vascular 2020, 28, 465–474. [Google Scholar] [CrossRef]
- Peng, Y.-Q.; Xiong, D.; Lin, X.; Cui, R.-R.; Xu, F.; Zhong, J.-Y.; Zhu, T.; Wu, F.; Mao, M.-Z.; Liao, X.-B.; et al. Oestrogen Inhibits Arterial Calcification by Promoting Autophagy. Sci. Rep. 2017, 7, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nanao-Hamai, M.; Son, B.-K.; Hashizume, T.; Ogawa, S.; Akishita, M. Protective effects of estrogen against vascular calcification via estrogen receptor α-dependent growth arrest-specific gene 6 transactivation. Biochem. Biophys. Res. Commun. 2016, 480, 429–435. [Google Scholar] [CrossRef]
- Balica, M.; Bostrom, K.I.; Shin, V.; Tillisch, K.; Demer, L.L. Calcifying Subpopulation of Bovine Aortic Smooth Muscle Cells Is Responsive to 17 & beta-Estradiol. Circulation 1997, 95, 1954–1960. [Google Scholar] [CrossRef]
- McRobb, L.S.; McGrath, K.C.; Tsatralis, T.; Liong, E.C.; Tan, J.T.; Hughes, G.; Handelsman, D.J.; Heather, A.K. Estrogen Receptor Control of Atherosclerotic Calcification and Smooth Muscle Cell Osteogenic Differentiation. Arter. Thromb. Vasc. Biol. 2017, 37, 1127–1137. [Google Scholar] [CrossRef] [Green Version]
- Harada, N.; Sasano, H.; Murakami, H.; Ohkuma, T.; Nagura, H.; Takagi, Y. Localized expression of aromatase in human vascular tissues. Circ. Res. 1999, 84, 1285–1291. [Google Scholar] [CrossRef]
- Park, B.-J.; Shim, J.-Y.; Lee, Y.-J.; Lee, J.-H.; Lee, H.-R. Inverse relationship between bioavailable testosterone and subclinical coronary artery calcification in non-obese Korean men. Asian J. Androl. 2012, 14, 612–615. [Google Scholar] [CrossRef] [Green Version]
- Travison, T.G.; O’Donnell, C.J.; Bhasin, S.; Massaro, J.M.; Hoffmann, U.; Vasan, R.S.; D’Agostino, R.B.; Basaria, S. Circulating Sex Steroids and Vascular Calcification in Community-Dwelling Men: The Framingham Heart Study. J. Clin. Endocrinol. Metab. 2016, 101, 2160–2167. [Google Scholar] [CrossRef] [Green Version]
- Subramanya, V.; Zhao, D.; Ouyang, P.; Ying, W.; Vaidya, D.; Ndumele, C.E.; Heckbert, S.R.; Budoff, M.J.; Post, W.S.; Michos, E.D. Association of endogenous sex hormone levels with coronary artery calcium progression among post-menopausal women in the Multi-Ethnic Study of Atherosclerosis (MESA). J. Cardiovasc. Comput. Tomogr. 2019, 13, 41–47. [Google Scholar] [CrossRef] [PubMed]
- McRobb, L.; Handelsman, D.J.; Heather, A.K. Androgen-Induced Progression of Arterial Calcification in Apolipoprotein E-Null Mice Is Uncoupled from Plaque Growth and Lipid Levels. Endocrinology 2009, 150, 841–848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, D.; Hadoke, P.W.F.; Wu, J.; Vesey, A.T.; Lerman, D.A.; Dweck, M.R.; Newby, D.E.; Smith, L.B.; Macrae, V.E. Ablation of the androgen receptor from vascular smooth muscle cells demonstrates a role for testosterone in vascular calcification. Sci. Rep. 2016, 6, 24807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Son, B.-K.; Akishita, M.; Iijima, K.; Ogawa, S.; Maemura, K.; Yu, J.; Takeyama, K.; Kato, S.; Eto, M.; Ouchi, Y. Androgen Receptor-dependent Transactivation of Growth Arrest-specific Gene 6 Mediates Inhibitory Effects of Testosterone on Vascular Calcification. J. Biol. Chem. 2010, 285, 7537–7544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akishita, M.; Hashimoto, M.; Ohike, Y.; Ogawa, S.; Iijima, K.; Eto, M.; Ouchi, Y. Low testosterone level as a predictor of cardiovascular events in Japanese men with coronary risk factors. Atherosclerosis 2010, 210, 232–236. [Google Scholar] [CrossRef]
- Achar, S.; Rostamian, A.; Narayan, S.M. Cardiac and Metabolic Effects of Anabolic-Androgenic Steroid Abuse on Lipids, Blood Pressure, Left Ventricular Dimensions, and Rhythm. Am. J. Cardiol. 2010, 106, 893–901. [Google Scholar] [CrossRef] [Green Version]
- Moisan, M.-P.; Castanon, N. Emerging Role of Corticosteroid-Binding Globulin in Glucocorticoid-Driven Metabolic Disorders. Front. Endocrinol. 2016, 7, 160. [Google Scholar] [CrossRef] [Green Version]
- Kuiper, G.G.; Enmark, E.; Pelto-Huikko, M.; Nilsson, S.; Gustafsson, J.A. Cloning of a novel receptor expressed in rat prostate and ovary. Proc. Natl. Acad. Sci. USA 1996, 93, 5925–5930. [Google Scholar] [CrossRef] [Green Version]
- Filardo, E.J.; Quinn, J.A.; Frackelton, A.R.; Bland, K.I. Estrogen Action Via the G Protein-Coupled Receptor, GPR30: Stimulation of Adenylyl Cyclase and cAMP-Mediated Attenuation of the Epidermal Growth Factor Receptor-to-MAPK Signaling Axis. Mol. Endocrinol. 2002, 16, 70–84. [Google Scholar] [CrossRef]
- Murphy, E. Estrogen Signaling and Cardiovascular Disease. Circ. Res. 2011, 109, 687–696. [Google Scholar] [CrossRef] [Green Version]
- Jakacka, M.; Ito, M.; Weiss, J.; Chien, P.-Y.; Gehm, B.D.; Jameson, J.L. Estrogen Receptor Binding to DNA Is Not Required for Its Activity through the Nonclassical AP1 Pathway. J. Biol. Chem. 2001, 276, 13615–13621. [Google Scholar] [CrossRef] [Green Version]
- Burns, K.A.; Li, Y.; Arao, Y.; Petrovich, R.M.; Korach, K.S. Selective Mutations in Estrogen Receptor α D-domain Alters Nuclear Translocation and Non-estrogen Response Element Gene Regulatory Mechanisms. J. Biol. Chem. 2011, 286, 12640–12649. [Google Scholar] [CrossRef] [Green Version]
- Curtis, S.W.; Washburn, T.F.; Sewall, C.; DiAugustine, R.P.; Lindzey, J.; Couse, J.F.; Korach, K.S. Physiological coupling of growth factor and steroid receptor signaling pathways: Estrogen receptor knockout mice lack estrogen-like response to epidermal growth factor. Proc. Natl. Acad. Sci. USA 1996, 93, 12626–12630. [Google Scholar] [CrossRef] [Green Version]
- Simoncini, T.; Hafezi-Moghadam, A.; Brazil, D.P.; Ley, K.; Chin, W.W.; Liao, J.K. Interaction of oestrogen receptor with the regulatory subunit of phosphatidylinositol-3-OH kinase. Nat. Cell Biol. 2000, 407, 538–541. [Google Scholar] [CrossRef]
- Mendelsohn, M.E.; Karas, R.H. The Protective Effects of Estrogen on the Cardiovascular System. N. Engl. J. Med. 1999, 340, 1801–1811. [Google Scholar] [CrossRef]
- Ueda, K.; Adachi, Y.; Liu, P.; Fukuma, N.; Takimoto, E. Regulatory Actions of Estrogen Receptor Signaling in the Cardiovascular System. Front. Endocrinol. 2020, 10, 909. [Google Scholar] [CrossRef]
- Teoh, J.-P.; Li, X.; Simoncini, T.; Zhu, D.; Fu, X. Estrogen-Mediated Gaseous Signaling Molecules in Cardiovascular Disease. Trends Endocrinol. Metab. 2020, 31, 773–784. [Google Scholar] [CrossRef]
- Kloner, R.A.; Carson, C.; Dobs, A.; Kopecky, S.; Mohler, E.R. Testosterone and Cardiovascular Disease. J. Am. Coll. Cardiol. 2016, 67, 545–557. [Google Scholar] [CrossRef]
- Tostes, R.C.; Carneiro, F.S.; Carvalho, M.H.C.; Reckelhoff, J.F. Reactive oxygen species: Players in the cardiovascular effects of testosterone. Am. J. Physiol. Integr. Comp. Physiol. 2016, 310, R1–R14. [Google Scholar] [CrossRef] [Green Version]
- Thomas, P. Membrane Androgen Receptors Unrelated to Nuclear Steroid Receptors. Endocrinology 2019, 160, 772–781. [Google Scholar] [CrossRef] [PubMed]
- Gonzales, R.J.; Ansar, S.; Duckles, S.P.; Krause, D.N. Androgenic/Estrogenic Balance in the Male Rat Cerebral Circulation: Metabolic Enzymes and Sex Steroid Receptors. Br. J. Pharmacol. 2007, 27, 1841–1852. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brinkmann, A.O. Molecular Mechanisms of Androgen Action—A Historical Perspective. Adv. Struct. Saf. Stud. 2011, 776, 3–24. [Google Scholar] [CrossRef]
- Fujimoto, R.; Morimoto, I.; Morita, E.; Sugimoto, H.; Ito, Y.; Eto, S. Androgen receptors, 5 alpha-reductase activity and androgen-dependent proliferation of vascular smooth muscle cells. J. Steroid Biochem. Mol. Biol. 1994, 50, 169–174. [Google Scholar] [CrossRef]
- Mukherjee, T.K.; Dinh, H.; Chaudhuri, G.; Nathan, L. Testosterone attenuates expression of vascular cell adhesion molecule-1 by conversion to estradiol by aromatase in endothelial cells: Implications in atherosclerosis. Proc. Natl. Acad. Sci. USA 2002, 99, 4055–4060. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beery, A.K.; Zucker, I. Sex bias in neuroscience and biomedical research. Neurosci. Biobehav. Rev. 2011, 35, 565–572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beery, A.K. Inclusion of females does not increase variability in rodent research studies. Curr. Opin. Behav. Sci. 2018, 23, 143–149. [Google Scholar] [CrossRef] [PubMed]
- Becher, O.J.; Holland, E.C. Genetically Engineered Models Have Advantages over Xenografts for Preclinical Studies. Cancer Res. 2006, 66, 3355–3359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sider, K.L.; Blaser, M.C.; Simmons, C.A. Animal Models of Calcific Aortic Valve Disease. Int. J. Inflamm. 2011, 2011, 1–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rattazzi, M.; Bennett, B.J.; Bea, F.; Kirk, E.A.; Ricks, J.L.; Speer, M.; Schwartz, S.M.; Giachelli, C.M.; Rosenfeld, M.E. Calcification of Advanced Atherosclerotic Lesions in the Innominate Arteries of ApoE-Deficient Mice. Arter. Thromb. Vasc. Biol. 2005, 25, 1420–1425. [Google Scholar] [CrossRef] [Green Version]
- Awan, Z.; Denis, M.; Bailey, D.; Giaid, A.; Prat, A.; Goltzman, D.; Seidah, N.G.; Genest, J. The LDLR deficient mouse as a model for aortic calcification and quantification by micro-computed tomography. Atherosclerosis 2011, 219, 455–462. [Google Scholar] [CrossRef]
- Watanabe, R.; Fujita, N.; Sato, Y.; Kobayashi, T.; Morita, M.; Oike, T.; Miyamoto, K.; Kuro-O, M.; Michigami, T.; Fukumoto, S.; et al. Enpp1 is an anti-aging factor that regulates Klotho under phosphate overload conditions. Sci. Rep. 2017, 7, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Kauffenstein, G.; Pizard, A.; Le Corre, Y.; Vessières, E.; Grimaud, L.; Toutain, B.; Labat, C.; Mauras, Y.; Gorgels, T.G.; Bergen, A.A.; et al. Disseminated arterial calcification and enhanced myogenic response are associated with abcc6 deficiency in a mouse model of pseudoxanthoma elasticum. Arter. Thromb. Vasc. Biol. 2014, 34, 1045–1056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herrmann, J.; Babic, M.; Tölle, M.; Van Der Giet, M.; Schuchardt, M. Research Models for Studying Vascular Calcification. Int. J. Mol. Sci. 2020, 21, 2204. [Google Scholar] [CrossRef] [Green Version]
- Scatena, M.; Jackson, M.F.; Speer, M.Y.; Leaf, E.M.; Wallingford, M.C.; Giachelli, C.M. Increased Calcific Aortic Valve Disease in response to a diabetogenic, procalcific diet in the LDLr −/− ApoB 100/100 mouse model. Cardiovasc. Pathol. 2018, 34, 28–37. [Google Scholar] [CrossRef]
- Liao, R.; Wang, L.; Li, J.; Sun, S.; Xiong, Y.; Li, Y.; Han, M.; Jiang, H.; Anil, M.; Su, B. Vascular calcification is associated with Wnt-signaling pathway and blood pressure variability in chronic kidney disease rats. Nephrology 2019, 25, 264–272. [Google Scholar] [CrossRef]
- Price, P.A.; Faus, S.A.; Williamson, M.K. Warfarin Causes Rapid Calcification of the Elastic Lamellae in Rat Arteries and Heart Valves. Arter. Thromb. Vasc. Biol. 1998, 18, 1400–1407. [Google Scholar] [CrossRef] [Green Version]
- Shobeiri, N.; Adams, M.; Holden, R. Vascular Calcification in Animal Models of CKD: A Review. Am. J. Nephrol. 2010, 31, 471–481. [Google Scholar] [CrossRef] [PubMed]
- Karas, R.H.; Schulten, H.; Pare, G.; Aronovitz, M.J.; Ohlsson, C.; Gustafsson, J.-A.; Mendelsohn, M.E. Effects of Estrogen on the Vascular Injury Response in Estrogen Receptor α,β (Double) Knockout Mice. Circ. Res. 2001, 89, 534–539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.; Kuhn, G.; Schirmer, M.; Müller, R.; Ruffoni, D. Impaired bone formation in ovariectomized mice reduces implant integration as indicated by longitudinal in vivo micro-computed tomography. PLoS ONE 2017, 12, e0184835. [Google Scholar] [CrossRef] [PubMed]
- Marek, I.; Canu, M.; Cordasic, N.; Rauh, M.; Volkert, G.; Fahlbusch, F.B.; Rascher, W.; Hilgers, K.F.; Hartner, A.; Menendez-Castro, C. Sex differences in the development of vascular and renal lesions in mice with a simultaneous deficiency of Apoe and the integrin chain Itga8. Biol. Sex Differ. 2017, 8, 1–13. [Google Scholar] [CrossRef]
- Ferencik, M. Insights Into Coronary Plaque Microstructure Differences Between Women and Men. Circ. Cardiovasc. Imaging 2016, 9, 005343. [Google Scholar] [CrossRef] [Green Version]
- Tang, Y.; Shah, T.A.; Yurkow, E.J.; Rogers, M.B. Post-transcriptional regulation of aortic calcification in KLOTHO deficient mice: Impact of miR-145 and miR-378. Biorivex. 2020. pre-print. [Google Scholar] [CrossRef]
- Shah, T.A.; Tang, Y.; Yurkow, E.J.; Rogers, M. B Post-Transcriptional Bone Morphogenetic Protein 2 (BMP2) Gene Regulation in Aorta. Biorivex. 2019. pre-print. [Google Scholar] [CrossRef]
- Pereira, T.M.; Nogueira, B.V.; Lima, L.C.; Porto, M.L.; Arruda, J.A.; Vasquez, E.C.; Meyrelles, S.S. Cardiac and vascular changes in elderly atherosclerotic mice: The influence of gender. Lipids Health Dis. 2010, 9, 87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiao, J.H.; Xie, P.Z.; Fishbein, M.C.; Kreuzer, J.; Drake, T.A.; Demer, L.L.; Lusis, A.J. Pathology of atheromatous lesions in inbred and genetically engineered mice. Genetic determination of arterial calcification. Arterioscler Thromb. 1994, 14, 1480–1497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, S.S.; Martin, L.J.; Schadt, E.E.; Meng, H.; Wang, X.; Zhao, W.; Ingram-Drake, L.; Nebohacova, M.; Mehrabian, M.; Drake, T.A.; et al. Disruption of the aortic elastic lamina and medial calcification share genetic determinants in mice. Circ Cardiovasc Genet. 2009, 2, 573–582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakamura, T.; Akishita, M.; Kozaki, K.; Toba, K.; Orimo, H.; Ouchi, Y. Influence of sex and estrogen on vitamin D-induced arterial calcification in rats. Geriatr Gerontol Int. 2002, 3, 145–149. [Google Scholar] [CrossRef]
- Kohn, M.H.; Price, R.E.; Pelz, H.J. A cardiovascular phenotype in warfarin-resistant Vkorc1 mutant rats. Artery Res. 2008, 2, 138–147. [Google Scholar] [CrossRef] [Green Version]
- Ng, K.; Hildreth, C.M.; Avolio, A.P.; Phillips, J.K. Angiotensin-converting enzyme inhibitor limits pulse-wave velocity and aortic calcification in a rat model of cystic renal disease. Am J Physiol Renal Physiol. 2011, 301, F959–F966. [Google Scholar] [CrossRef] [Green Version]
- Dixon, J.A.; Spinale, F.G. Large Animal Models of Heart Failure. Circ. Heart Fail. 2009, 2, 262–271. [Google Scholar] [CrossRef] [Green Version]
- Warren, H.S.; Fitting, C.; Hoff, E.; Adib-Conquy, M.; Beasley-Topliffe, L.; Tesini, B.; Liang, X.; Valentine, C.; Hellman, J.; Hayden, D.; et al. Resilience to Bacterial Infection: Difference between Species Could Be Due to Proteins in Serum. J. Infect. Dis. 2010, 201, 223–232. [Google Scholar] [CrossRef] [Green Version]
- Cimini, M.; Boughner, D.R.; Ronald, J.A.; Aldington, L.; Rogers, K.A. Development of aortic valve sclerosis in a rabbit model of atherosclerosis: An immunohistochemical and histological study. J. Heart Valve Dis. 2005, 14, 365–375. [Google Scholar]
- Rajamannan, N.M.; Subramaniam, M.; Caira, F.; Stock, S.R.; Spelsberg, T.C. Atorvastatin inhibits hypercholesterolemia-induced calcification in the aortic valves via the Lrp5 receptor pathway. Circulation 2005, 112, I229–I234. [Google Scholar]
- Marechaux, S.; Corseaux, D.; Vincentelli, A.; Richardson, M.; Ung, A.; Susen, S.; Zawadzki, C.; Beregi, J.-P.; Ezekowitz, M.D.; Jude, B.; et al. Identification of tissue factor in experimental aortic valve sclerosis. Cardiovasc. Pathol. 2009, 18, 67–76. [Google Scholar] [CrossRef] [PubMed]
- Choi, B.; Lee, S.; Song, J.-K.; Kim, S.-M.; Lee, E.-J.; Lee, S.R.; Kim, D.-H.; Jang, J.Y.; Kang, S.-W.; Lee, K.-U.; et al. Dipeptidyl Peptidase-4 Induces Aortic Valve Calcification by Inhibiting Insulin-Like Growth Factor-1 Signaling in Valvular Interstitial Cells. Circulation 2017, 135, 1935–1950. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Nie, M.; Li, J.; Xu, Z.; Zhang, M.; Yan, Y.; Feng, T.; Zhao, X.; Zhao, Q. Effect of pioglitazone on inflammation and calcification in atherosclerotic rabbits. Herz 2017, 43, 733–740. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Waqar, A.B.; Nishijima, K.; Ning, B.; Kitajima, S.; Matsuhisa, F.; Chen, L.; Liu, E.; Koike, T.; Yu, Y.; et al. Macrophage-derived MMP-9 enhances the progression of atherosclerotic lesions and vascular calcification in transgenic rabbits. J. Cell. Mol. Med. 2020, 24, 4261–4274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beute, T.J.; Goehler, M.; Parker, J.; Boeve, T.; Heiser, J.; Murphy, E.; Timek, T.; Willekes, C.L. Long-Term Outcomes of Mosaic Versus Perimount Mitral Replacements: 17-Year Follow-Up of 940 Implants. Ann. Thorac. Surg. 2020, 110, 508–515. [Google Scholar] [CrossRef] [PubMed]
- Badin, J.K.; Progar, V.; Pareddy, A.; Cagle, J.; Alloosh, M.; Sturek, M. Effect of Age on Diabetogenicity of Alloxan in Ossabaw Miniature Swine. Comp. Med. 2019, 69, 114–122. [Google Scholar] [CrossRef] [PubMed]
- Van Ditzhuijzen, N.S.; Heuvel, M.V.D.; Nieman, K.; Mulder, M.T.; Zijlstra, F.; Duncker, D.J.; Van Beusekom, H.M.; Regar, E.; Sorop, O.; Rossi, A.; et al. Serial Coronary Imaging of Early Atherosclerosis Development in Fast-Food-Fed Diabetic and Nondiabetic Swine. JACC Basic Transl. Sci. 2016, 1, 449–460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sider, K.L.; Zhu, C.; Kwong, A.V.; Mirzaei, Z.; De Langé, C.F.; Simmons, C.A. Evaluation of a porcine model of early aortic valve sclerosis. Cardiovasc. Pathol. 2014, 23, 289–297. [Google Scholar] [CrossRef]
- Simmons, C.A.; Grant, G.R.; Manduchi, E.; Davies, P.F. Spatial Heterogeneity of Endothelial Phenotypes Correlates With Side-Specific Vulnerability to Calcification in Normal Porcine Aortic Valves. Circ. Res. 2005, 96, 792–799. [Google Scholar] [CrossRef] [Green Version]
- McCoy, C.M.; Nicholas, D.Q.; Masters, K.S. Sex-Related Differences in Gene Expression by Porcine Aortic Valvular Interstitial Cells. PLoS ONE 2012, 7, e39980. [Google Scholar] [CrossRef]
- Masjedi, S.; Lei, Y.; Patel, J.; Ferdous, Z. Sex-related differences in matrix remodeling and early osteogenic markers in aortic valvular interstitial cells. Heart Vessel. 2016, 32, 217–228. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Model | Method of Calcification Induction | Type of Calcification | Sex Differences in Calcification | Treatment | Ref |
|---|---|---|---|---|---|
| Mouse | |||||
| ApoE−/− | Aged to 36 weeks | Vascular and valvular | ↑ calcification in females (aortic sinus) | 17β-estradiol | [93] |
| ApoE−/− | Crossed with Itga8−/− | Vascular | ↑ calcification in females | [93] | |
| Klotho−/− | Vascular | None | MicroRNA-145 and microRNA-378a | [138] | |
| Klotho−/− | Vascular | None | None | [139] | |
| ApoE−/− | Vascular, aortic sinus | ↑ calcification in females | Testosterone and DHT | [98] | |
| ApoE−/− | 18 months | Vascular | More vascular calcification in males | [140] | |
| ApoE−/− | Uraemia | Vascular | ↑ calcification in females | [141] | |
| C57BL/6J | Vascular | None | [141] | ||
| ApoA-II | Vascular | None | [141] | ||
| ApoE−/− | Vascular | ↑ calcification in females | [141] | ||
| ApoE−/− | Hyperlipidaemic diet | Vascular | ↑ calcification in females (medial arteries) | [142] | |
| Rat | |||||
| Fisher | 1α-Hydroxyvitamin D3 | Vascular | ↑ calcification in males | [143] | |
| Wistar | Vascular | ↑ calcification in males | [144] | ||
| Lewis Polycystic Kidney | Vascular | None | Perindopril | [145] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Woodward, H.J.; Zhu, D.; Hadoke, P.W.F.; MacRae, V.E. Regulatory Role of Sex Hormones in Cardiovascular Calcification. Int. J. Mol. Sci. 2021, 22, 4620. https://doi.org/10.3390/ijms22094620
Woodward HJ, Zhu D, Hadoke PWF, MacRae VE. Regulatory Role of Sex Hormones in Cardiovascular Calcification. International Journal of Molecular Sciences. 2021; 22(9):4620. https://doi.org/10.3390/ijms22094620
Chicago/Turabian StyleWoodward, Holly J., Dongxing Zhu, Patrick W. F. Hadoke, and Victoria E. MacRae. 2021. "Regulatory Role of Sex Hormones in Cardiovascular Calcification" International Journal of Molecular Sciences 22, no. 9: 4620. https://doi.org/10.3390/ijms22094620
APA StyleWoodward, H. J., Zhu, D., Hadoke, P. W. F., & MacRae, V. E. (2021). Regulatory Role of Sex Hormones in Cardiovascular Calcification. International Journal of Molecular Sciences, 22(9), 4620. https://doi.org/10.3390/ijms22094620