
Mitochondria Turnover and Lysosomal Function in Hematopoietic Stem Cell Metabolism



{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Glycolysis and Oxidative Phosphorylation (OXPHOS) during Hematopoietic Differentiation
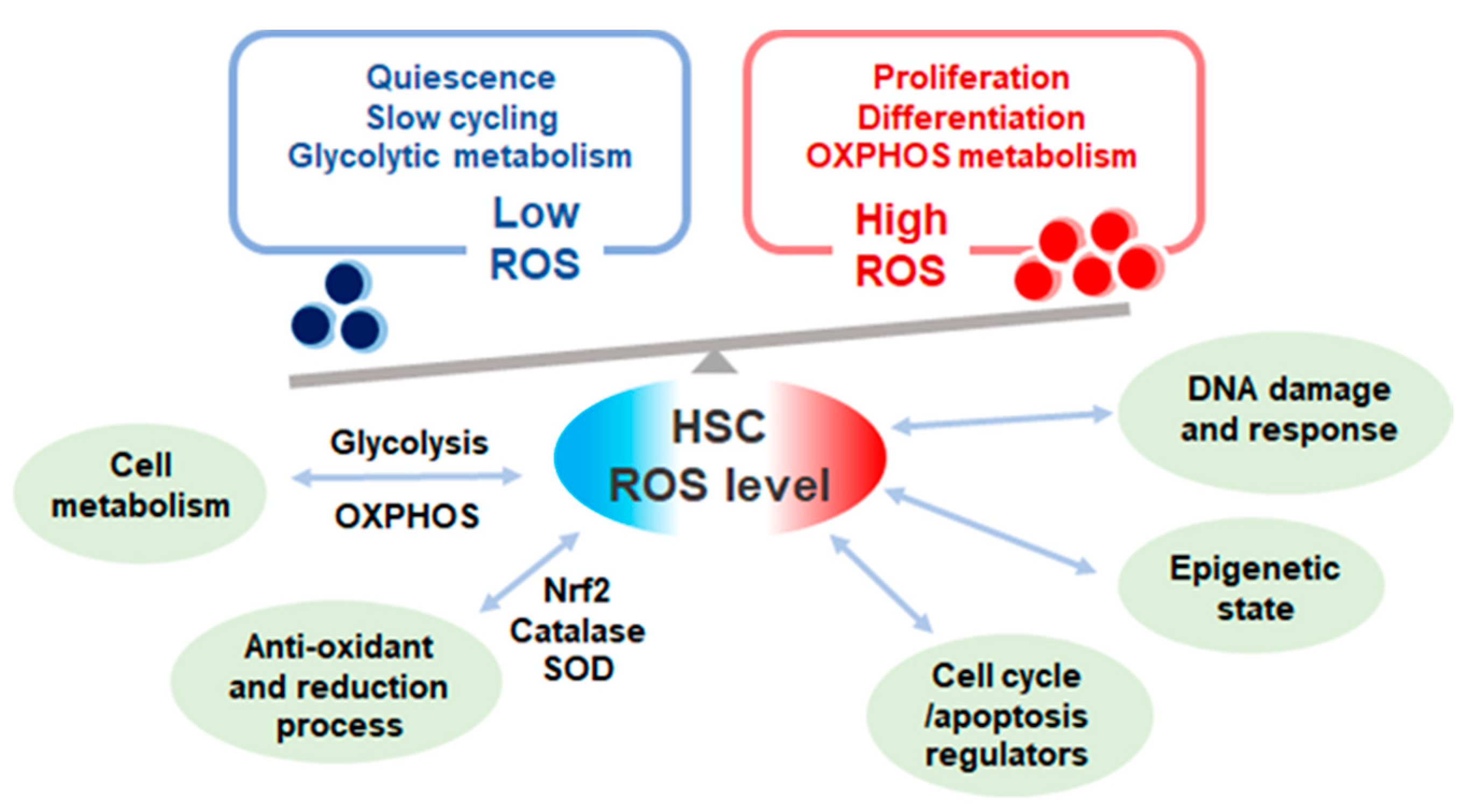
3. Reactive Oxygen Species (ROS) Production in Hematopoietic Stem Cells (HSCs)
4. Reduction of ROS through Redox Regulation in HSCs
5. The Effect of ROS on HSC Fate
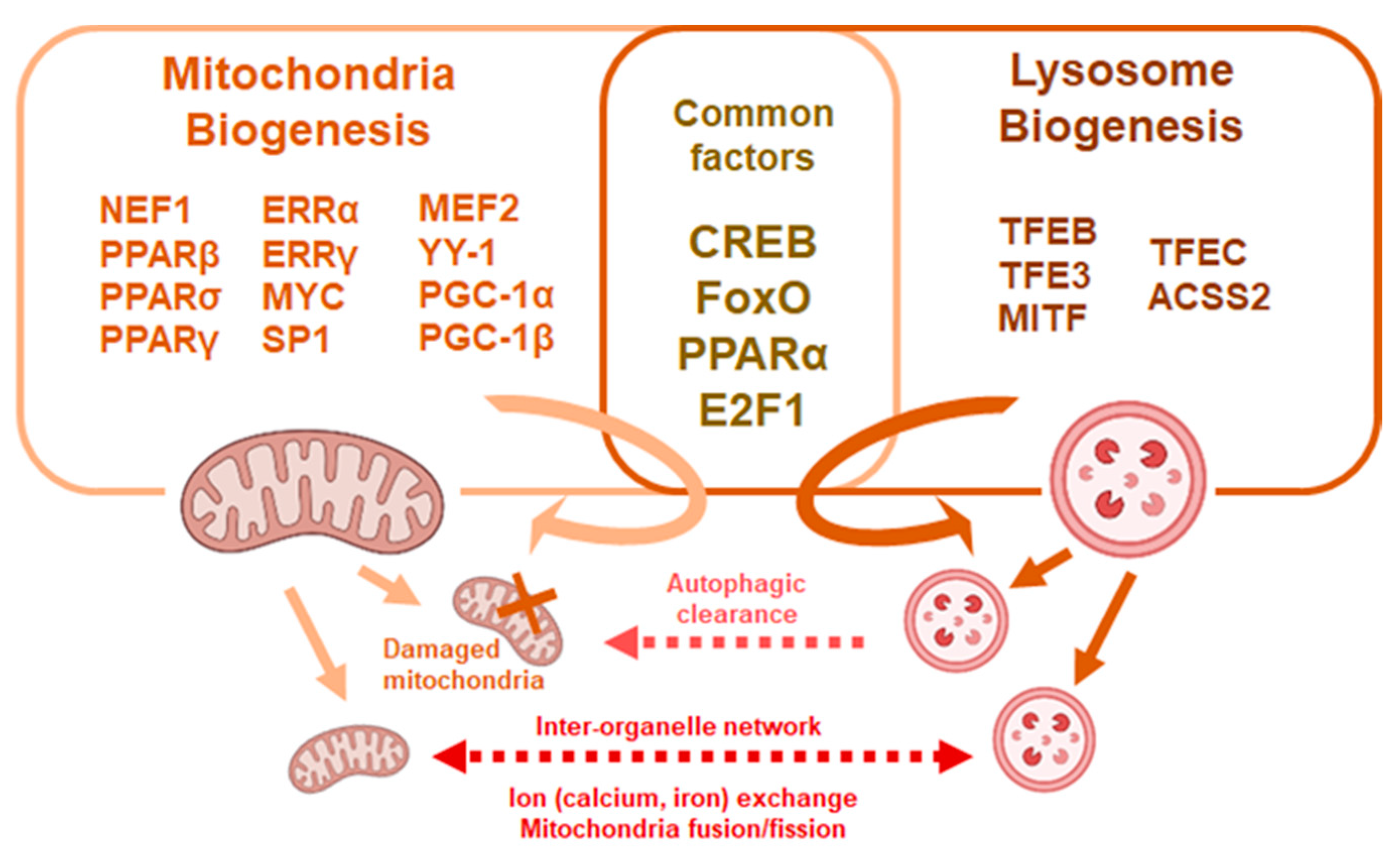
6. Mitochondria Volume and Turnover in HSCs
7. The Role of Lysosome and Mitochondria Network in HSCs
8. Regulation of Cellular Metabolism through Folliculin Signal
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Haas, S.; Trumpp, A.; Milsom, M.D. Causes and Consequences of Hematopoietic Stem Cell Heterogeneity. Cell Stem Cell 2018, 22, 627–638. [Google Scholar] [CrossRef] [Green Version]
- Ema, H.; Morita, Y.; Suda, T. Heterogeneity and hierarchy of hematopoietic stem cells. Exp. Hematol. 2014, 42, 74–82.e2. [Google Scholar] [CrossRef] [PubMed]
- Nakamura-Ishizu, A.; Takizawa, H.; Suda, T. The analysis, roles and regulation of quiescence in hematopoietic stem cells. Development 2014, 141, 4656–4666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spencer, J.A.; Ferraro, F.; Roussakis, E.; Klein, A.; Wu, J.; Runnels, J.M.; Zaher, W.; Mortensen, L.J.; Alt, C.; Turcotte, R.; et al. Direct measurement of local oxygen concentration in the bone marrow of live animals. Nature 2014, 508, 269–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mattes, K.; Vellenga, E.; Schepers, H. Differential redox-regulation and mitochondrial dynamics in normal and leukemic hematopoietic stem cells: A potential window for leukemia therapy. Crit. Rev. Oncol. 2019, 144, 102814. [Google Scholar] [CrossRef] [PubMed]
- Suda, T.; Takubo, K.; Semenza, G.L. Metabolic Regulation of Hematopoietic Stem Cells in the Hypoxic Niche. Cell Stem Cell 2011, 9, 298–310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kubota, Y.; Takubo, K.; Suda, T. Bone marrow long label-retaining cells reside in the sinusoidal hypoxic niche. Biochem. Biophys. Res. Commun. 2008, 366, 335–339. [Google Scholar] [CrossRef]
- Takubo, K.; Goda, N.; Yamada, W.; Iriuchishima, H.; Ikeda, E.; Kubota, Y.; Shima, H.; Johnson, R.S.; Hirao, A.; Suematsu, M.; et al. Regulation of the HIF-1α Level Is Essential for Hematopoietic Stem Cells. Cell Stem Cell 2010, 7, 391–402. [Google Scholar] [CrossRef] [Green Version]
- Simsek, T.; Kocabas, F.; Zheng, J.; DeBerardinis, R.J.; Mahmoud, A.I.; Olson, E.N.; Schneider, J.W.; Zhang, C.C.; Sadek, H.A. The Distinct Metabolic Profile of Hematopoietic Stem Cells Reflects Their Location in a Hypoxic Niche. Cell Stem Cell 2010, 7, 380–390. [Google Scholar] [CrossRef] [Green Version]
- Kocabas, F.; Zheng, J.; Thet, S.; Copeland, N.G.; Jenkins, N.A.; DeBerardinis, R.J.; Zhang, C.; Sadek, H.A. Meis1 regulates the metabolic phenotype and oxidant defense of hematopoietic stem cells. Blood 2012, 120, 4963–4972. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, H.; Morikawa, T.; Okinaga, A.; Hamano, F.; Hashidate-Yoshida, T.; Watanuki, S.; Hishikawa, D.; Shindou, H.; Arai, F.; Kabe, Y.; et al. Environmental Optimization Enables Maintenance of Quiescent Hematopoietic Stem Cells Ex Vivo. Cell Rep. 2019, 28, 145–158.e9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilkinson, A.C.; Ishida, R.; Kikuchi, M.; Sudo, K.; Morita, M.; Crisostomo, R.V.; Yamamoto, R.; Loh, K.M.; Nakamura, Y.; Watanabe, M.; et al. Long-term ex vivo haematopoietic-stem-cell expansion allows nonconditioned transplantation. Nature 2019, 571, 117–121. [Google Scholar] [CrossRef] [PubMed]
- Unwin, R.D.; Smith, D.L.; Blinco, D.; Wilson, C.L.; Miller, C.J.; Evans, C.A.; Jaworska, E.; Baldwin, S.A.; Barnes, K.; Pierce, A.; et al. Quantitative proteomics reveals posttranslational control as a regulatory factor in primary hematopoietic stem cells. Blood 2006, 107, 4687–4694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takubo, K.; Nagamatsu, G.; Kobayashi, C.I.; Nakamura-Ishizu, A.; Kobayashi, H.; Ikeda, E.; Goda, N.; Rahimi, Y.; Johnson, R.S.; Soga, T.; et al. Regulation of Glycolysis by Pdk Functions as a Metabolic Checkpoint for Cell Cycle Quiescence in Hematopoietic Stem Cells. Cell Stem Cell 2013, 12, 49–61. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.P.; Zhou, L.S.; Zhao, Y.Z.; Wang, S.W.; Chen, L.L.; Liu, L.X.; Ling, Z.Q.; Hu, F.J.; Sun, Y.P.; Zhang, J.Y.; et al. Regulation of G6PD acetylation by SIRT2 and KAT9 modulates NADPH homeostasis and cell survival during oxidative stress. EMBO J. 2014, 33, 1304–1320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baldwin, J.E.; Krebs, H. The evolution of metabolic cycles. Nature 1981, 291, 381–382. [Google Scholar] [CrossRef]
- Spinelli, J.B.; Haigis, M.C. The multifaceted contributions of mitochondria to cellular metabolism. Nat. Cell Biol. 2018, 20, 745–754. [Google Scholar] [CrossRef]
- Yu, W.-M.; Liu, X.; Shen, J.; Jovanovic, O.; Pohl, E.E.; Gerson, S.L.; Finkel, T.; Broxmeyer, H.E.; Qu, C.-K. Metabolic Regulation by the Mitochondrial Phosphatase PTPMT1 Is Required for Hematopoietic Stem Cell Differentiation. Cell Stem Cell 2013, 12, 62–74. [Google Scholar] [CrossRef] [Green Version]
- Ansó, E.; Weinberg, S.E.; Diebold, L.P.; Thompson, B.J.; Malinge, S.; Schumacker, P.T.; Liu, X.; Zhang, Y.; Shao, Z.; Steadman, M.; et al. The mitochondrial respiratory chain is essential for haematopoietic stem cell function. Nat. Cell Biol. 2017, 19, 614–625. [Google Scholar] [CrossRef]
- Norddahl, G.L.; Pronk, C.J.; Wahlestedt, M.; Sten, G.; Nygren, J.M.; Ugale, A.; Sigvardsson, M.; Bryder, D. Accumulating Mitochondrial DNA Mutations Drive Premature Hematopoietic Aging Phenotypes Distinct from Physiological Stem Cell Aging. Cell Stem Cell 2011, 8, 499–510. [Google Scholar] [CrossRef] [Green Version]
- Tan, D.Q.; Suda, T. Reactive Oxygen Species and Mitochondrial Homeostasis as Regulators of Stem Cell Fate and Function. Antioxid. Redox Signal. 2018, 29, 149–168. [Google Scholar] [CrossRef]
- Sommerkamp, P.; Altamura, S.; Renders, S.; Narr, A.; Ladel, L.; Zeisberger, P.; Eiben, P.L.; Fawaz, M.; Rieger, M.A.; Cabezas-Wallscheid, N.; et al. Differential Alternative Polyadenylation Landscapes Mediate Hematopoietic Stem Cell Activation and Regulate Glutamine Metabolism. Cell Stem Cell 2020, 26, 722–738. [Google Scholar] [CrossRef] [PubMed]
- Oburoglu, L.; Tardito, S.; Fritz, V.; de Barros, S.C.; Merida, P.; Craveiro, M.; Mamede, J.; Cretenet, G.; Mongellaz, C.; An, X.; et al. Glucose and Glutamine Metabolism Regulate Human Hematopoietic Stem Cell Lineage Specification. Cell Stem Cell 2014, 15, 169–184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, S.K.; Singh, S.; Gadomski, S.; Sun, L.; Pfannenstein, A.; Magidson, V.; Chen, X.; Kozlov, S.; Tessarollo, L.; Klarmann, K.D.; et al. Id1 Ablation Protects Hematopoietic Stem Cells from Stress-Induced Exhaustion and Aging. Cell Stem Cell 2018, 23, 252–265.e8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tahara, E.B.; Navarete, F.D.; Kowaltowski, A.J. Tissue-, substrate-, and site-specific characteristics of mitochondrial reactive oxygen species generation. Free Radic. Biol. Med. 2009, 46, 1283–1297. [Google Scholar] [CrossRef] [PubMed]
- Ito, K.; Hirao, A.; Arai, F.; Matsuoka, S.; Takubo, K.; Hamaguchi, I.; Nomiyama, K.; Hosokawa, K.; Sakurada, K.; Nakagata, N.; et al. Regulation of oxidative stress by ATM is required for self-renewal of haematopoietic stem cells. Nature 2004, 431, 997–1002. [Google Scholar] [CrossRef] [PubMed]
- Ito, K.; Hirao, A.; Arai, F.; Takubo, K.; Matsuoka, S.; Miyamoto, K.; Ohmura, M.; Naka, K.; Hosokawa, K.; Ikeda, Y.; et al. Reactive oxygen species act through p38 MAPK to limit the lifespan of hematopoietic stem cells. Nat. Med. 2006, 12, 446–451. [Google Scholar] [CrossRef] [PubMed]
- Lagadinou, E.D.; Sach, A.; Callahan, K.; Rossi, R.M.; Neering, S.J.; Minhajuddin, M.; Ashton, J.M.; Pei, S.; Grose, V.; O’Dwyer, K.M.; et al. BCL-2 Inhibition Targets Oxidative Phosphorylation and Selectively Eradicates Quiescent Human Leukemia Stem Cells. Cell Stem Cell 2013, 12, 329–341. [Google Scholar] [CrossRef] [Green Version]
- Pei, S.; Minhajuddin, M.; Adane, B.; Khan, N.; Stevens, B.M.; Mack, S.C.; Lai, S.; Rich, J.N.; Inguva, A.; Shannon, K.M.; et al. AMPK/FIS1-Mediated Mitophagy Is Required for Self-Renewal of Human AML Stem Cells. Cell Stem Cell 2018, 23, 86–100.e6. [Google Scholar] [CrossRef] [Green Version]
- Wu, Z.; Puigserver, P.; Andersson, U.; Zhang, C.; Adelmant, G.; Mootha, V.; Troy, A.; Cinti, S.; Lowell, B.; Scarpulla, R.C.; et al. Mechanisms Controlling Mitochondrial Biogenesis and Respiration through the Thermogenic Coactivator PGC-1. Cell 1999, 98, 115–124. [Google Scholar] [CrossRef] [Green Version]
- Maryanovich, M.; Oberkovitz, G.; Niv, H.; Vorobiyov, L.; Zaltsman, Y.; Brenner, O.; Lapidot, T.; Jung, S.; Gross, A. The ATM–BID pathway regulates quiescence and survival of haematopoietic stem cells. Nat. Cell Biol. 2012, 14, 535–541. [Google Scholar] [CrossRef]
- Zhang, Y.; Dépond, M.; He, L.; Foudi, A.; Kwarteng, E.O.; Lauret, E.; Plo, I.; Desterke, C.; Dessen, P.; Fujii, N.; et al. CXCR4/CXCL12 axis counteracts hematopoietic stem cell exhaustion through selective protection against oxidative stress. Sci. Rep. 2016, 6, 37827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karigane, D.; Kobayashi, H.; Morikawa, T.; Ootomo, Y.; Sakai, M.; Nagamatsu, G.; Kubota, Y.; Goda, N.; Matsumoto, M.; Nishimura, E.K.; et al. p38α Activates Purine Metabolism to Initiate Hematopoietic Stem/Progenitor Cell Cycling in Response to Stress. Cell Stem Cell 2016, 19, 192–204. [Google Scholar] [CrossRef] [Green Version]
- Walter, D.; Lier, A.; Geiselhart, A.; Thalheimer, F.B.; Huntscha, S.; Sobotta, M.C.; Moehrle, B.; Brocks, D.; Bayindir, I.; Kaschutnig, P.; et al. Exit from dormancy provokes DNA-damage-induced attrition in haematopoietic stem cells. Nat. Cell Biol. 2015, 520, 549–552. [Google Scholar] [CrossRef] [PubMed]
- Mantel, C.R.; O’Leary, H.A.; Chitteti, B.R.; Huang, X.; Cooper, S.; Hangoc, G.; Brustovetsky, N.; Srour, E.F.; Lee, M.R.; Messina-Graham, S.; et al. Enhancing Hematopoietic Stem Cell Transplantation Efficacy by Mitigating Oxygen Shock. Cell 2015, 161, 1553–1565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riganti, C.; Gazzano, E.; Polimeni, M.; Aldieri, E.; Ghigo, D. The pentose phosphate pathway: An antioxidant defense and a crossroad in tumor cell fate. Free. Radic. Biol. Med. 2012, 53, 421–436. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.-N.; Wang, T.-S.; Li, X.; Wang, Y.-P. SIRT2 activates G6PD to enhance NADPH production and promote leukaemia cell proliferation. Sci. Rep. 2016, 6, 32734. [Google Scholar] [CrossRef] [Green Version]
- Wu, L.E.; Sinclair, D.A. SIRT 2 controls the pentose phosphate switch. EMBO J. 2014, 33, 1287–1288. [Google Scholar] [CrossRef] [Green Version]
- Luo, H.; Mu, W.C.; Karki, R.; Chiang, H.H.; Mohrin, M.; Shin, J.J.; Ohkubo, R.; Ito, K.; Kanneganti, T.-D.; Chen, D. Mitochondrial Stress-Initiated Aberrant Activation of the NLRP3 Inflammasome Regulates the Functional Deterioration of Hematopoietic Stem Cell Aging. Cell Rep. 2019, 26, 945–954.e4. [Google Scholar] [CrossRef] [Green Version]
- Paglialunga, F.; Fico, A.; Iaccarino, I.; Notaro, R.; Luzzatto, L.; Martini, G.; Filosa, S. G6PD is indispensable for erythropoiesis after the embryonic-adult hemoglobin switch. Blood 2004, 104, 3148–3152. [Google Scholar] [CrossRef] [Green Version]
- Mccord, J.M.; Keele, B.B.; Fridovich, I. An Enzyme-Based Theory of Obligate Anaerobiosis: The Physiological Function of Superoxide Dismutase. Proc. Natl. Acad. Sci. USA 1971, 68, 1024–1027. [Google Scholar] [CrossRef] [Green Version]
- Miyamoto, K.; Araki, K.Y.; Naka, K.; Arai, F.; Takubo, K.; Yamazaki, S.; Matsuoka, S.; Miyamoto, T.; Ito, K.; Ohmura, M.; et al. Foxo3a Is Essential for Maintenance of the Hematopoietic Stem Cell Pool. Cell Stem Cell 2007, 1, 101–112. [Google Scholar] [CrossRef] [Green Version]
- Tothova, Z.; Kollipara, R.; Huntly, B.J.; Lee, B.H.; Castrillon, D.H.; Cullen, D.E.; McDowell, E.P.; Lazo-Kallanian, S.; Williams, I.R.; Sears, C.; et al. FoxOs Are Critical Mediators of Hematopoietic Stem Cell Resistance to Physiologic Oxidative Stress. Cell 2007, 128, 325–339. [Google Scholar] [CrossRef] [Green Version]
- Yalcin, S.; Zhang, X.; Luciano, J.P.; Mungamuri, S.K.; Marinkovic, D.; Vercherat, C.; Sarkar, A.; Grisotto, M.; Taneja, R.; Ghaffari, S. Foxo3 Is Essential for the Regulation of Ataxia Telangiectasia Mutated and Oxidative Stress-mediated Homeostasis of Hematopoietic Stem Cells. J. Biol. Chem. 2008, 283, 25692–25705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bräutigam, L.; Zhang, J.; Dreij, K.; Spahiu, L.; Holmgren, A.; Abe, H.; Tew, K.D.; Townsend, D.M.; Kelner, M.J.; Morgenstern, R.; et al. MGST1, a GSH transferase/peroxidase essential for development and hematopoietic stem cell differentiation. Redox Biol. 2018, 17, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Hu, L.; Zhang, Y.; Miao, W.; Cheng, T. Reactive Oxygen Species and Nrf2: Functional and Transcriptional Regulators of Hematopoiesis. Oxidative Med. Cell. Longev. 2019, 2019, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Murakami, S.; Suzuki, T.; Harigae, H.; Romeo, P.-H.; Yamamoto, M.; Motohashi, H. NRF2 Activation Impairs Quiescence and Bone Marrow Reconstitution Capacity of Hematopoietic Stem Cells. Mol. Cell. Biol. 2017, 37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Merchant, A.A.; Singh, A.; Matsui, W.; Biswal, S. The redox-sensitive transcription factor Nrf2 regulates murine hematopoietic stem cell survival independently of ROS levels. Blood 2011, 118, 6572–6579. [Google Scholar] [CrossRef] [Green Version]
- Shinohara, A.; Imai, Y.; Nakagawa, M.; Takahashi, T.; Ichikawa, M.; Kurokawa, M. Intracellular Reactive Oxygen Species Mark and Influence the Megakaryocyte-Erythrocyte Progenitor Fate of Common Myeloid Progenitors. Stem Cells 2014, 32, 548–557. [Google Scholar] [CrossRef]
- Sattler, M.; Winkler, T.; Verma, S.; Byrne, C.H.; Shrikhande, G.; Salgia, R.; Griffin, J.D. Hematopoietic Growth Factors Signal Through the Formation of Reactive Oxygen Species. Blood 1999, 93, 2928–2935. [Google Scholar] [CrossRef] [PubMed]
- Rönn, R.E.; Guibentif, C.; Saxena, S.; Woods, N.-B. Reactive Oxygen Species Impair the Function of CD90+ Hematopoietic Progenitors Generated from Human Pluripotent Stem Cells. Stem Cells 2017, 35, 197–206. [Google Scholar] [CrossRef]
- Li, J.; Du, W.; Maynard, S.; Andreassen, P.R.; Pang, Q. Oxidative stress–specific interaction between FANCD2 and FOXO3a. Blood 2010, 115, 1545–1548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Li, J.; Wilson, A.; Sipple, J.; Schick, J.; Pang, Q. Fancd2 Is Required for Nuclear Retention of Foxo3a in Hematopoietic Stem Cell Maintenance. J. Biol. Chem. 2015, 290, 2715–2727. [Google Scholar] [CrossRef] [Green Version]
- Rodrigues, C.P.; Shvedunova, M.; Akhtar, A. Epigenetic Regulators as the Gatekeepers of Hematopoiesis. Trends Genet. 2021, 37, 125–142. [Google Scholar] [CrossRef]
- Kietzmann, T.; Petry, A.; Shvetsova, A.; Gerhold, J.M.; Görlach, A. The epigenetic landscape related to reactive oxygen species formation in the cardiovascular system. Br. J. Pharmacol. 2017, 174, 1533–1554. [Google Scholar] [CrossRef] [PubMed]
- Han, D.; Lu, X.; Shih, A.H.; Nie, J.; You, Q.; Xu, M.M.; Melnick, A.M.; Levine, R.L.; He, C. A Highly Sensitive and Robust Method for Genome-wide 5hmC Profiling of Rare Cell Populations. Mol. Cell 2016, 63, 711–719. [Google Scholar] [CrossRef] [Green Version]
- Taya, Y.; Ota, Y.; Wilkinson, A.C.; Kanazawa, A.; Watarai, H.; Kasai, M.; Nakauchi, H.; Yamazaki, S. Depleting dietary valine permits nonmyeloablative mouse hematopoietic stem cell transplantation. Science 2016, 354, 1152–1155. [Google Scholar] [CrossRef] [PubMed]
- Raffel, S.; Falcone, M.; Kneisel, N.; Hansson, J.; Wang, W.; Lutz, C.; Bullinger, L.; Poschet, G.; Nonnenmacher, Y.; Barnert, A.; et al. BCAT1 restricts αKG levels in AML stem cells leading to IDHmut-like DNA hypermethylation. Nature 2017, 551, 384–388. [Google Scholar] [CrossRef]
- Takihara, Y.; Nakamura-Ishizu, A.; Tan, D.Q.; Fukuda, M.; Matsumura, T.; Endoh, M.; Arima, Y.; Chin, D.W.L.; Umemoto, T.; Hashimoto, M.; et al. High mitochondrial mass is associated with reconstitution capacity and quiescence of hematopoietic stem cells. Blood Adv. 2019, 3, 2323–2327. [Google Scholar] [CrossRef]
- de Almeida, M.J.; Luchsinger, L.L.; Corrigan, D.J.; Williams, L.J.; Snoeck, H.-W. Dye-Independent Methods Reveal Elevated Mitochondrial Mass in Hematopoietic Stem Cells. Cell Stem Cell 2017, 21, 725–729.e4. [Google Scholar] [CrossRef] [Green Version]
- Bonora, M.; Ito, K.; Morganti, C.; Pinton, P.; Ito, K. Membrane-potential compensation reveals mitochondrial volume expansion during HSC commitment. Exp. Hematol. 2018, 68, 30–37.e1. [Google Scholar] [CrossRef]
- Vannini, N.; Girotra, M.; Naveiras, O.; Nikitin, G.; Campos, V.; Giger, S.; Roch, A.; Auwerx, J.; Lutolf, M.P. Specification of haematopoietic stem cell fate via modulation of mitochondrial activity. Nat. Commun. 2016, 7, 13125. [Google Scholar] [CrossRef]
- Sukumar, M.; Liu, J.; Mehta, G.U.; Patel, S.J.; Roychoudhuri, R.; Crompton, J.G.; Klebanoff, C.A.; Ji, Y.; Li, P.; Yu, Z.; et al. Mitochondrial Membrane Potential Identifies Cells with Enhanced Stemness for Cellular Therapy. Cell Metab. 2016, 23, 63–76. [Google Scholar] [CrossRef] [Green Version]
- Wagner-Souza, K.; Diamond, H.R.; Ornellas, M.H.; Gomes, B.E.; Almeida-Oliveira, A.; Abdelhay, E.; Bouzas, L.F.; Rumjanek, V.M. Rhodamine 123 efflux in human subpopulations of hematopoietic stem cells: Comparison between bone marrow, umbilical cord blood and mobilized peripheral blood CD34+ cells. Int. J. Mol. Med. 2008, 22, 237–242. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.; Cooper, D.D.; Hayes, S.F.; Spangrude, G.J. Rhodamine-123 Staining in Hematopoietic Stem Cells of Young Mice Indicates Mitochondrial Activation Rather Than Dye Efflux. Blood J. Am. Soc. Hematol. 1998, 91, 4106–4117. [Google Scholar]
- Morganti, C.; Bonora, M.; Ito, K. Improving the Accuracy of Flow Cytometric Assessment of Mitochondrial Membrane Potential in Hematopoietic Stem and Progenitor Cells Through the Inhibition of Efflux Pumps. J. Vis. Exp. 2019, e60057. [Google Scholar] [CrossRef] [PubMed]
- Filippi, M.D.; Ghaffari, S. Mitochondria in the maintenance of hematopoietic stem cells: New perspectives and opportunities. Blood 2019, 133, 1943–1952. [Google Scholar] [CrossRef] [PubMed]
- Hinge, A.; He, J.; Bartram, J.; Javier, J.; Xu, J.; Fjellman, E.; Sesaki, H.; Li, T.; Yu, J.; Wunderlich, M.; et al. Asymmetrically Segregated Mitochondria Provide Cellular Memory of Hematopoietic Stem Cell Replicative History and Drive HSC Attrition. Cell Stem Cell 2020, 26, 420–430. [Google Scholar] [CrossRef] [PubMed]
- Mortensen, M.; Soilleux, E.J.; Djordjevic, G.; Tripp, R.; Lutteropp, M.; Sadighi-Akha, E.; Stranks, A.J.; Glanville, J.; Knight, S.; Jacobsen, S.E.W.; et al. The autophagy protein Atg7 is essential for hematopoietic stem cell maintenance. J. Exp. Med. 2011, 208, 455–467. [Google Scholar] [CrossRef]
- Ho, T.T.; Warr, M.R.; Adelman, E.R.; Lansinger, O.M.; Flach, J.; Verovskaya, E.V.; Figueroa, M.E.; Passegué, E. Autophagy maintains the metabolism and function of young and old stem cells. Nature 2017, 543, 205–210. [Google Scholar] [CrossRef] [Green Version]
- Jung, H.E.; Shim, Y.R.; Oh, J.E.; Oh, D.S.; Lee, H.K. The autophagy Protein Atg5 Plays a Crucial Role in the Maintenance and Reconstitution Ability of Hematopoietic Stem Cells. Immune Netw. 2019, 19, e12. [Google Scholar] [CrossRef]
- Joshi, A.; Kundu, M. Mitophagy in Hematopoietic Stem Cells: The Case for Exploration. Autophagy 2013, 9, 1737–1749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ito, K.; Turcotte, R.; Cui, J.; Zimmerman, S.E.; Pinho, S.; Mizoguchi, T.; Arai, F.; Runnels, J.M.; Alt, C.; Teruya-Feldstein, J.; et al. Self-renewal of a purified Tie2+ hematopoietic stem cell population relies on mitochondrial clearance. Science 2016, 354, 1156–1160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ito, K.; Ito, K. Hematopoietic stem cell fate through metabolic control. Exp. Hematol. 2018, 64, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Narendra, D.P.; Jin, S.M.; Tanaka, A.; Suen, D.-F.; Gautier, C.A.; Shen, J.; Cookson, M.R.; Youle, R.J. PINK1 Is Selectively Stabilized on Impaired Mitochondria to Activate Parkin. PLoS Biol. 2010, 8, e1000298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Warr, M.R.; Binnewies, M.; Flach, J.; Reynaud, D.; Garg, T.; Malhotra, R.; Debnath, J.; Passegué, E. FOXO3A directs a protective autophagy program in haematopoietic stem cells. Nature 2013, 494, 323–327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Nartiss, Y.; Steipe, B.; McQuibban, G.A.; Kim, P.K. ROS-induced mitochondrial depolarization initiates PARK2/PARKIN-dependent mitochondrial degradation by autophagy. Autophagy 2012, 8, 1462–1476. [Google Scholar] [CrossRef] [Green Version]
- Ashrafi, G.H.; Schwarz, T.L. The pathways of mitophagy for quality control and clearance of mitochondria. Cell Death Differ. 2013, 20, 31–42. [Google Scholar] [CrossRef] [Green Version]
- Liang, R.; Arif, T.; Kalmykova, S.; Kasianov, A.; Lin, M.; Menon, V.; Qiu, J.; Bernitz, J.M.; Moore, K.; Lin, F.; et al. Restraining Lysosomal Activity Preserves Hematopoietic Stem Cell Quiescence and Potency. Cell Stem Cell 2020, 26, 359–376. [Google Scholar] [CrossRef]
- Loeffler, D.; Wehling, A.; Schneiter, F.; Zhang, Y.; Müller-Bötticher, N.; Hoppe, P.S.; Hilsenbeck, O.; Kokkaliaris, K.D.; Endele, M.; Schroeder, T. Asymmetric lysosome inheritance predicts activation of haematopoietic stem cells. Nature 2019, 573, 426–429. [Google Scholar] [CrossRef] [Green Version]
- Deus, C.M.; Yambire, K.F.; Oliveira, P.J.; Raimundo, N. Mitochondria–Lysosome Crosstalk: From Physiology to Neurodegeneration. Trends Mol. Med. 2020, 26, 71–88. [Google Scholar] [CrossRef] [Green Version]
- Wong, Y.C.; Ysselstein, D.; Krainc, D. Mitochondria–lysosome contacts regulate mitochondrial fission via RAB7 GTP hydrolysis. Nature 2018, 554, 382–386. [Google Scholar] [CrossRef]
- Wong, Y.C.; Kim, S.; Peng, W.; Krainc, D. Regulation and Function of Mitochondria–Lysosome Membrane Contact Sites in Cellular Homeostasis. Trends Cell Biol. 2019, 29, 500–513. [Google Scholar] [CrossRef]
- Schmidt, L.S.; Linehan, W.M. FLCN: The causative gene for Birt-Hogg-Dubé syndrome. Gene 2018, 640, 28–42. [Google Scholar] [CrossRef]
- Nickerson, M.L.; Warren, M.B.; Toro, J.R.; Matrosova, V.; Glenn, G.; Turner, M.L.; Duray, P.; Merino, M.; Choyke, P.; Pavlovich, C.P.; et al. Mutations in a novel gene lead to kidney tumors, lung wall defects, and benign tumors of the hair follicle in patients with the Birt-Hogg-Dubé syndrome. Cancer Cell 2002, 2, 157–164. [Google Scholar] [CrossRef] [Green Version]
- Napolitano, G.; Di Malta, C.; Esposito, A.; de Araujo, M.E.G.; Pece, S.; Bertalot, G.; Matarese, M.; Benedetti, V.; Zampelli, A.; Stasyk, T.; et al. A substrate-specific mTORC1 pathway underlies Birt–Hogg–Dubé syndrome. Nature 2020, 585, 597–602. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, R.E.; Fromm, S.A.; Fu, Y.; Yokom, A.L.; Kim, D.J.; Thelen, A.M.; Young, L.N.; Lim, C.-Y.; Samelson, A.J.; Hurley, J.H.; et al. Structural mechanism of a Rag GTPase activation checkpoint by the lysosomal folliculin complex. Science 2019, 366, 971–977. [Google Scholar] [CrossRef]
- de Martín Garrido, N.; Aylett, C.H. Nutrient Signaling and Lysosome Positioning Crosstalk Through a Multifunctional Protein, Folliculin. Front. Cell Dev. Biol. 2020, 8, 108. [Google Scholar] [CrossRef] [Green Version]
- Abu-Remaileh, M.; Wyant, G.A.; Kim, C.; Laqtom, N.N.; Abbasi, M.; Chan, S.H.; Freinkman, E.; Sabatini, D.M. Lysosomal metabolomics reveals V-ATPase- and mTOR-dependent regulation of amino acid efflux from lysosomes. Science 2017, 358, 807–813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Endoh, M.; Baba, M.; Endoh, T.; Hirayama, A.; Nakamura-Ishizu, A.; Umemoto, T.; Hashimoto, M.; Nagashima, K.; Soga, T.; Lang, M.; et al. A FLCN-TFE3 Feedback Loop Prevents Excessive Glycogenesis and Phagocyte Activation by Regulating Lysosome Activity. Cell Rep. 2020, 30, 1823–1834.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kennedy, B.K.; Lamming, D.W. The Mechanistic Target of Rapamycin: The Grand ConducTOR of Metabolism and Aging. Cell Metab. 2016, 23, 990–1003. [Google Scholar] [CrossRef] [Green Version]
- Baba, M.; Toyama, H.; Sun, L.; Takubo, K.; Suh, H.-C.; Hasumi, H.; Nakamura-Ishizu, A.; Hasumi, Y.; Klarmann, K.D.; Nakagata, N.; et al. Loss of Folliculin Disrupts Hematopoietic Stem Cell Quiescence and Homeostasis Resulting in Bone Marrow Failure. Stem Cells 2016, 34, 1068–1082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, C.; Zhao, C.; Zhang, F.; Kesarwani, M.; Tu, Z.; Cai, X.; Davis, A.K.; Xu, L.; Hochstetler, C.L.; Chen, X.; et al. Adaptive responses tomTORgene targeting in hematopoietic stem cells reveal a proliferative mechanism evasive to mTOR inhibition. Proc. Natl. Acad. Sci. USA 2021, 118, e2020102118. [Google Scholar] [CrossRef] [PubMed]
- Gan, B.; Depinho, R.A. mTORC1 signaling governs hematopoietic stem cell quiescence. Cell Cycle 2009, 8, 1003–1006. [Google Scholar] [CrossRef] [PubMed]
- Kalaitzidis, D.; Lee, D.; Efeyan, A.; Kfoury, Y.; Nayyar, N.; Sykes, D.B.; Mercier, F.E.; Papazian, A.; Baryawno, N.; Victora, G.D.; et al. Amino acid-insensitive mTORC1 regulation enables nutritional stress resilience in hematopoietic stem cells. J. Clin. Investig. 2017, 127, 1405–1413. [Google Scholar] [CrossRef] [Green Version]
- Hasumi, H.; Baba, M.; Hasumi, Y.; Huang, Y.; Oh, H.; Hughes, R.M.; Klein, M.E.; Takikita, S.; Nagashima, K.; Schmidt, L.S.; et al. Regulation of Mitochondrial Oxidative Metabolism by Tumor Suppressor FLCN. J. Natl. Cancer Inst. 2012, 104, 1750–1764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manford, A.G.; Rodríguez-Pérez, F.; Shih, K.Y.; Shi, Z.; Berdan, C.A.; Choe, M.; Titov, D.V.; Nomura, D.K.; Rape, M. A Cellular Mechanism to Detect and Alleviate Reductive Stress. Cell 2020, 183, 46–61.e21. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mochizuki-Kashio, M.; Shiozaki, H.; Suda, T.; Nakamura-Ishizu, A. Mitochondria Turnover and Lysosomal Function in Hematopoietic Stem Cell Metabolism. Int. J. Mol. Sci. 2021, 22, 4627. https://doi.org/10.3390/ijms22094627
Mochizuki-Kashio M, Shiozaki H, Suda T, Nakamura-Ishizu A. Mitochondria Turnover and Lysosomal Function in Hematopoietic Stem Cell Metabolism. International Journal of Molecular Sciences. 2021; 22(9):4627. https://doi.org/10.3390/ijms22094627
Chicago/Turabian StyleMochizuki-Kashio, Makiko, Hiroko Shiozaki, Toshio Suda, and Ayako Nakamura-Ishizu. 2021. "Mitochondria Turnover and Lysosomal Function in Hematopoietic Stem Cell Metabolism" International Journal of Molecular Sciences 22, no. 9: 4627. https://doi.org/10.3390/ijms22094627
APA StyleMochizuki-Kashio, M., Shiozaki, H., Suda, T., & Nakamura-Ishizu, A. (2021). Mitochondria Turnover and Lysosomal Function in Hematopoietic Stem Cell Metabolism. International Journal of Molecular Sciences, 22(9), 4627. https://doi.org/10.3390/ijms22094627