
Species-Specific Regulation of TRPM2 by PI(4,5)P2 via the Membrane Interfacial Cavity


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
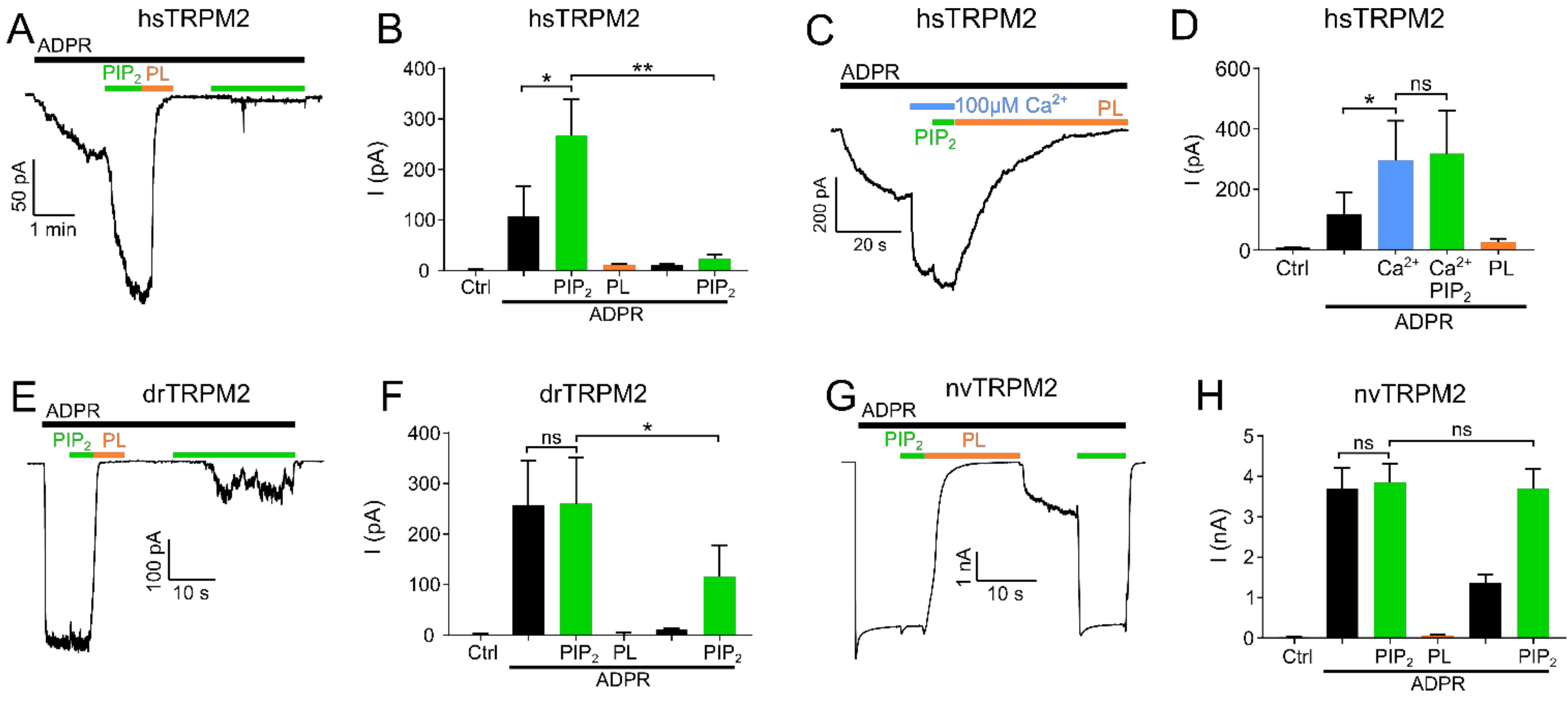
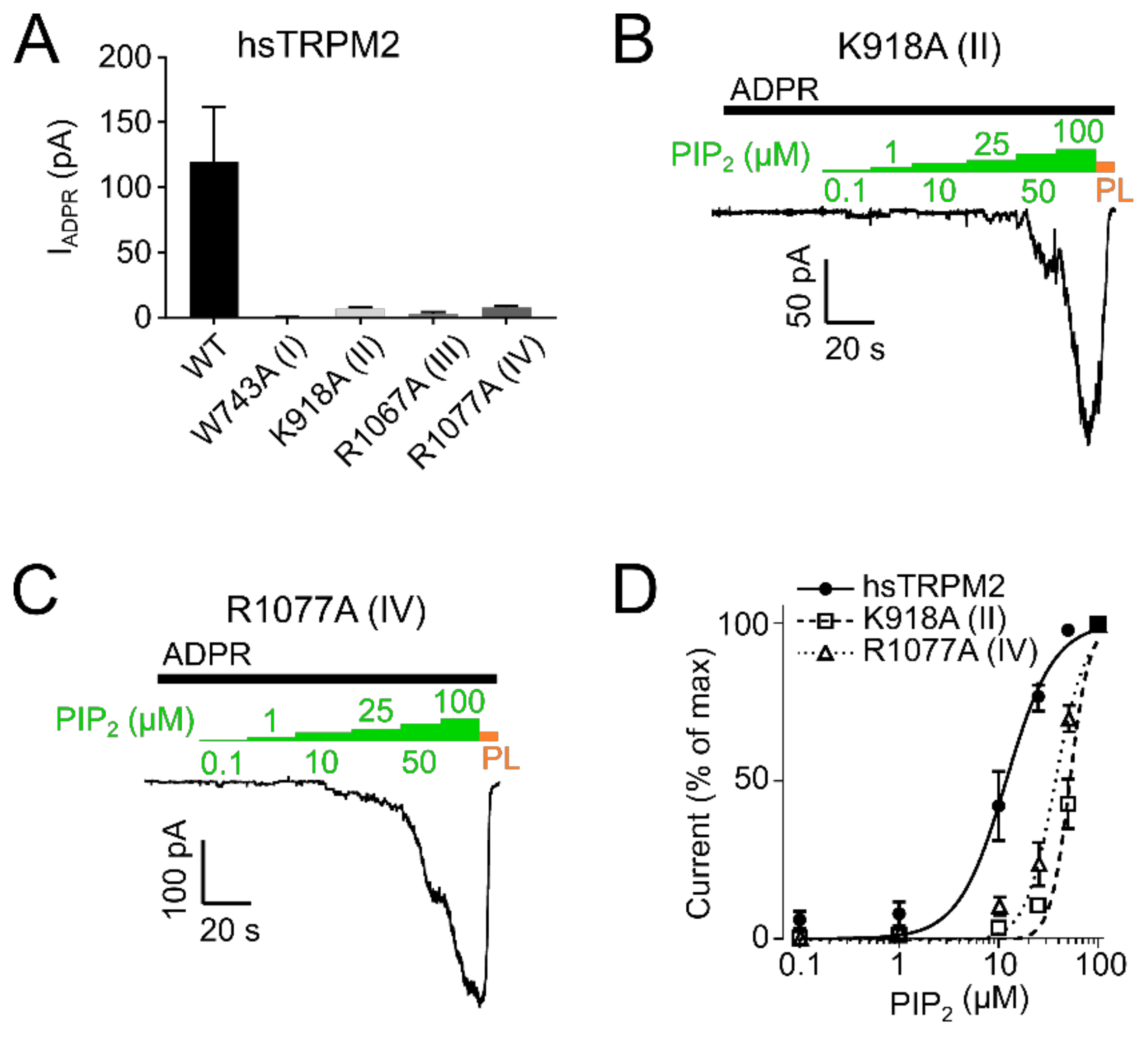
2.1. PIP2 Is a Necessary Cofactor for TRPM2 Activation in Mammalian Cells
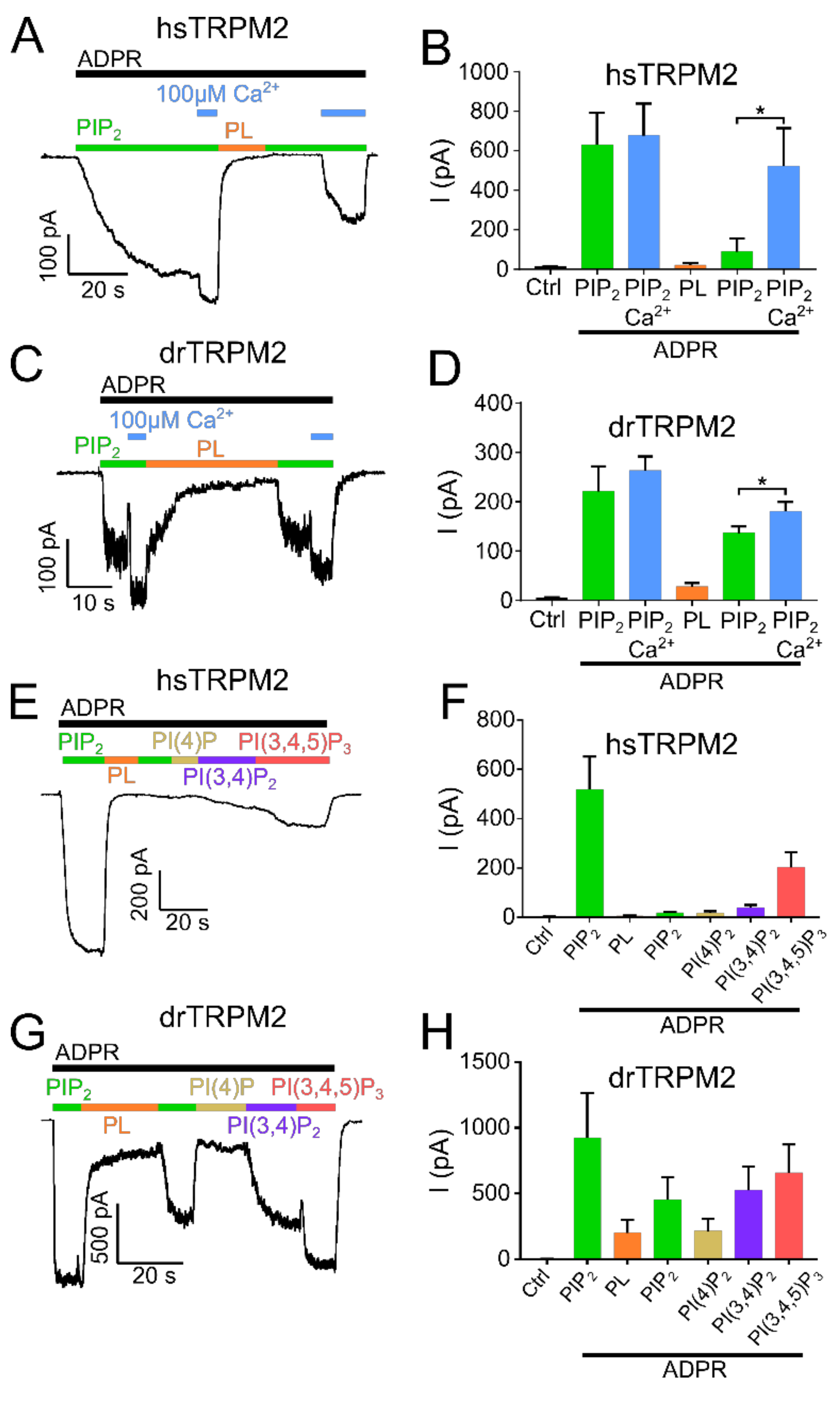
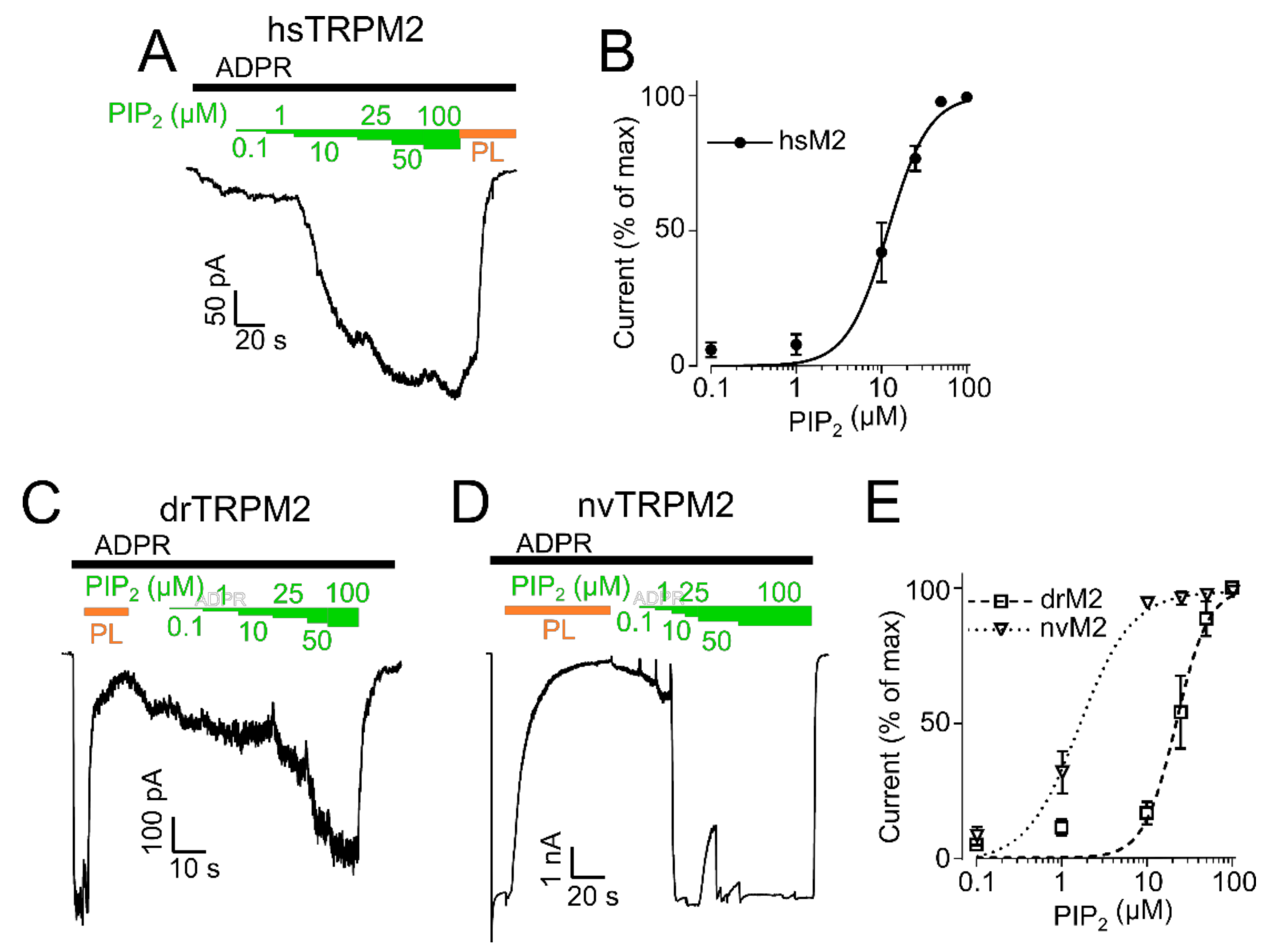
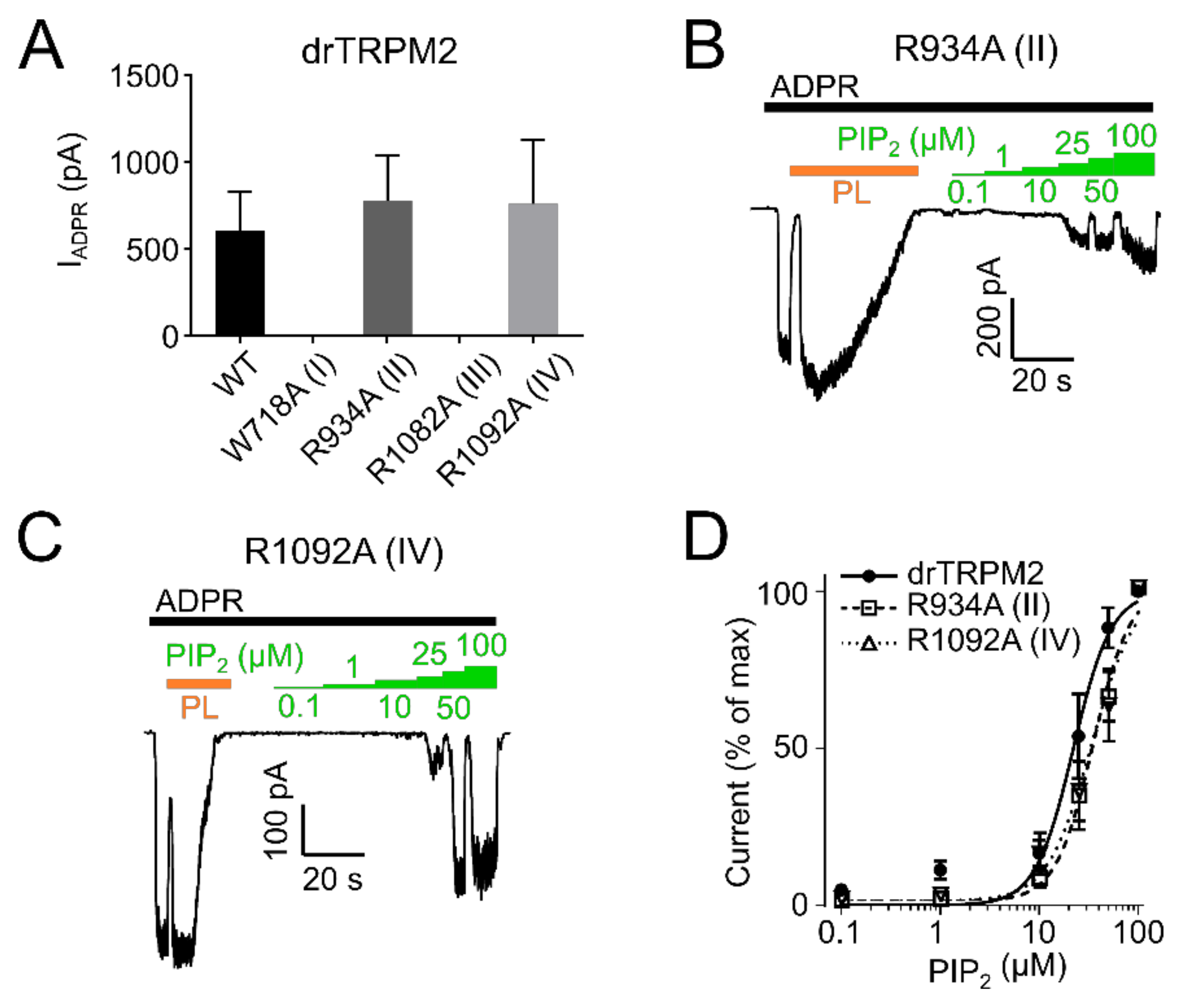
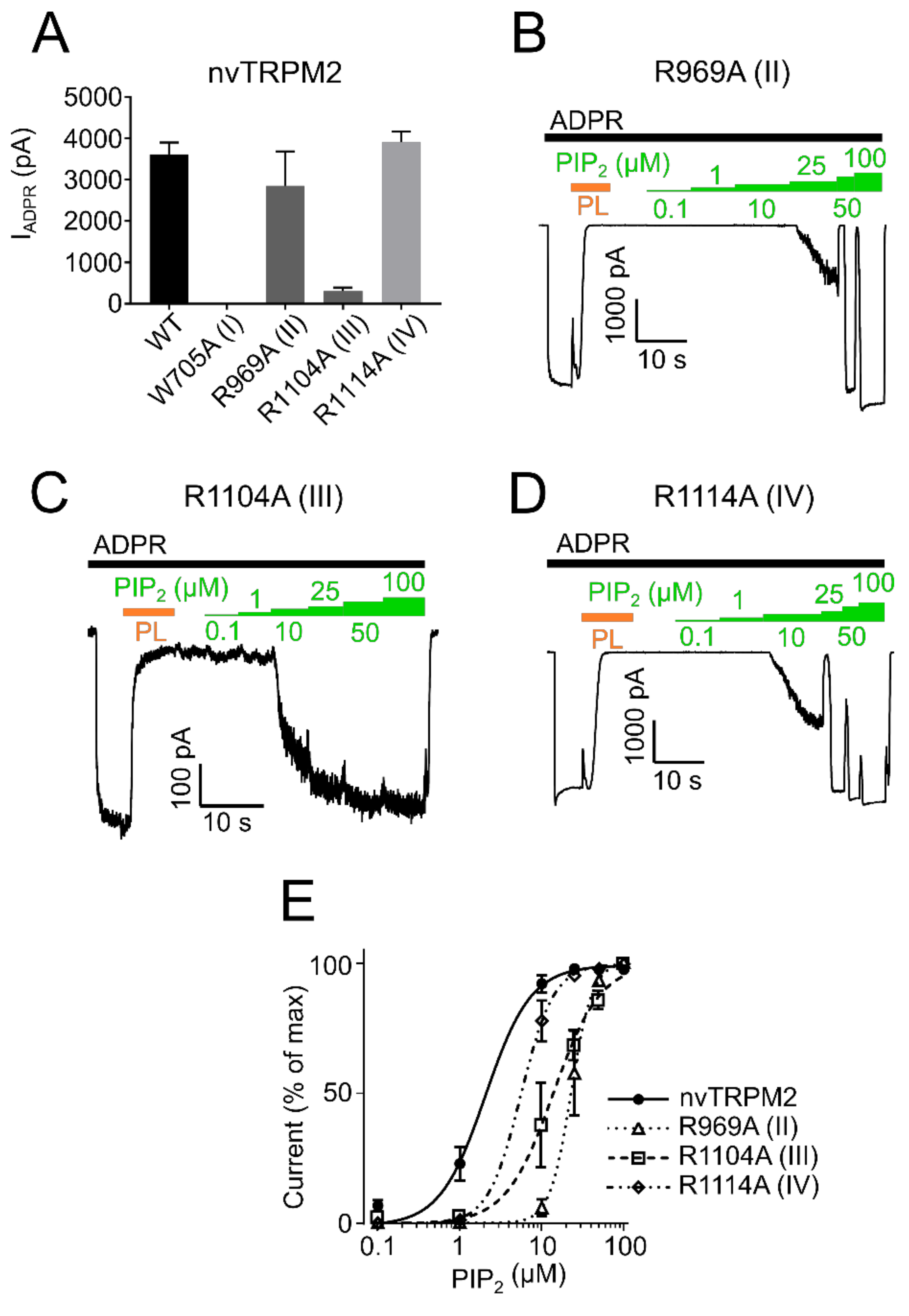
2.2. PIP2 Sensitivity Varies among TRPM2 Species
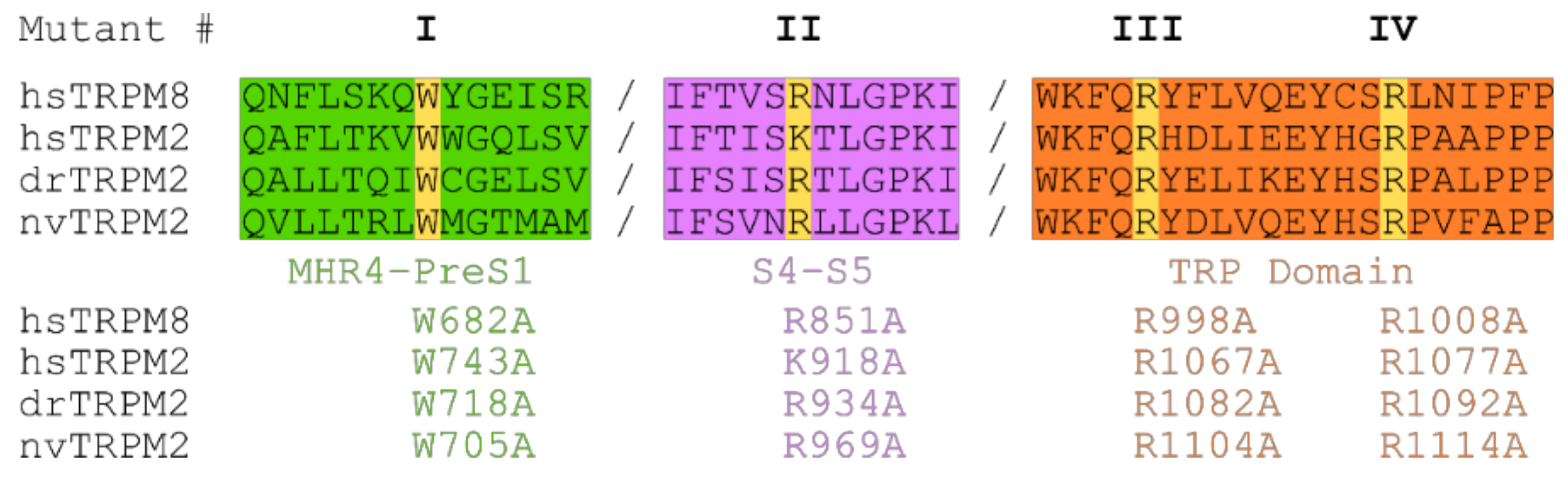
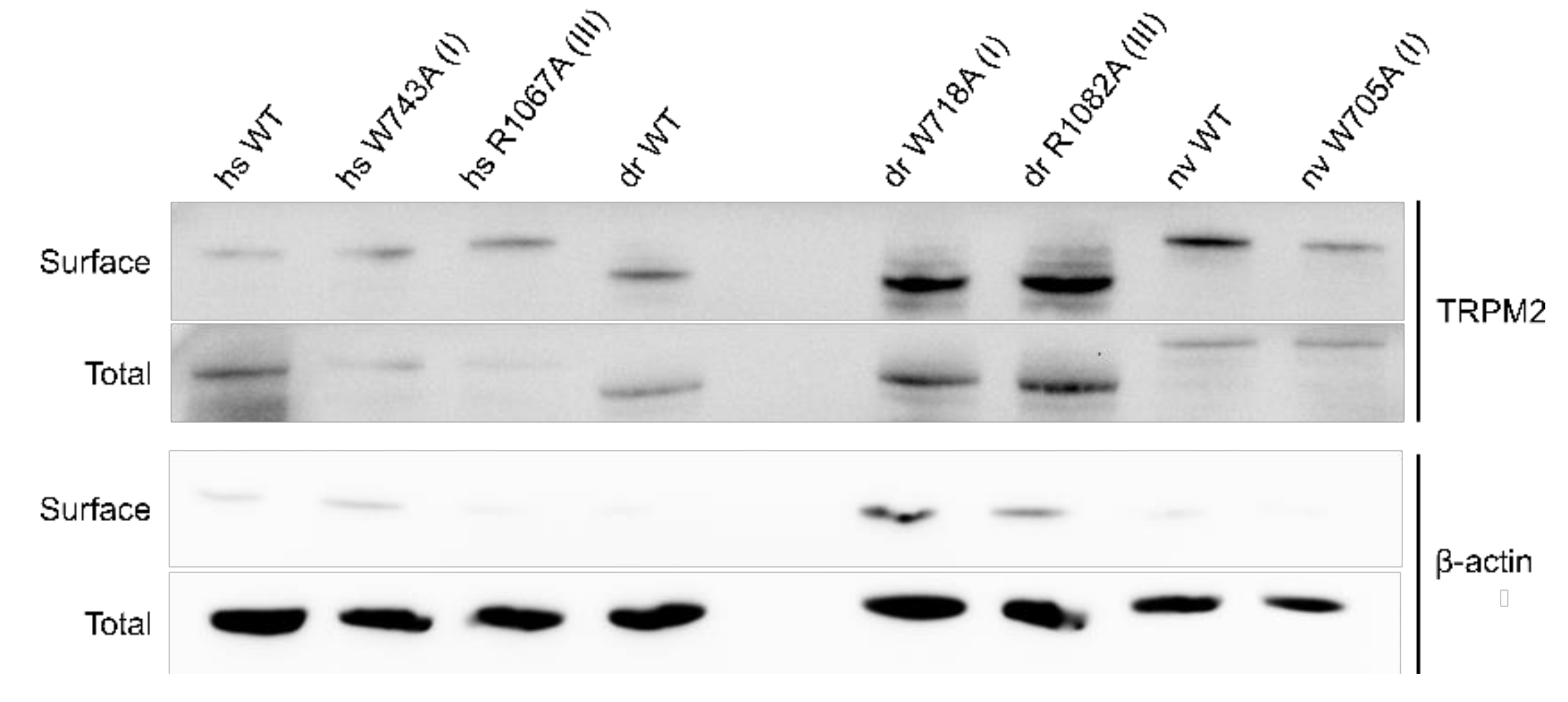
2.3. Residues within the Membrane Interfacial Cavity Are Putative PIP2-Interaction Sites
3. Discussion
3.1. PIP2 Is a Necessary Cofactor for TRPM2 Function
3.2. Structural Elements of TRPM2 for PIP2 Interaction
3.3. Physiological Role of PIP2 Interaction with TRPM2
4. Materials and Methods
4.1. Molecular Biology
4.2. Biotinylation
4.3. Cell Culture and Transfection
4.4. Patch Clamp
4.5. Data Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kuhn, F.J.P. Structure-function relationship of TRPM2: Recent advances, contradictions, and open questions. Int. J. Mol. Sci. 2020, 21, 6481. [Google Scholar] [CrossRef] [PubMed]
- Sano, Y.; Inamura, K.; Miyake, A.; Mochizuki, S.; Yokoi, H.; Matsushime, H.; Furuichi, K. Immunocyte Ca2+ influx system mediated by LTRPC2. Science 2001, 293, 1327–1330. [Google Scholar] [CrossRef] [PubMed]
- Perraud, A.L.; Fleig, A.; Dunn, C.A.; Bagley, L.A.; Launay, P.; Schmitz, C.; Stokes, A.J.; Zhu, Q.; Bessman, M.J.; Penner, R.; et al. ADP-ribose gating of the calcium-permeable LTRPC2 channel revealed by Nudix motif homology. Nature 2001, 411, 595–599. [Google Scholar] [CrossRef] [PubMed]
- Csanady, L.; Torocsik, B. Four Ca2+ ions activate TRPM2 channels by binding in deep crevices near the pore but intracellularly of the gate. J. Gen. Physiol. 2009, 133, 189–203. [Google Scholar] [CrossRef]
- McHugh, D.; Flemming, R.; Xu, S.Z.; Perraud, A.L.; Beech, D.J. Critical intracellular Ca2+ dependence of transient receptor potential melastatin 2 (TRPM2) cation channel activation. J. Biol. Chem. 2003, 278, 11002–11006. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, S.; Shimizu, S.; Kiyonaka, S.; Takahashi, N.; Wajima, T.; Hara, Y.; Negoro, T.; Hiroi, T.; Kiuchi, Y.; Okada, T.; et al. TRPM2-mediated Ca2+influx induces chemokine production in monocytes that aggravates inflammatory neutrophil infiltration. Nat. Med. 2008, 14, 738–747. [Google Scholar] [CrossRef]
- Sun, L.; Yau, H.Y.; Wong, W.Y.; Li, R.A.; Huang, Y.; Yao, X. Role of TRPM2 in H(2)O(2)-induced cell apoptosis in endothelial cells. PLoS ONE 2012, 7, e43186. [Google Scholar] [CrossRef]
- Inamura, K.; Sano, Y.; Mochizuki, S.; Yokoi, H.; Miyake, A.; Nozawa, K.; Kitada, C.; Matsushime, H.; Furuichi, K. Response to ADP-ribose by activation of TRPM2 in the CRI-G1 insulinoma cell line. J. Membr. Biol. 2003, 191, 201–207. [Google Scholar] [CrossRef]
- Togashi, K.; Hara, Y.; Tominaga, T.; Higashi, T.; Konishi, Y.; Mori, Y.; Tominaga, M. TRPM2 activation by cyclic ADP-ribose at body temperature is involved in insulin secretion. EMBO J. 2006, 25, 1804–1815. [Google Scholar] [CrossRef]
- Tan, C.H.; McNaughton, P.A. TRPM2 and warmth sensation. Pflug. Arch. 2018, 470, 787–798. [Google Scholar] [CrossRef] [Green Version]
- Toth, B.; Iordanov, I.; Csanady, L. Selective profiling of N- and C-terminal nucleotide-binding sites in a TRPM2 channel. J. Gen. Physiol. 2020, 152. [Google Scholar] [CrossRef] [Green Version]
- Kuhn, F.J.P.; Watt, J.M.; Potter, B.V.L.; Luckhoff, A. Different substrate specificities of the two ADPR binding sites in TRPM2 channels of Nematostella vectensis and the role of IDPR. Sci. Rep. 2019, 9, 4985. [Google Scholar] [CrossRef]
- Kuhn, F.J.P.; Ehrlich, W.; Barth, D.; Kuhn, C.; Luckhoff, A. Functional importance of NUDT9H domain and N-terminal ADPR-binding pocket in two species variants of vertebrate TRPM2 channels. Sci. Rep. 2019, 9, 19224. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.; Roth, B.; Lu, W.; Du, J. Ligand recognition and gating mechanism through three ligand-binding sites of human TRPM2 channel. Elife 2019, 8. [Google Scholar] [CrossRef]
- Huang, Y.; Winkler, P.A.; Sun, W.; Lü, W.; Du, J. Architecture of the TRPM2 channel and its activation mechanism by ADP-ribose and calcium. Nature 2018. [Google Scholar] [CrossRef]
- Kuhn, F.J.; Kuhn, C.; Winking, M.; Hoffmann, D.C.; Luckhoff, A. ADP-Ribose Activates the TRPM2 Channel from the Sea Anemone Nematostella vectensis independently of the NUDT9H domain. PLoS ONE 2016, 11, e0158060. [Google Scholar] [CrossRef]
- Iordanov, I.; Toth, B.; Szollosi, A.; Csanady, L. Enzyme activity and selectivity filter stability of ancient TRPM2 channels were simultaneously lost in early vertebrates. Elife 2019, 8. [Google Scholar] [CrossRef]
- Toth, B.; Iordanov, I.; Csanady, L. Putative chanzyme activity of TRPM2 cation channel is unrelated to pore gating. Proc. Natl. Acad. Sci. USA 2014, 111, 16949–16954. [Google Scholar] [CrossRef] [Green Version]
- Perraud, A.L.; Takanishi, C.L.; Shen, B.; Kang, S.; Smith, M.K.; Schmitz, C.; Knowles, H.M.; Ferraris, D.; Li, W.; Zhang, J.; et al. Accumulation of free ADP-ribose from mitochondria mediates oxidative stress-induced gating of TRPM2 cation channels. J. Biol. Chem. 2005, 280, 6138–6148. [Google Scholar] [CrossRef] [Green Version]
- Kuhn, F.J.; Luckhoff, A. Sites of the NUDT9-H domain critical for ADP-ribose activation of the cation channel TRPM2. J. Biol. Chem. 2004, 279, 46431–46437. [Google Scholar] [CrossRef] [Green Version]
- Perraud, A.L.; Schmitz, C.; Scharenberg, A.M. TRPM2 Ca2+ permeable cation channels: From gene to biological function. Cell Calcium 2003, 33, 519–531. [Google Scholar] [CrossRef]
- Wang, L.; Fu, T.M.; Zhou, Y.; Xia, S.; Greka, A.; Wu, H. Structures and gating mechanism of human TRPM2. Science 2018, 362. [Google Scholar] [CrossRef] [Green Version]
- Kuhn, F.J.; Kuhn, C.; Luckhoff, A. Functional characterisation of a TRPM2 orthologue from the sea anemone Nematostella vectensis in human cells. Sci. Rep. 2015, 5, 8032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duncan, A.L.; Song, W.; Sansom, M.S.P. Lipid-dependent regulation of ion channels and G protein-coupled receptors: Insights from structures and simulations. Ann. Rev. Pharmacol. Toxicol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Choquette, D.; Hakim, G.; Filoteo, A.G.; Plishker, G.A.; Bostwick, J.R.; Penniston, J.T. Regulation of plasma membrane Ca2+ ATPases by lipids of the phosphatidylinositol cycle. Biochem. Biophys. Res. Commun. 1984, 125, 908–915. [Google Scholar] [CrossRef]
- Rohacs, T. Phosphoinositide regulation of TRP channels. Handb. Exp. Pharmacol. 2014, 223, 1143–1176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, W.; Cai, R.; Hofmann, L.; Nesin, V.; Hu, Q.; Long, W.; Fatehi, M.; Liu, X.; Hussein, S.; Kong, T.; et al. Direct binding between Pre-S1 and TRP-like domains in TRPP channels mediates gating and functional regulation by PIP2. Cell. Rep. 2018, 22, 1560–1573. [Google Scholar] [CrossRef] [Green Version]
- Otsuguro, K.; Tang, J.; Tang, Y.; Xiao, R.; Freichel, M.; Tsvilovskyy, V.; Ito, S.; Flockerzi, V.; Zhu, M.X.; Zholos, A.V. Isoform-specific inhibition of TRPC4 channel by phosphatidylinositol 4,5-bisphosphate. J. Biol. Chem. 2008, 283, 10026–10036. [Google Scholar] [CrossRef] [Green Version]
- Ma, R.; Li, W.P.; Rundle, D.; Kong, J.; Akbarali, H.I.; Tsiokas, L. PKD2 functions as an epidermal growth factor-activated plasma membrane channel. Mol. Cell. Biol. 2005, 25, 8285–8298. [Google Scholar] [CrossRef] [Green Version]
- Toth, B.; Csanady, L. Pore collapse underlies irreversible inactivation of TRPM2 cation channel currents. Proc. Natl. Acad Sci. USA 2012, 109, 13440–13445. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Toth, B.; Szollosi, A.; Chen, J.; Csanady, L. Structure of a TRPM2 channel in complex with Ca(2+) explains unique gating regulation. Elife 2018, 7. [Google Scholar] [CrossRef]
- Yin, Y.; Le, S.C.; Hsu, A.L.; Borgnia, M.J.; Yang, H.; Lee, S.Y. Structural basis of cooling agent and lipid sensing by the cold-activated TRPM8 channel. Science 2019, 363. [Google Scholar] [CrossRef]
- Rohacs, T.; Lopes, C.M.; Michailidis, I.; Logothetis, D.E. PI(4,5)P2 regulates the activation and desensitization of TRPM8 channels through the TRP domain. Nat. Neurosci. 2005, 8, 626–634. [Google Scholar] [CrossRef]
- Zhang, H.; Craciun, L.C.; Mirshahi, T.; Rohacs, T.; Lopes, C.M.; Jin, T.; Logothetis, D.E. PIP(2) activates KCNQ channels, and its hydrolysis underlies receptor-mediated inhibition of M currents. Neuron 2003, 37, 963–975. [Google Scholar] [CrossRef] [Green Version]
- Lopes, C.M.; Zhang, H.; Rohacs, T.; Jin, T.; Yang, J.; Logothetis, D.E. Alterations in conserved Kir channel-PIP2 interactions underlie channelopathies. Neuron 2002, 34, 933–944. [Google Scholar] [CrossRef] [Green Version]
- Suh, B.C.; Hille, B. PIP2 is a necessary cofactor for ion channel function: How and why? Annu. Rev. Biophys. 2008, 37, 175–195. [Google Scholar] [CrossRef] [Green Version]
- Hilgemann, D.W.; Feng, S.; Nasuhoglu, C. The complex and intriguing lives of PIP2 with ion channels and transporters. Sci. STKE 2001, 2001, re19. [Google Scholar] [CrossRef]
- Hille, B.; Dickson, E.J.; Kruse, M.; Vivas, O.; Suh, B.C. Phosphoinositides regulate ion channels. Biochim. Biophys. Acta 2015, 1851, 844–856. [Google Scholar] [CrossRef]
- Berridge, M.J. Inositol trisphosphate and diacylglycerol as second messengers. Biochem. J. 1984, 220, 345–360. [Google Scholar] [CrossRef] [Green Version]
- Ostapchenko, V.G.; Chen, M.; Guzman, M.S.; Xie, Y.F.; Lavine, N.; Fan, J.; Beraldo, F.H.; Martyn, A.C.; Belrose, J.C.; Mori, Y.; et al. The transient receptor potential melastatin 2 (TRPM2) channel contributes to beta-amyloid oligomer-related neurotoxicity and memory impairment. J. Neurosci. 2015, 35, 15157–15169. [Google Scholar] [CrossRef] [Green Version]
- Fonfria, E.; Marshall, I.C.; Boyfield, I.; Skaper, S.D.; Hughes, J.P.; Owen, D.E.; Zhang, W.; Miller, B.A.; Benham, C.D.; McNulty, S. Amyloid beta-peptide(1-42) and hydrogen peroxide-induced toxicity are mediated by TRPM2 in rat primary striatal cultures. J. Neurochem. 2005, 95, 715–723. [Google Scholar] [CrossRef] [PubMed]
- Shimohama, S.; Sasaki, Y.; Fujimoto, S.; Kamiya, S.; Taniguchi, T.; Takenawa, T.; Kimura, J. Phospholipase C isozymes in the human brain and their changes in Alzheimer's disease. Neuroscience 1998, 82, 999–1007. [Google Scholar] [CrossRef]
- Shimohama, S.; Homma, Y.; Suenaga, T.; Fujimoto, S.; Taniguchi, T.; Araki, W.; Yamaoka, Y.; Takenawa, T.; Kimura, J. Aberrant accumulation of phospholipase C-delta in Alzheimer brains. Am. J. Pathol. 1991, 139, 737–742. [Google Scholar] [PubMed]
- Stokes, C.E.; Hawthorne, J.N. Reduced phosphoinositide concentrations in anterior temporal cortex of Alzheimer-diseased brains. J. Neurochem. 1987, 48, 1018–1021. [Google Scholar] [CrossRef]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barth, D.; Lückhoff, A.; Kühn, F.J.P. Species-Specific Regulation of TRPM2 by PI(4,5)P2 via the Membrane Interfacial Cavity. Int. J. Mol. Sci. 2021, 22, 4637. https://doi.org/10.3390/ijms22094637
Barth D, Lückhoff A, Kühn FJP. Species-Specific Regulation of TRPM2 by PI(4,5)P2 via the Membrane Interfacial Cavity. International Journal of Molecular Sciences. 2021; 22(9):4637. https://doi.org/10.3390/ijms22094637
Chicago/Turabian StyleBarth, Daniel, Andreas Lückhoff, and Frank J. P. Kühn. 2021. "Species-Specific Regulation of TRPM2 by PI(4,5)P2 via the Membrane Interfacial Cavity" International Journal of Molecular Sciences 22, no. 9: 4637. https://doi.org/10.3390/ijms22094637
APA StyleBarth, D., Lückhoff, A., & Kühn, F. J. P. (2021). Species-Specific Regulation of TRPM2 by PI(4,5)P2 via the Membrane Interfacial Cavity. International Journal of Molecular Sciences, 22(9), 4637. https://doi.org/10.3390/ijms22094637