
Histone Methylation Regulation in Neurodegenerative Disorders



Abstract
:1. Introduction
2. Epigenetic Changes: Environmental Conditions and Agents That Induce ND
3. PTM of DNA-Associated Histone Proteins
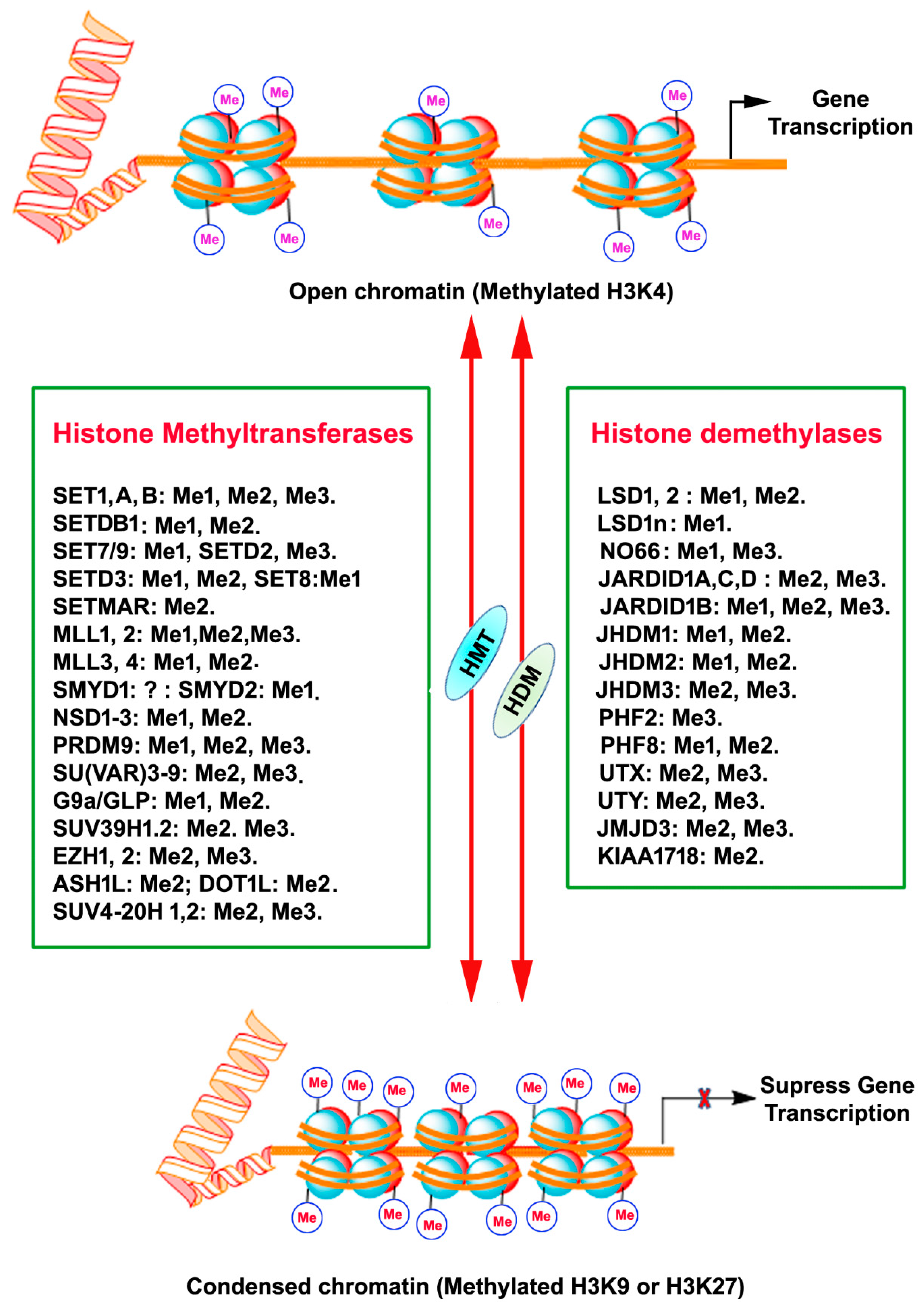
4. Histone Methylation
5. Histone Demethylation
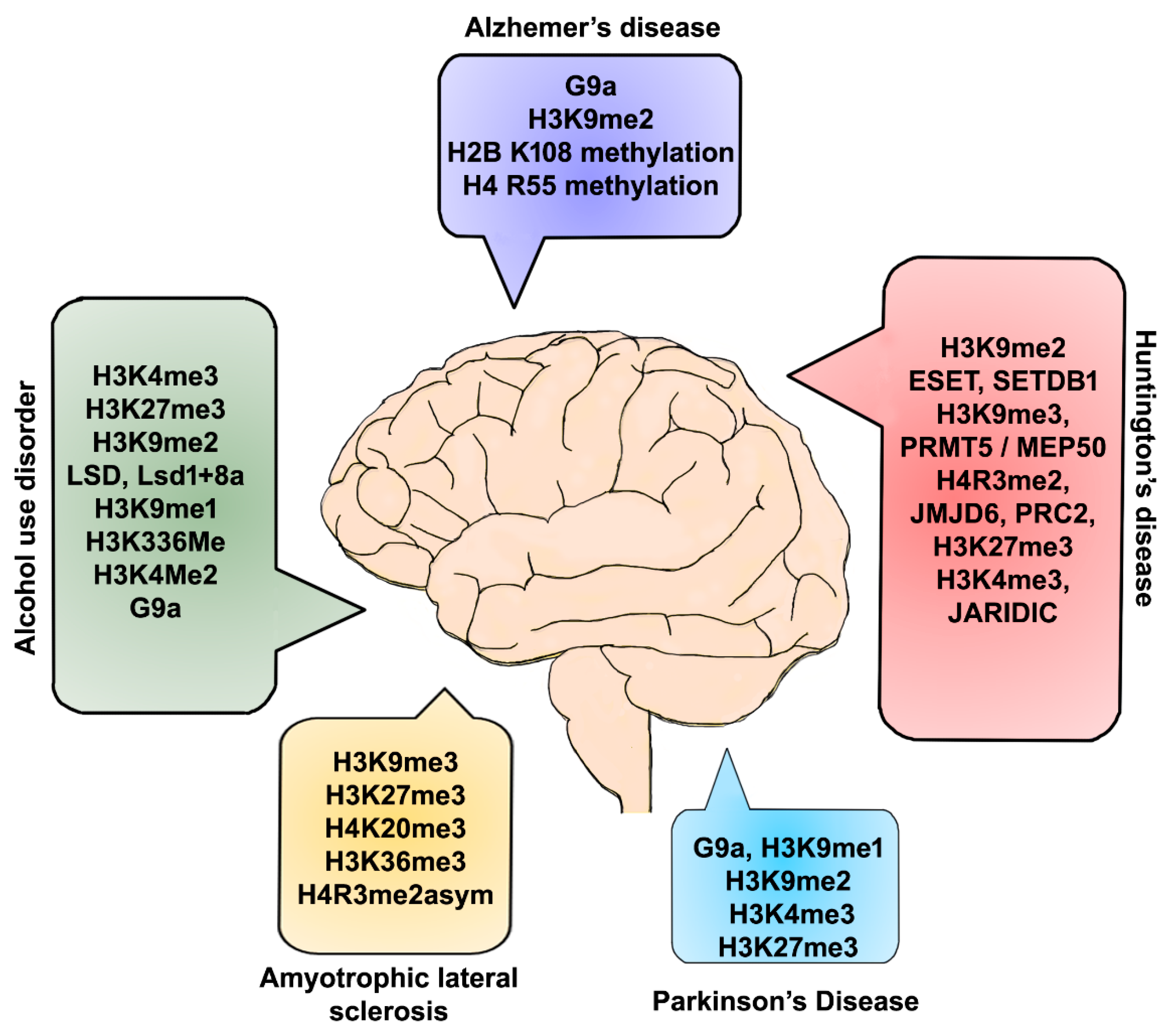
6. Alzheimer’s Disease (AD)
7. Huntington’s Disease (HD)
8. Parkinson’s Disease (PD)
9. Amyotrophic Lateral Sclerosis (ALS)
10. Alcohol Use Disorders (AUD)
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AD | Alzheimer’s disease |
| PD | Parkinson’s disease |
| HD | Huntington’s disease |
| ALS | Amyotrophic lateral sclerosis |
| ND | Neurodegenerative |
| miRNAs | micro RNAs |
| K | Lysine |
| HMT | Histone methyltransferases |
| HMD | Histone demethylases |
| H3K4me3 | Trimethylated Histone3 Lysine 4 |
| MLL2 | H3K4-specific histone methyltransferase |
| H3K9me2 | Dimethylated histone H3 lysine 9 |
| GLP/G9a | Histone 3 Lysine9-specific histone methyltransferase |
| H2B K108 | Histone 2B lysine 108 |
| H4 | Histone 4 |
| FAD | Fetal alcohol spectrum disorder |
| Htt | Huntingtin |
| ESET | ERG-associated protein with SET domain |
| SETDB1 | Histone-lysine N-methyltransferase domain bifurcated 1 |
| CREB | Cyclic AMP response element-binding |
| CBP | Cyclic AMP response element-binding protein |
| Ac | Acetylcholine |
| CHRM1 | Cholinergic Receptor Muscarinic 1 |
| PRMT5 | Protein arginine methyltransferase 5 |
| H2A | Histone 2A |
| H4 | Histone 4 |
| H4R3Me2 | Dimethylated histone 4 arginine3 |
| JMJD6 | H4R3Me2 demethylase, PRC2, Polycomb group protein |
| H3K27Me3 | Trimethylated Histone 3 lysine 2 |
| Hox | homeobox |
| Ezh2 | Enhancer of Zeste 2 |
| SUZ12 | Polycomb repressive complex 2 subunit |
| Utx | H3K27me3 demethylases |
| PFC | Prefrontal cortex |
| CAG | Cystine–Alanine–Guanine |
| REST/NRSF | RE-1 silencing transcription factor/neuron-restrictive silencer factor |
| REST | RE-1 silencing transcription factor |
| BDNF | Brain derived neurotropic factor |
| JARID1C | Jumonji AT-rich interactive domain 1C |
| REM | Rapid eye movement |
| H3K27 | Histone 3 Lysine 27 |
| FUS | fused in sarcoma |
| HDAC | Histone deacetylase |
| SNCA | alpha synuclein gene |
| α-SYN | alpha Synuclein |
| NRF2 | Nuclear Factor erythroid-2-Related Factor 2 |
| Hmox1 | Heme oxygenase decycling 1 |
| Ngf | Nerve growth factor |
| Vgf | Nerve growth factor inducible |
| SYN | Synaptophysin |
| SH-SY5Y | Human Bone Marrow Neuroblast Cell Line |
| H3K9me1 | Mono methylated Histone 3 lysine 9 |
| iPSCs | Induced Pluripotent Stem Cells |
| NeuN | Neuronal nuclei |
| FAS | Fetal alcohol syndrome |
| FASD | Fetal alcohol spectrum disorders |
| AUD | Alcohol use disorder |
| HP | Hippocampus |
| NC | neocortex PDYN, Prodynorphin |
| PNOC | Pronociceptin |
| CIE | Chronic intermittent ethanol |
| NMDA | N-methyl-D-aspartate |
| NR2B | N-methyl-D-aspartate receptor 2B subunit |
| Setd1a | SET Domain Containing 1A, Histone Lysine Methyltransferase |
| Setd1b | SET Domain Containing 1b, Histone Lysine Methyltransferase |
| Setdb2 | SET Domain Containing b2, Histone Lysine Methyltransferase |
| Suv39 h1 | suppressor of variegation 3–9 homolog 1 |
| Setd4 | SET Domain Containing 4 |
| Setd6 | SET domain containing 6 |
| Setdb1 | SET Domain Bifurcated Histone Lysine Methyltransferase 1 |
| Prmt6 | Protein Arginine Methyltransferase 6 |
| AIE | adolescent intermittent ethanol |
| c-FOS | Fos-related antigen 1 |
| Cdk5 | Cyclin Dependent Kinase 5 |
| FosB | Proto-Oncogene, AP-1 Transcription Factor Subunit |
| TDP-43 | transactive response DNA binding protein with Mr 43 kD (TDP-43) |
| LSD1 | lysine demethylase 1 |
| CeA | Central nucleus of the amygdala |
| MeA | medial amygdala |
| PRDM2) | Histone methyltransferase PR domain containing 2, with ZNF domain |
| Mag | Myelin-associated glycoprotein |
| mab | Myelin basic protein |
| Mobp | Myelin-associated oligodendrocytic basic protein |
| Plp | Myelin proteolipid protein |
| PAE | Prenatal alcohol exposure |
| ARC | β-endorphin protein in the arcuate |
| PEE | postnatal ethanol exposure |
| VGLUT2 | vesicular glutamate transporter |
| CNS | central nervous system |
| Kmt2 | lysine methyltransferase 2E |
References
- Basavarajappa, B.S.; Shivakumar, M.; Joshi, V.; Subbanna, S. Endocannabinoid system in neurodegenerative disorders. J. Neurochem. 2017, 142, 624–648. [Google Scholar] [CrossRef]
- Ferrari, R.; Kapogiannis, D.; Huey, E.D.; Momeni, P. FTD and ALS: A tale of two diseases. Curr. Alzheimer Res. 2011, 8, 273–294. [Google Scholar] [CrossRef] [PubMed]
- Gibson, S.B.; Figueroa, K.P.; Bromberg, M.B.; Pulst, S.M.; Cannon-Albright, L. Familial clustering of ALS in a population-based resource. Neurology 2014, 82, 17–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, S.; Al Khleifat, A.; Al-Chalabi, A. What causes amyotrophic lateral sclerosis? F1000Research 2017, 6, 371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hely, M.A.; Morris, J.G.; Reid, W.G.; Trafficante, R. Sydney Multicenter Study of Parkinson’s disease: Non-L-dopa-responsive problems dominate at 15 years. Mov. Disord. 2005, 20, 190–199. [Google Scholar] [CrossRef] [PubMed]
- Reid, W.G.; Hely, M.A.; Morris, J.G.; Loy, C.; Halliday, G.M. Dementia in Parkinson’s disease: A 20-year neuropsychological study (Sydney Multicentre Study). J. Neurol. Neurosurg. Psychiatry 2011, 82, 1033–1037. [Google Scholar] [CrossRef] [PubMed]
- Panza, F.; Lozupone, M.; Seripa, D.; Daniele, A.; Watling, M.; Giannelli, G.; Imbimbo, B.P. Development of disease-modifying drugs for frontotemporal dementia spectrum disorders. Nat. Rev. Neurol. 2020, 16, 213–228. [Google Scholar] [CrossRef]
- Peng, C.; Trojanowski, J.Q.; Lee, V.M. Protein transmission in neurodegenerative disease. Nat. Rev. Neurol. 2020, 16, 199–212. [Google Scholar] [CrossRef]
- Bennett, S.A.; Tanaz, R.; Cobos, S.N.; Torrente, M.P. Epigenetics in amyotrophic lateral sclerosis: A role for histone post-translational modifications in neurodegenerative disease. Transl. Res. 2019, 204, 19–30. [Google Scholar] [CrossRef]
- Deans, C.; Maggert, K.A. What do you mean, “epigenetic”? Genetics 2015, 199, 887–896. [Google Scholar] [CrossRef] [Green Version]
- Jablonka, E.; Lamb, M.J. Epigenetic inheritance. In International Encyclopedia of the Social & Behavioral Sciences; Baltes, P.B., Smelser, N.J., Eds.; Pergamon: Oxford, UK, 2001; pp. 4706–4710. [Google Scholar]
- Burggren, W. Epigenetic Inheritance and Its Role in Evolutionary Biology: Re-Evaluation and New Perspectives. Biology 2016, 5, 24. [Google Scholar] [CrossRef]
- Peaston, A.E.; Whitelaw, E. Epigenetics and phenotypic variation in mammals. Mamm. Genome 2006, 17, 365–374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Basavarajappa, B.S.; Subbanna, S. Epigenetic Mechanisms in Developmental Alcohol-Induced Neurobehavioral Deficits. Brain Sci. 2016, 6, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, Y.; Langley, B.; Lubin, F.D.; Renthal, W.; Wood, M.A.; Yasui, D.H.; Kumar, A.; Nestler, E.J.; Akbarian, S.; Beckel-Mitchener, A.C. Epigenetics in the nervous system. J. Neurosci. 2008, 28, 11753–11759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luger, K.; Dechassa, M.L.; Tremethick, D.J. New insights into nucleosome and chromatin structure: An ordered state or a disordered affair? Nat. Rev. Mol. Cell. Biol. 2012, 13, 436–447. [Google Scholar] [CrossRef] [Green Version]
- Smolle, M.; Workman, J.L. Transcription-associated histone modifications and cryptic transcription. Biochim. Biophys. Acta 2013, 1829, 84–97. [Google Scholar] [CrossRef]
- Barski, A.; Cuddapah, S.; Kairong, C.; Roh, T.-Y.; Schones, D.E.; Wang, Z.; Wei, G.; Chepelev, I.; Zhao, K. High-resolution profiling of histone methylations in the human genome. Cell 2007, 129, 823–837. [Google Scholar] [CrossRef] [Green Version]
- Heintzman, N.D.; Stuart, R.K.; Hon, G.; Fu, Y.; Ching, C.W.; Hawkins, R.D.; Barrera, L.O.; Calcar, S.V.; Qu, C.; Ching, K.A.; et al. Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nat. Genet. 2007, 39, 311–318. [Google Scholar] [CrossRef]
- Shi, X.; Hong, T.; Walter, K.L.; Ewalt, M.; Michishita, E.; Hung, T.; Carney, D.; Pena, P.; Lan, F.; Kaadige, M.R.; et al. ING2 PHD domain links histone H3 lysine 4 methylation to active gene repression. Nature 2006, 442, 96–99. [Google Scholar] [CrossRef]
- Shi, X.; Kachirskaia, I.; Walter, K.L.; Kuo, J.-H.A.; Lake, A.; Davrazou, F.; Chan, S.M.; Martin, D.G.E.; Fingerman, I.M.; Briggs, S.D.; et al. Proteome-wide analysis in Saccharomyces cerevisiae identifies several PHD fingers as novel direct and selective binding modules of histone H3 methylated at either lysine 4 or lysine 36. J. Biol. Chem. 2007, 282, 2450–2455. [Google Scholar] [CrossRef] [Green Version]
- Wysocka, J.; Swigut, T.; Xiao, H.; Milne, T.A.; Kwon, S.Y.; Landry, J.; Kauer, M.; Tackett, A.J.; Chait, B.T.; Badenhorst, P.; et al. A PHD finger of NURF couples histone H3 lysine 4 trimethylation with chromatin remodelling. Nature 2006, 442, 86–90. [Google Scholar] [CrossRef] [PubMed]
- Xiao, B.; Jing, C.; Wilson, J.R.; Walker, P.A.; Vasisht, N.; Kelly, G.; Howell, S.; Taylor, I.A.; Blackburn, G.M.; Gamblin, S.J. Structure and catalytic mechanism of the human histone methyltransferase SET7/9. Nature 2003, 421, 652–656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allis, C.D.; Berger, S.L.; Cote, J.; Dent, S.; Jenuwien, T.; Kouzarides, T.; Pillus, L.; Reinberg, D.; Shi, Y.; Shiekhattar, R.; et al. New nomenclature for chromatin-modifying enzymes. Cell 2007, 131, 633–636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qian, C.; Zhou, M.M. SET domain protein lysine methyltransferases: Structure, specificity and catalysis. Cell. Mol. Life Sci. 2006, 63, 2755–2763. [Google Scholar] [CrossRef] [PubMed]
- Aravind, L.; Abhiman, S.; Iyer, L.M. Natural history of the eukaryotic chromatin protein methylation system. Prog. Mol. Biol. Transl. Sci. 2011, 101, 105–176. [Google Scholar]
- Mozzetta, C.; Boyarchuk, E.; Pontis, J.; Ait-Si-Ali, S. Sound of silence: The properties and functions of repressive Lys methyltransferases. Nat. Rev. Mol. Cell. Biol. 2015, 16, 499–513. [Google Scholar] [CrossRef]
- Glaser, S.; Shaft, J.; Lubitz, S.; Vintersten, K.; van der Hoeven, F.; Tufteland, K.R.; Aasland, R.; Anastassiadis, K.; Ang, S.-L.; Stewart, A.F. Multiple epigenetic maintenance factors implicated by the loss of Mll2 in mouse development. Development 2006, 133, 1423–1432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goo, Y.H.; Sohn, Y.C.; Kim, D.H.; Kim, S.W.; Kang, M.J.; Jung, D.J.; Kwak, E.; Barlev, N.A.; Berger, S.L.; Chow, V.T.; et al. Activating signal cointegrator 2 belongs to a novel steady-state complex that contains a subset of trithorax group proteins. Mol. Cell. Biol. 2003, 23, 140–149. [Google Scholar] [CrossRef] [Green Version]
- Milne, T.A.; Briggs, S.D.; Brock, H.W.; Martin, M.E.; Gibbs, D.; Allis, C.D.; Hess, J.L. MLL targets SET domain methyltransferase activity to Hox gene promoters. Mol. Cell. 2002, 10, 1107–1117. [Google Scholar] [CrossRef]
- Bernstein, B.E.; Humphrey, E.L.; Erlich, R.L.; Schneider, R.; Bouman, P.; Liu, J.S.; Kouzarides, T.; Schreiber, S.L. Methylation of histone H3 Lys 4 in coding regions of active genes. Proc. Natl. Acad. Sci. USA 2002, 99, 8695–8700. [Google Scholar] [CrossRef] [Green Version]
- Greer, E.L.; Shi, Y. Histone methylation: A dynamic mark in health, disease and inheritance. Nat. Rev. Genet. 2012, 13, 343–357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peters, A.H.; O’Carroll, D.; Scherthan, H.; Mechtler, K.; Sauer, S.; Schöfer, C.; Weipoltshammer, K.; Pagani, M.; Lachner, M.; Kohlmaier, A.; et al. Loss of the Suv39h histone methyltransferases impairs mammalian heterochromatin and genome stability. Cell 2001, 107, 323–337. [Google Scholar] [CrossRef] [Green Version]
- Subbanna, S.; Shivakumar, M.; Umapathy, N.S.; Saito, M.; Mohan, P.S.; Kumar, A.; Nixon, R.A.; Verin, A.D.; Psychoyos, D.; Basavarajappa, B.S. G9a-Mediated Histone Methylation Regulates Ethanol-Induced Neurodegeneration in the Neonatal Mouse Brain. Neurobiol. Dis. 2013, 54, 475–485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tachibana, M.; Shivakumar, M.; Umapathy, N.S.; Saito, M.; Mohan, P.S.; Kumar, A.; Nixon, R.A.; Verin, A.D.; Psychoyos, D.; Basavarajappa, B.S. G9a histone methyltransferase plays a dominant role in euchromatic histone H3 lysine 9 methylation and is essential for early embryogenesis. Genes. Dev. 2002, 16, 1779–1791. [Google Scholar] [CrossRef] [Green Version]
- Tachibana, M.; Ueda, J.; Fukuda, M.; Takeda, N.; Ohta, T.; Iwanari, H.; Sakihama, T.; Kodama, T.; Hamakubo, T.; Shinkai, Y. Histone methyltransferases G9a and GLP form heteromeric complexes and are both crucial for methylation of euchromatin at H3-K9. Genes. Dev. 2005, 19, 815–826. [Google Scholar] [CrossRef] [Green Version]
- Schultz, D.C.; Ayyanathan, K.; Negorev, D.; Maul, G.G.; Rauscher, F.J. 3rd. SETDB1: A novel KAP-1-associated histone H3, lysine 9-specific methyltransferase that contributes to HP1-mediated silencing of euchromatic genes by KRAB zinc-finger proteins. Genes. Dev. 2002, 16, 919–932. [Google Scholar] [CrossRef] [Green Version]
- Strahl, B.D.; Grant, P.A.; Briggs, S.D.; Sun, Z.W.; Bone, J.R.; Caldwell, J.A.; Mollah, S.; Cook, R.G.; Shabanowitz, J.; Hunt, D.F. Set2 is a nucleosomal histone H3-selective methyltransferase that mediates transcriptional repression. Mol. Cell. Biol. 2002, 22, 1298–1306. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.; Perez-Burgos, L.; Placek, B.J.; Sengupta, R.; Richter, M.; Dorsey, J.A.; Kubicek, S.; Opravil, S.; Jenuwein, T.; Berger, S.L. Repression of p53 activity by Smyd2-mediated methylation. Nature 2006, 444, 629–632. [Google Scholar] [CrossRef]
- Lu, T.; Jackson, M.W.; Wang, B.; Yang, M.; Chance, M.R.; Miyagi, M.; Gudkov, A.V.; Stark, G.R. Regulation of NF-kappaB by NSD1/FBXL11-dependent reversible lysine methylation of p65. Proc. Natl. Acad. Sci. USA 2010, 107, 46–51. [Google Scholar] [CrossRef] [Green Version]
- Saddic, L.A.; West, L.E.; Aslanian, A.; Yates, J.R., 3rd; Rubin, S.M.; Gozani, O.; Sage, J. Methylation of the retinoblastoma tumor suppressor by SMYD2. J. Biol. Chem. 2010, 285, 37733–37740. [Google Scholar] [CrossRef] [Green Version]
- Steger, D.J.; Lefterova, M.I.; Ying, L.; Stonestrom, A.J.; Schupp, M.; Zhuo, D.; Vakoc, A.L.; Kim, J.E.; Chen, J.; Lazar, M.A.; et al. DOT1L/KMT4 recruitment and H3K79 methylation are ubiquitously coupled with gene transcription in mammalian cells. Mol. Cell. Biol. 2008, 28, 2825–2839. [Google Scholar] [CrossRef] [Green Version]
- Karachentsev, D.; Sarma, K.; Reinberg, D.; Steward, R. PR-Set7-dependent methylation of histone H4 Lys 20 functions in repression of gene expression and is essential for mitosis. Genes. Dev. 2005, 19, 431–435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Joshi, P.; Miller, E.L.; Higgins, L.; Slattery, M.; Simon, J.A. A Role for Monomethylation of Histone H3-K27 in Gene Activity in Drosophila. Genetics 2018, 208, 1023–1036. [Google Scholar] [CrossRef] [Green Version]
- Jacob, Y.; Feng, S.; LeBlanc, C.A.; Bernatavichute, Y.V.; Stroud, H.; Cokus, S.; Johnson, L.M.; Pellegrini, M.; Jacobsen, S.E.; Michaels, S.D. ATXR5 and ATXR6 are H3K27 monomethyltransferases required for chromatin structure and gene silencing. Nat. Struct. Mol. Biol. 2009, 16, 763–768. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Molascon, A.J.; Gao, S.; Liu, Y.; Andrews, P.C. Quantitative proteomics reveals that the specific methyltransferases Txr1p and Ezl2p differentially affect the mono-, di- and trimethylation states of histone H3 lysine 27 (H3K27). Mol. Cell. Proteomics 2013, 12, 1678–1688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, M.G.; Villa, R.; Trojer, P.; Norman, J.; Yan, K.P.; Reinberg, D.; Di Croce, L.; Shiekhattar, R. Demethylation of H3K27 regulates polycomb recruitment and H2A ubiquitination. Science 2007, 318, 447–450. [Google Scholar] [CrossRef] [PubMed]
- Lund, A.H.; van Lohuizen, M. Polycomb complexes and silencing mechanisms. Curr. Opin. Cell. Biol. 2004, 16, 239–246. [Google Scholar] [CrossRef] [PubMed]
- Otte, A.P.; Kwaks, T.H. Gene repression by Polycomb group protein complexes: A distinct complex for every occasion? Curr. Opin. Genet. Dev. 2003, 13, 448–454. [Google Scholar] [CrossRef]
- Ciccone, D.N.; Su, H.; Hevi, S.; Gay, F.; Lei, H.; Bajko, J.; Xu, G.; Li, E.; Chen, T. KDM1B is a histone H3K4 demethylase required to establish maternal genomic imprints. Nature 2009, 461, 415–418. [Google Scholar] [CrossRef]
- Jin, L.; Hanigan, C.L.; Wu, Y.; Wang, W.; Park, B.H.; Woster, P.M.; Casero, R.A. Loss of LSD1 (lysine-specific demethylase 1) suppresses growth and alters gene expression of human colon cancer cells in a p53- and DNMT1(DNA methyltransferase 1)-independent manner. Biochem. J. 2013, 449, 459–468. [Google Scholar] [CrossRef]
- Van Essen, D.; Zhu, Y.; Saccani, S. A feed-forward circuit controlling inducible NF-kappaB target gene activation by promoter histone demethylation. Mol. Cell. 2010, 39, 750–760. [Google Scholar] [CrossRef]
- Seward, D.J.; Cubberley, G.; Kim, S.; Schonewald, M.; Zhang, L.; Tripet, B.; Bentley, D.L. Demethylation of trimethylated histone H3 Lys4 in vivo by JARID1 JmjC proteins. Nat. Struct. Mol. Biol. 2007, 14, 240–242. [Google Scholar] [CrossRef]
- Xiang, Y.; Zhu, Z.; Han, G.; Ye, X.; Xu, B.; Peng, Z.; Ma, Y.; Yu, Y.; Lin, H.; Chen, A.P. JARID1B is a histone H3 lysine 4 demethylase up-regulated in prostate cancer. Proc. Natl. Acad. Sci. USA 2007, 104, 19226–19231. [Google Scholar] [CrossRef] [Green Version]
- Tsukada, Y.; Fang, J.; Erdjument-Bromage, H.; Warren, M.E.; Borchers, C.H.; Tempst, P.; Zhang, Y. Histone demethylation by a family of JmjC domain-containing proteins. Nature 2006, 439, 811–816. [Google Scholar] [CrossRef]
- Yamane, K.; Toumazou, C.; Tsukada, Y.; Erdjument-Bromage, H.; Tempst, P.; Wong, J.; Zhang, Y. JHDM2A, a JmjC-containing H3K9 demethylase, facilitates transcription activation by androgen receptor. Cell 2006, 125, 483–495. [Google Scholar] [CrossRef] [Green Version]
- Fodor, B.D.; Kubicek, S.; Yonezawa, M.; O’Sullivan, R.J.; Sengupta, R.; Perez-Burgos, L.; Opravil, S.; Mechtler, K. Jmjd2b antagonizes H3K9 trimethylation at pericentric heterochromatin in mammalian cells. Genes. Dev. 2006, 20, 1557–1562. [Google Scholar] [CrossRef] [Green Version]
- Klose, R.J.; Yamane, K.; Bae, Y.; Zhang, D.; Erdjument-Bromage, H.; Tempst, P.; Wong, J.; Zhang, Y. The transcriptional repressor JHDM3A demethylates trimethyl histone H3 lysine 9 and lysine 36. Nature 2006, 442, 312–316. [Google Scholar] [CrossRef]
- Whetstine, J.R.; Nottke, A.; Lan, F.; Huarte, M.; Smolikov, S.; Chen, Z.; Spooner, E.; Li, E.; Zhang, G.; Colaiacovo, M. Reversal of histone lysine trimethylation by the JMJD2 family of histone demethylases. Cell 2006, 125, 467–481. [Google Scholar] [CrossRef] [Green Version]
- Adams-Cioaba, M.A.; Min, J. Structure and function of histone methylation binding proteins. Biochem. Cell. Biol. 2009, 87, 93–105. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Fang, J.; Bedford, M.T.; Zhang, Y.; Xu, R.M. Recognition of histone H3 lysine-4 methylation by the double tudor domain of JMJD2A. Science 2006, 312, 748–751. [Google Scholar] [CrossRef]
- Christensen, J.; Agger, K.; Cloos, P.A.; Pasini, D.; Rose, S.; Sennels, L.; Rappsilber, J.; Hansen, K.H.; Salcini, A.E.; Helin, K. RBP2 belongs to a family of demethylases, specific for tri-and dimethylated lysine 4 on histone 3. Cell 2007, 128, 1063–1076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iwase, S.; Lan, F.; Bayliss, P.; de la Torre-Ubieta, L.; Huarte, M.; Qi, H.H.; Whetstine, J.R.; Bonni, A.; Roberts, T.M.; Shi, Y. The X-linked mental retardation gene SMCX/JARID1C defines a family of histone H3 lysine 4 demethylases. Cell 2007, 128, 1077–1088. [Google Scholar] [CrossRef] [Green Version]
- Lee, M.G.; Norman, J.; Shilatifard, A.; Shiekhattar, R. Physical and functional association of a trimethyl H3K4 demethylase and Ring6a/MBLR, a polycomb-like protein. Cell 2007, 128, 877–887. [Google Scholar] [CrossRef] [Green Version]
- Tahiliani, M.; Mei, P.; Fang, R.; Leonor, T.; Rutenberg, M.; Shimizu, F.; Li, J.; Rao, A.; Shi, Y. The histone H3K4 demethylase SMCX links REST target genes to X-linked mental retardation. Nature 2007, 447, 601–605. [Google Scholar] [CrossRef] [PubMed]
- Yamane, K.; Tateishi, K.; Klose, R.J.; Fang, J.; Fabrizio, L.A.; Erdjument-Bromage, H.; Taylor-Papadimitriou, J.; Tempst, P.; Zhang, Y. PLU-1 is an H3K4 demethylase involved in transcriptional repression and breast cancer cell proliferation. Mol. Cell. 2007, 25, 801–812. [Google Scholar] [CrossRef]
- Wang, G.G.; Song, J.; Wang, Z.; Dormann, H.L.; Casadio, F.; Li, H.; Luo, J.L.; Patel, D.J.; Allis, C.D. Haematopoietic malignancies caused by dysregulation of a chromatin-binding PHD finger. Nature 2009, 459, 847–851. [Google Scholar] [CrossRef] [Green Version]
- Agger, K.; Cloos, P.A.; Christensen, J.; Pasini, D.; Rose, S.; Rappsilber, J.; Canaani, E.; Salcini, A.E.; Helin, K. UTX and JMJD3 are histone H3K27 demethylases involved in HOX gene regulation and development. Nature 2007, 449, 731–734. [Google Scholar] [CrossRef]
- De Santa, F.; Cloos, P.A.; Christensen, J.; Pasini, D.; Rose, S.; Rappsilber, J.; Issaeva, I.; Canaani, E.; Salcini, A.E.; Helin, K. The histone H3 lysine-27 demethylase Jmjd3 links inflammation to inhibition of polycomb-mediated gene silencing. Cell 2007, 130, 1083–1094. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, S.; Broun, A.; Ge, K. Identification of JmjC domain-containing UTX and JMJD3 as histone H3 lysine 27 demethylases. Proc. Natl. Acad. Sci. USA 2007, 104, 18439–18444. [Google Scholar] [CrossRef] [Green Version]
- Xiang, Y.; Zhu, Z.; Han, G.; Lin, H.; Xu, L.; Chen, C.D. JMJD3 is a histone H3K27 demethylase. Cell Res. 2007, 17, 850–857. [Google Scholar] [CrossRef] [Green Version]
- Baba, A.; Ohtake, F.; Okuno, Y.; Yokota, K.; Okada, M.; Imai, Y.; Ni, M.; Meyer, C.A.; Igarashi, K.; Kanno, J.; et al. PKA-dependent regulation of the histone lysine demethylase complex PHF2-ARID5B. Nat. Cell. Biol. 2011, 13, 668–675. [Google Scholar] [CrossRef]
- Feng, W.; Yonezawa, M.; Ye, J.; Jenuwein, T.; Grummt, I. PHF8 activates transcription of rRNA genes through H3K4me3 binding and H3K9me1/2 demethylation. Nat. Struct. Mol. Biol. 2010, 17, 445–450. [Google Scholar] [CrossRef] [PubMed]
- Fortschegger, K.; de Graaf, P.; Outchkourov, N.S.; van Schaik, F.M.; Timmers, H.T.; Shiekhattar, R. PHF8 targets histone methylation and RNA polymerase II to activate transcription. Mol. Cell. Biol. 2010, 30, 3286–3298. [Google Scholar] [CrossRef] [Green Version]
- Kleine-Kohlbrecher, D.; Christensen, J.; Vandamme, J.; Abarrategui, I.; Bak, M.; Tommerup, N.; Shi, X.; Gozani, O.; Rappsilber, J.; Salcini, A.E.; et al. A functional link between the histone demethylase PHF8 and the transcription factor ZNF711 in X-linked mental retardation. Mol. Cell. 2010, 38, 165–178. [Google Scholar] [CrossRef] [Green Version]
- Qiu, J.; Shi, G.; Jia, Y.; Li, J.; Wu, M.; Li, J.; Dong, S.; Wong, J. The X-linked mental retardation gene PHF8 is a histone demethylase involved in neuronal differentiation. Cell Res. 2010, 20, 908–918. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Sun, L.; Li, Q.; Liang, J.; Yu, W.; Yi, X.; Yang, X.; Li, Y.; Han, X.; Zhang, Y.; et al. Histone demethylase JMJD2B coordinates H3K4/H3K9 methylation and promotes hormonally responsive breast carcinogenesis. Proc. Natl. Acad. Sci. USA 2011, 108, 7541–7546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wen, H.; Li, J.; Song, T.; Lu, M.; Kan, P.Y.; Lee, M.G.; Sha, B.; Shi, X. Recognition of histone H3K4 trimethylation by the plant homeodomain of PHF2 modulates histone demethylation. J. Biol. Chem. 2010, 285, 9322–9326. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Z.; Wang, Y.; Li, X.; Wang, Y.; Xu, L.; Wang, X.; Sun, T.; Dong, X.; Chen, L.; Mao, H.; et al. PHF8 is a histone H3K9me2 demethylase regulating rRNA synthesis. Cell Res. 2010, 20, 794–801. [Google Scholar] [CrossRef]
- Liu, W.; Tanasa, B.; Tyurina, O.V.; Zhou, T.Y.; Gassmann, R.; Liu, W.T.; Ohgi, K.A.; Benner, C.; Garcia-Bassets, I.; Aggarwal, A.K.; et al. PHF8 mediates histone H4 lysine 20 demethylation events involved in cell cycle progression. Nature 2010, 466, 508–512. [Google Scholar] [CrossRef] [Green Version]
- Horton, J.R.; Upadhyay, A.K.; Qi, H.H.; Zhang, X.; Shi, Y.; Cheng, X. Enzymatic and structural insights for substrate specificity of a family of jumonji histone lysine demethylases. Nat. Struct. Mol. Biol. 2010, 17, 38–43. [Google Scholar] [CrossRef]
- Qi, H.H.; Sarkissian, M.; Hu, G.-Q.; Wang, Z.; Bhattacharjee, A.; Gordon, D.B.; Gonzales, M.; Lan, F.; Ongusaha, P.P.; Huarte, M.; et al. Histone H4K20/H3K9 demethylase PHF8 regulates zebrafish brain and craniofacial development. Nature 2010, 466, 503–507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, L.; Wang, Y.; Huang, S.; Wang, J.; Deng, Z.; Zhang, Q.; Wu, W.; Zhang, X.; Liu, Z.; Gong, W.; et al. Structural insights into a novel histone demethylase PHF8. Cell Res. 2010, 20, 166–173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collins, B.E.; Greer, C.B.; Coleman, B.C.; Sweatt, J.D. Histone H3 lysine K4 methylation and its role in learning and memory. Epigenetics Chromatin 2019, 12, 7. [Google Scholar] [CrossRef]
- Cruz, C.; Della Rosa, M.; Krueger, C.; Gao, Q.; Horkai, D.; King, M.; Field, L.; Houseley, J. Tri-methylation of histone H3 lysine 4 facilitates gene expression in ageing cells. Elife 2018, 7, e34081. [Google Scholar] [CrossRef]
- Lardenoije, R.; Iatrou, A.; Kenis, G.; Kompotis, K.; Steinbusch, H.W.; Mastroeni, D.; Coleman, P.; Lemere, C.A.; Hof, P.R.; Hove, D.L.V.D.; et al. The epigenetics of aging and neurodegeneration. Prog. Neurobiol. 2015, 131, 21–64. [Google Scholar]
- Mehler, M.F. Epigenetic principles and mechanisms underlying nervous system functions in health and disease. Prog. Neurobiol. 2008, 86, 305–341. [Google Scholar] [CrossRef] [Green Version]
- Mitchell, A.C.; Roussos, P.; Peter, C.J.; Tsankova, N.; Akbarian, S. The future of neuroepigenetics in the human brain. Prog. Mol. Biol. Transl. Sci. 2014, 128, 199–228. [Google Scholar] [PubMed] [Green Version]
- Woldemichael, B.T.; Bohacek, J.; Gapp, K.; Mansuy, I.M. Epigenetics of memory and plasticity. Prog. Mol. Biol. Transl. Sci. 2014, 122, 305–340. [Google Scholar]
- Fischer, A. Targeting histone-modifications in Alzheimer’s disease. What is the evidence that this is a promising therapeutic avenue? Neuropharmacology 2014, 80, 95–102. [Google Scholar] [CrossRef] [PubMed]
- Anderson, K.W.; Turko, I.V. Histone post-translational modifications in frontal cortex from human donors with Alzheimer’s disease. Clin. Proteomics. 2015, 12, 26. [Google Scholar] [CrossRef] [Green Version]
- Lithner, C.U.; Lacor, P.N.; Zhao, W.Q.; Mustafiz, T.; Klein, W.L.; Sweatt, J.D.; Hernandez, C.M. Disruption of neocortical histone H3 homeostasis by soluble Abeta: Implications for Alzheimer’s disease. Neurobiol. Aging 2013, 34, 2081–2090. [Google Scholar] [CrossRef] [PubMed]
- Sharma, M.; Dierkes, T.; Sajikumar, S. Epigenetic regulation by G9a/GLP complex ameliorates amyloid-beta 1-42 induced deficits in long-term plasticity and synaptic tagging/capture in hippocampal pyramidal neurons. Aging Cell 2017, 16, 1062–1072. [Google Scholar] [CrossRef] [Green Version]
- Zheng, Y.; Liu, A.; Wang, Z.J.; Cao, Q.; Wang, W.; Lin, L.; Ma, K.; Zhang, F.; Wei, J.; Matas, E.; et al. Inhibition of EHMT1/2 rescues synaptic and cognitive functions for Alzheimer’s disease. Brain 2019, 142, 787–807. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.; Liu, A.; Li, H.; Feng, J.; Yan, Z. Inhibition of Histone Methyltransferases EHMT1/2 Reverses Amyloid-beta-Induced Loss of AMPAR Currents in Human Stem Cell-Derived Cortical Neurons. J. Alzheimers. Dis. 2019, 70, 1175–1185. [Google Scholar] [CrossRef] [PubMed]
- Calderón-Garcidueñas, L.; Herrera-Soto, A.; Jury, N.; Maher, B.A.; González-Maciel, A.; Reynoso-Robles, R.; Ruiz-Rudolph, P.; van Zundert, B.; Varela-Nallar, L. Reduced repressive epigenetic marks, increased DNA damage and Alzheimer’s disease hallmarks in the brain of humans and mice exposed to particulate urban air pollution. Environ. Res. 2020, 183, 109226. [Google Scholar] [CrossRef] [PubMed]
- Griñán-Ferré, C.; Marsal-García, L.; Bellver-Sanchis, A.; Kondengaden, S.M.; Turga, R.C.; Vázquez, S.; Pallàs, M. Pharmacological inhibition of G9a/GLP restores cognition and reduces oxidative stress, neuroinflammation and beta-Amyloid plaques in an early-onset Alzheimer’s disease mouse model. Aging 2019, 11, 11591–11608. [Google Scholar] [CrossRef]
- Mostafavi, S.; Gaiteri, C.; Sullivan, S.E.; White, C.C.; Tasaki, S.; Xu, J.; Taga, M.; Klein, H.-U.; Patrick, E.; Komashko, V.; et al. A molecular network of the aging human brain provides insights into the pathology and cognitive decline of Alzheimer’s disease. Nat. Neurosci. 2018, 21, 811–819. [Google Scholar] [CrossRef]
- Hernández-Ortega, K.; Garcia-Esparcia, P.; Gil, L.; Lucas, J.J.; Ferrer, I. Altered Machinery of Protein Synthesis in Alzheimer’s: From the Nucleolus to the Ribosome. Brain Pathol. 2016, 26, 593–605. [Google Scholar] [CrossRef]
- Wan, G.; Zhou, W.; Hu, Y.; Ma, R.; Jin, S.; Liu, G.; Jiang, Q. Transcriptional Regulation of lncRNA Genes by Histone Modification in Alzheimer’s Disease. Biomed. Res. Int. 2016, 2016, 3164238. [Google Scholar] [CrossRef] [Green Version]
- Smith, A.R.; Smith, R.G.; Macdonald, R.; Marzi, S.J.; Burrage, J.; Troakes, C.; Al-Sarraj, S.; Mill, J.; Lunnon, K. The histone modification H3K4me3 is altered at the ANK1 locus in Alzheimer’s disease brain. Future Sci. 2021, 7. [Google Scholar] [CrossRef]
- Dyer, M.; Phipps, A.J.; Mitew, S.; Taberlay, P.C.; Woodhouse, A. Age, but Not Amyloidosis, Induced Changes in Global Levels of Histone Modifications in Susceptible and Disease-Resistant Neurons in Alzheimer’s Disease Model Mice. Front. Aging Neurosci. 2019, 11, 68. [Google Scholar] [CrossRef] [Green Version]
- Mastroeni, D.; Delvaux, E.; Nolz, J.; Tan, Y.; Grover, A.; Oddo, S.; Coleman, P.D. Aberrant intracellular localization of H3k4me3 demonstrates an early epigenetic phenomenon in Alzheimer’s disease. Neurobiol. Aging 2015, 36, 3121–3129. [Google Scholar] [CrossRef] [Green Version]
- Cao, Q.; Wang, W.; Williams, J.B.; Yang, F.; Wang, Z.-J.; Yan, Z. Targeting histone K4 trimethylation for treatment of cognitive and synaptic deficits in mouse models of Alzheimer’s disease. Sci Adv. 2020, 6. [Google Scholar] [CrossRef] [PubMed]
- THsDCR Group. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. The Huntington’s Disease Collaborative. Res. Group Cell. 1993, 72, 971–983. [Google Scholar]
- MacDonald, M.E.; Gines, S.; Gusella, J.F.; Wheeler, V.C. Huntington’s disease. Neuromol. Med. 2003, 4, 7–20. [Google Scholar] [CrossRef]
- Vonsattel, J.P.; DiFiglia, M. Huntington disease. J. Neuropathol. Exp. Neurol. 1998, 57, 369–384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bassi, S.; Tripathi, T.; Monziani, A.; Di Leva, F.; Biagioli, M. Epigenetics of Huntington’s Disease. Adv. Exp. Med. Biol. 2017, 978, 277–299. [Google Scholar]
- Wang, F.; Fischhaber, P.L.; Guo, C.; Tang, T.-S. Epigenetic modifications as novel therapeutic targets for Huntington’s disease. Epigenomics 2014, 6, 287–297. [Google Scholar] [CrossRef]
- Ferrante, R.J.; Kubilus, J.K.; Lee, J.; Ryu, H.; Beesen, A.; Zucker, B.; Smith, K.; Kowall, N.W.; Ratan, R.R.; Luthi-Carter, R.; et al. Histone deacetylase inhibition by sodium butyrate chemotherapy ameliorates the neurodegenerative phenotype in Huntington’s disease mice. J. Neurosci. 2003, 23, 9418–9427. [Google Scholar] [CrossRef]
- Ferrante, R.J.; Ryu, H.; Kubilus, J.K.; D’Mello, S.; Sugars, K.L.; Lee, J.; Lu, P.; Smith, K.; Browne, S.; Beal, M.F.; et al. Chemotherapy for the brain: The antitumor antibiotic mithramycin prolongs survival in a mouse model of Huntington’s disease. J. Neurosci. 2004, 24, 10335–10342. [Google Scholar] [CrossRef]
- Gardian, G.; Browne, S.E.; Choi, D.-K.; Klivenyi, P.; Gregorio, J.; Kubilus, J.K.; Ryu, H.; Langley, B.; Ratan, R.R.; Ferrante, R.J.; et al. Neuroprotective effects of phenylbutyrate in the N171-82Q transgenic mouse model of Huntington’s disease. J. Biol. Chem. 2005, 280, 556–563. [Google Scholar] [CrossRef] [Green Version]
- Rea, S.; Eisenhaber, F.; O’Carroll, D.; Strahl, B.D.; Sun, Z.-W.; Schmid, M.; Opravil, S.; Mechtler, K.; Ponting, C.P.; Allis, C.D.; et al. Regulation of chromatin structure by site-specific histone H3 methyltransferases. Nature 2000, 406, 593–599. [Google Scholar] [CrossRef]
- Ryu, H.; Lee, J.; Hagerty, S.W.; Soh, B.Y.; McAlpin, S.E.; Cormier, K.A.; Smith, K.M.; Ferrante, R.J. ESET/SETDB1 gene expression and histone H3 (K9) trimethylation in Huntington’s disease. Proc. Natl. Acad. Sci. USA 2006, 103, 19176–19181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sadri-Vakili, G.; Cha, J.H. Mechanisms of disease: Histone modifications in Huntington’s disease. Nat. Clin. Pract. Neurol. 2006, 2, 330–338. [Google Scholar] [CrossRef]
- Stack, E.C.; Del Signore, S.J.; Luthi-Carter, R.; Soh, B.Y.; Goldstein, D.R.; Matson, S.; Goodrich, S.; Markey, A.L.; Cormier, K.; Hagerty, S.W.; et al. Modulation of nucleosome dynamics in Huntington’s disease. Hum. Mol. Genet. 2007, 16, 1164–1175. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.; Hagerty, S.; Cormier, K.A.; Kim, J.; Kung, A.L.; Ferrante, R.J.; Ryu, H. Monoallele deletion of CBP leads to pericentromeric heterochromatin condensation through ESET expression and histone H3 (K9) methylation. Hum. Mol. Genet. 2008, 17, 1774–1782. [Google Scholar] [CrossRef] [Green Version]
- Wu, R.; Terry, A.V.; Singh, P.B.; Gilbert, D.M. Differential subnuclear localization and replication timing of histone H3 lysine 9 methylation states. Mol. Biol. Cell 2005, 16, 2872–2881. [Google Scholar] [CrossRef] [Green Version]
- Ratovitski, T.; Arbez, N.; Stewart, J.C.; Chighladze, E.; A Ross, C. PRMT5- mediated symmetric arginine dimethylation is attenuated by mutant huntingtin and is impaired in Huntington’s disease (HD). Cell Cycle 2015, 14, 1716–1729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seong, I.S.; Woda, J.M.; Song, J.-J.; Lloret, A.; Abeyrathne, P.D.; Woo, C.J.; Gregory, G.; Lee, J.-M.; Wheeler, V.C.; Walz, T.; et al. Huntingtin facilitates polycomb repressive complex 2. Hum. Mol. Genet. 2010, 19, 573–583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aranda, S.; Mas, G.; Di Croce, L. Regulation of gene transcription by Polycomb proteins. Sci. Adv. 2015, 1, e1500737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Margueron, R.; Reinberg, D. The Polycomb complex PRC2 and its mark in life. Nature 2011, 469, 343–349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biagioli, M.; Ferrari, F.; Mendenhall, E.M.; Zhang, Y.; Erdin, S.; Vijayvargia, R.; Vallabh, S.M.; Solomos, N.; Manavalan, P.; Ragavendran, A.; et al. Htt CAG repeat expansion confers pleiotropic gains of mutant huntingtin function in chromatin regulation. Hum. Mol. Genet. 2015, 24, 2442–2457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.-H.; Sharma, A.; Dhar, S.S.; Lee, S.-H.; Gu, B.; Chan, C.-H.; Lin, H.-K.; Lee, M.G. UTX and MLL4 coordinately regulate transcriptional programs for cell proliferation and invasiveness in breast cancer cells. Cancer Res. 2014, 74, 1705–1717. [Google Scholar] [CrossRef] [Green Version]
- Dong, X.; Tsuji, J.; Labadorf, A.; Roussos, P.; Chen, J.-F.; Myers, R.H.; Akbarian, S.; Weng, Z. The Role of H3K4me3 in Transcriptional Regulation Is Altered in Huntington’s Disease. PLoS ONE 2015, 10, e0144398. [Google Scholar] [CrossRef]
- Hoss, A.G.; Kartha, V.K.; Dong, X.; Latourelle, J.C.; Dumitriu, A.; Hadzi, T.C.; Macdonald, M.E.; Gusella, J.F.; Akbarian, S.; Chen, J.-F.; et al. MicroRNAs located in the Hox gene clusters are implicated in huntington’s disease pathogenesis. PLoS Genet. 2014, 10, e1004188. [Google Scholar] [CrossRef] [PubMed]
- Labadorf, A.; Hoss, A.G.; Lagomarsino, V.; Latourelle, J.C.; Hadzi, T.C.; Bregu, J.; MacDonald, M.E.; Gusella, J.F.; Chen, J.F.; Akbarian, S.; et al. RNA Sequence Analysis of Human Huntington Disease Brain Reveals an Extensive Increase in Inflammatory and Developmental Gene Expression. PLoS ONE 2015, 10, e0143563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Labadorf, A.T.; Myers, R.H. Evidence of Extensive Alternative Splicing in Post Mortem Human Brain HTT Transcription by mRNA Sequencing. PLoS ONE 2015, 10, e0141298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- von Schimmelmann, M.; Feinberg, P.A.; Sullivan, J.M.; Ku, S.M.; Badimon, A.; Duff, M.K.; Wang, Z.; Lachmann, A.; Dewell, S.; Ma’ayan, A.; et al. Polycomb repressive complex 2 (PRC2) silences genes responsible for neurodegeneration. Nat. Neurosci. 2016, 19, 1321–1330. [Google Scholar] [CrossRef] [PubMed]
- Pasini, D.; Hansen, K.H.; Christensen, J.; Agger, K.; Cloos, P.A.; Helin, K. Coordinated regulation of transcriptional repression by the RBP2 H3K4 demethylase and Polycomb-Repressive Complex 2. Genes. Dev. 2008, 22, 1345–1355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vashishtha, M.; Ng, C.W.; Yildirim, F.; Gipson, T.A.; Kratter, I.H.; Bodai, L.; Song, W.; Lau, A.L.; Labadorf, A.; Vogel-Ciernia, A.; et al. Targeting H3K4 trimethylation in Huntington disease. Proc. Natl. Acad. Sci. USA 2013, 110, E3027–E3036. [Google Scholar] [CrossRef] [Green Version]
- Zuccato, C.; Cattaneo, E. Role of brain-derived neurotrophic factor in Huntington’s disease. Prog. Neurobiol. 2007, 81, 294–330. [Google Scholar] [CrossRef]
- Zuccato, C.; Ciammola, A.; Rigamonti, D.; Leavitt, B.R.; Goffredo, D.; Conti, L.; Macdonald, M.E.; Friedlander, R.M.; Silani, V.; Hayden, M.R.; et al. Loss of huntingtin-mediated BDNF gene transcription in Huntington’s disease. Science 2001, 293, 493–498. [Google Scholar] [CrossRef] [Green Version]
- Kuhn, A.; Goldstein, D.R.; Hodges, A.; Strand, A.D.; Sengstag, T.; Kooperberg, C.; Becanovic, K.; Pouladi, M.A.; Sathasivam, K.; Cha, J.J.; et al. Mutant huntingtin’s effects on striatal gene expression in mice recapitulate changes observed in human Huntington’s disease brain and do not differ with mutant huntingtin length or wild-type huntingtin dosage. Hum. Mol. Genet. 2007, 16, 1845–1861. [Google Scholar] [CrossRef] [Green Version]
- Bai, G.; Cheung, I.; Shulha, H.P.; Coelho, J.E.; Li, P.; Dong, X.; Jakovcevski, M.; Wang, Y.; Grigorenko, A.; Jiang, Y.; et al. Epigenetic dysregulation of hairy and enhancer of split 4 (HES4) is associated with striatal degeneration in postmortem Huntington brains. Hum. Mol. Genet. 2015, 24, 1441–1456. [Google Scholar] [CrossRef] [Green Version]
- Fitzgerald, J.C.; Plun-Favreau, H. Emerging pathways in genetic Parkinson’s disease: Autosomal-recessive genes in Parkinson’s disease—A common pathway? FEBS J. 2008, 275, 5758–5766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jankovic, J. Parkinson’s disease: Clinical features and diagnosis. J. Neurol. Neurosurg. Psychiatry. 2008, 79, 368–376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mori, F.; Tanji, K.; Zhang, H.; Kakita, A.; Takahashi, H.; Wakabayashi, K. Alpha-Synuclein pathology in the neostriatum in Parkinson’s disease. Acta Neuropathol. 2008, 115, 453–459. [Google Scholar] [CrossRef]
- Wakabayashi, K.; Tanji, K.; Mori, F.; Takahashi, H. The Lewy body in Parkinson’s disease: Molecules implicated in the formation and degradation of alpha-synuclein aggregates. Neuropathology 2007, 27, 494–506. [Google Scholar] [CrossRef]
- Chatterjee, P.; Roy, D.; Bhattacharyya, M.; Bandyopadhyay, S. Biological networks in Parkinson’s disease: An insight into the epigenetic mechanisms associated with this disease. BMC Genom. 2017, 18, 721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, Y.; Jankovic, J.; Wu, Y.C. Epigenetic mechanisms in Parkinson’s disease. J. Neurol. Sci. 2015, 349, 3–9. [Google Scholar] [CrossRef]
- Labbe, C.; Lorenzo-Betancor, O.; Ross, O.A. Epigenetic regulation in Parkinson’s disease. Acta Neuropathol. 2016, 132, 515–530. [Google Scholar] [CrossRef]
- Van Heesbeen, H.J.; Smidt, M.P. Entanglement of Genetics and Epigenetics in Parkinson’s Disease. Front. Neurosci. 2019, 13, 277. [Google Scholar] [CrossRef] [PubMed]
- Sugeno, N.; Jäckel, S.; Voigt, A.; Wassouf, Z.; Schulze-Hentrich, J.; Kahle, P.J. Alpha-Synuclein enhances histone H3 lysine-9 dimethylation and H3K9me2-dependent transcriptional responses. Sci. Rep. 2016, 6, 36328. [Google Scholar] [CrossRef] [PubMed]
- Guhathakurta, S.; Kim, J.; Adams, L.; Basu, S.; Song, M.K.; Adler, E.; Je, G.; Fiadeiro, M.B.; Kim, Y. Targeted attenuation of elevated histone marks at SNCA alleviates alpha-synuclein in Parkinson’s disease. EMBO Mol. Med. 2021, 13, e12188. [Google Scholar] [CrossRef] [PubMed]
- Mu, M.-D.; Qian, Z.-M.; Yang, S.-X.; Rong, K.-L.; Yung, W.-H.; Ke, Y. Therapeutic effect of a histone demethylase inhibitor in Parkinson’s disease. Cell Death Dis. 2020, 11, 927. [Google Scholar] [CrossRef] [PubMed]
- Heinemann, B.; Nielsen, J.M.; Hudlebusch, H.R.; Lees, M.J.; Larsen, D.V.; Boesen, T.; Labelle, M.; Gerlach, L.-O.; Birk, P.; Helin, K. Inhibition of demethylases by GSK-J1/J4. Nature 2014, 514, E1–E2. [Google Scholar] [CrossRef] [PubMed]
- Kruidenier, L.; Chung, C.-W.; Cheng, Z.; Liddle, J.; Che, K.; Joberty, G.; Bantscheff, M.; Bountra, C.; Bridges, A.; Diallo, H.; et al. Kruidenier et al. reply. Nature 2014, 514, E2. [Google Scholar] [CrossRef] [PubMed]
- Pang, S.Y.-Y.; Hsu, J.S.; Teo, K.-C.; Li, Y.; Kung, M.H.; Cheah, K.S.; Chan, D.; Cheung, K.M.; Li, M.; Sham, P.-C.; et al. Burden of rare variants in ALS genes influences survival in familial and sporadic ALS. Neurobiol. Aging 2017, 58, 238.e9–238.e15. [Google Scholar] [CrossRef]
- Swinnen, B.; Robberecht, W. The phenotypic variability of amyotrophic lateral sclerosis. Nat. Rev. Neurol. 2014, 10, 661–670. [Google Scholar] [CrossRef] [Green Version]
- Zarei, S.; Carr, K.; Reiley, L.; Diaz, K.; Guerra, O.; Altamirano, P.F.; Pagani, W.; Lodin, D.; Orozco, G.; Chinea, A. A comprehensive review of amyotrophic lateral sclerosis. Surg. Neurol. Int. 2015, 6, 171. [Google Scholar] [CrossRef]
- Stetkarova, I.; Ehler, E. Diagnostics of Amyotrophic Lateral Sclerosis: Up to Date. Diagnostics 2021, 11, 231. [Google Scholar] [CrossRef]
- Belzil, V.V.; Katzman, R.B.; Petrucelli, L. ALS and FTD: An epigenetic perspective. Acta Neuropathol. 2016, 132, 487–502. [Google Scholar] [CrossRef]
- Ahmed, A.; Wicklund, M.P. Amyotrophic lateral sclerosis: What role does environment play? Neurol. Clin. 2011, 29, 689–711. [Google Scholar] [CrossRef] [PubMed]
- Al-Chalabi, A.; Kwak, S.; Mehler, M.; Rouleau, G.; Siddique, T.; Strong, M.; Leigh, P.N. Genetic and epigenetic studies of amyotrophic lateral sclerosis. Amyotroph. Lateral. Scler. Frontotemporal. Degener. 2013, 14, 44–52. [Google Scholar] [CrossRef] [Green Version]
- DeJesus-Hernandez, M.; Mackenzie, I.R.; Boeve, B.F.; Boxer, A.L.; Baker, M.; Rutherford, N.J.; Nicholson, A.M.; Finch, N.A.; Flynn, H.; Adamson, J.; et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 2011, 72, 245–256. [Google Scholar] [CrossRef] [Green Version]
- Renton, A.E.; Majounie, E.; Waite, A.; Simón-Sánchez, J.; Rollinson, S.; Gibbs, J.R.; Schymick, J.C.; Laaksovirta, H.; van Swieten, J.C.; Myllykangas, L.; et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 2011, 72, 257–268. [Google Scholar] [CrossRef] [Green Version]
- He, F.; Todd, P.K. Epigenetics in nucleotide repeat expansion disorders. Semin. Neurol. 2011, 31, 470–483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ebbert, M.T.W.; Lank, R.J.; Belzil, V.V. An Epigenetic Spin to ALS and FTD. Adv. Neurobiol. 2018, 20, 1–29. [Google Scholar] [PubMed]
- Belzil, V.V.; Bauer, P.O.; Prudencio, M.; Gendron, T.F.; Stetler, C.T.; Yan, I.K.; Pregent, L.; Daughrity, L.; Baker, M.C.; Rademakers, R.; et al. Reduced C9orf72 gene expression in c9FTD/ALS is caused by histone trimethylation, an epigenetic event detectable in blood. Acta Neuropathol. 2013, 126, 895–905. [Google Scholar] [CrossRef] [Green Version]
- Esanov, R.; Cabrera, G.T.; Andrade, N.S.; Gendron, T.F.; Brown, R.H.; Benatar, M.; Wahlestedt, C.; Mueller, C.; Zeier, Z. A C9ORF72 BAC mouse model recapitulates key epigenetic perturbations of ALS/FTD. Mol. Neurodegener. 2017, 12, 46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jury, N.; Abarzua, S.; Diaz, I.; Guerra, M.V.; Ampuero, E.; Cubillos, P.; Martinez, P.; Herrera-Soto, A.; Arredondo, C.; Rojas, F.; et al. Widespread loss of the silencing epigenetic mark H3K9me3 in astrocytes and neurons along with hippocampal-dependent cognitive impairment in C9orf72 BAC transgenic mice. Clin. Epigenetics. 2020, 12, 32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gary, J.D.; Lin, W.-J.; Yang, M.C.; Herschman, H.R.; Clarke, S. The predominant protein-arginine methyltransferase from Saccharomyces cerevisiae. J. Biol. Chem. 1996, 271, 12585–12594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carrozza, M.J.; Li, B.; Florens, L.; Suganuma, T.; Swanson, S.K.; Lee, K.K.; Shia, W.-J.; Anderson, S.; Yates, J.; Washburn, M.P.; et al. Histone H3 methylation by Set2 directs deacetylation of coding regions by Rpd3S to suppress spurious intragenic transcription. Cell 2005, 123, 581–592. [Google Scholar] [CrossRef] [Green Version]
- Joshi, A.A.; Struhl, K. Eaf3 chromodomain interaction with methylated H3-K36 links histone deacetylation to Pol II elongation. Mol. Cell 2005, 20, 971–978. [Google Scholar] [CrossRef] [PubMed]
- Jun, M.H.; Ryu, H.H.; Jun, Y.W.; Liu, T.; Li, Y.; Lim, C.S.; Lee, Y.S.; Kaang, B.K.; Jang, D.J.; Lee, J.A. Sequestration of PRMT1 and Nd1-L mRNA into ALS-linked FUS mutant R521C-positive aggregates contributes to neurite degeneration upon oxidative stress. Sci Rep. 2017, 7, 40474. [Google Scholar] [CrossRef]
- Tibshirani, M.; Tradewell, M.L.; Mattina, K.R.; Minotti, S.; Yang, W.; Zhou, H.; Strong, M.J.; Hayward, L.J.; Durham, H.D. Cytoplasmic sequestration of FUS/TLS associated with ALS alters histone marks through loss of nuclear protein arginine methyltransferase 1. Hum. Mol. Genet. 2015, 24, 773–786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scotter, E.L.; Chen, H.J.; Shaw, C.E. TDP-43 Proteinopathy and ALS: Insights into Disease Mechanisms and Therapeutic Targets. Neurotherapeutics 2015, 12, 352–363. [Google Scholar] [CrossRef] [Green Version]
- Boyadjieva, N.I.; Sarkar, D.K. Role of microglia in ethanol’s apoptotic action on hypothalamic neuronal cells in primary cultures. Alcohol Clin. Exp. Res. 2010, 34, 1835–1842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Byun, K.; Bayarsaikhan, D.; Bayarsaikhan, E.; Son, M.; Oh, S.; Lee, J.; Son, H.-I.; Won, M.-H.; Kim, S.U.; Song, B.-J.; et al. Microglial AGE-albumin is critical in promoting alcohol-induced neurodegeneration in rats and humans. PLoS ONE 2014, 9, e104699. [Google Scholar] [CrossRef] [Green Version]
- Marshall, S.A.; McClain, J.A.; Kelso, M.L.; Hopkins, D.M.; Pauly, J.R.; Nixon, K. Microglial activation is not equivalent to neuroinflammation in alcohol-induced neurodegeneration: The importance of microglia phenotype. Neurobiol. Dis. 2013, 54, 239–251. [Google Scholar] [CrossRef] [Green Version]
- Subbanna, S.; Shivakumar, M.; Psychoyos, D.; Xie, S.; Basavarajappa, B.S. Anandamide-CB1 Receptor Signaling Contributes to Postnatal Ethanol-Induced Neonatal Neurodegeneration, Adult Synaptic and Memory Deficits. J. Neurosci. 2013, 33, 6350–6366. [Google Scholar] [CrossRef] [Green Version]
- Tateno, M.; Saito, T. Biological studies on alcohol-induced neuronal damage. Psychiatry Investig. 2008, 5, 21–27. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.-N.; Wang, F.; Fan, Y.-X.; Ping, G.-F.; Yang, J.-Y.; Wu, C.-F. Activated microglia are implicated in cognitive deficits, neuronal death, and successful recovery following intermittent ethanol exposure. Behav. Brain Res. 2013, 236, 270–282. [Google Scholar] [CrossRef] [PubMed]
- Berkel, T.D.; Pandey, S.C. Emerging Role of Epigenetic Mechanisms in Alcohol Addiction. Alcohol Clin. Exp. Res. 2017, 41, 666–680. [Google Scholar] [CrossRef] [Green Version]
- Ciafrè, S.; Carito, V.; Ferraguti, G.; Greco, A.; Chaldakov, G.N.; Fiore, M.; Ceccanti, M. How alcohol drinking affects our genes: An epigenetic point of view. Biochem. Cell. Biol. 2019, 97, 345–356. [Google Scholar] [CrossRef] [Green Version]
- Palmisano, M.; Pandey, S.C. Epigenetic mechanisms of alcoholism and stress-related disorders. Alcohol 2017, 60, 7–18. [Google Scholar] [CrossRef]
- Bohnsack, J.P.; Pandey, S.C. Histone modifications, DNA methylation, and the epigenetic code of alcohol use disorder. Int. Rev. Neurobiol. 2021, 156, 1–62. [Google Scholar]
- Zhou, Z.; Yuan, Q.; Mash, D.C.; Goldman, D. Substance-specific and shared transcription and epigenetic changes in the human hippocampus chronically exposed to cocaine and alcohol. Proc. Natl. Acad. Sci. USA 2011, 108, 6626–6631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ponomarev, I.; Wang, S.; Zhang, L.; Harris, R.A.; Mayfield, R.D. Gene coexpression networks in human brain identify epigenetic modifications in alcohol dependence. J. Neurosci. 2012, 32, 1884–1897. [Google Scholar] [CrossRef] [PubMed]
- Bohnsack, J.P.; Teppen, T.; Kyzar, E.J.; Dzitoyeva, S.; Pandey, S.C. The lncRNA BDNF-AS is an epigenetic regulator in the human amygdala in early onset alcohol use disorders. Transl. Psychiatry 2019, 9, 34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnstone, A.L.; Andrade, N.S.; Barbier, E.; Khomtchouk, B.B.; Rienas, C.A.; Lowe, K.; Van Booven, D.J.; Domi, E.; Esanov, R.; Vilca, S.; et al. Dysregulation of the histone demethylase KDM6B in alcohol dependence is associated with epigenetic regulation of inflammatory signaling pathways. Addict. Biol. 2021, 26, e12816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finegersh, A.; Homanics, G.A. Acute ethanol alters multiple histone modifications at model gene promoters in the cerebral cortex. Alcohol Clin. Exp. Res. 2014, 38, 1865–1873. [Google Scholar] [CrossRef]
- D’Addario, C.; Caputi, F.F.; Ekström, T.J.; Di Benedetto, M.; Maccarrone, M.; Romualdi, P.; Candeletti, S. Ethanol induces epigenetic modulation of prodynorphin and pronociceptin gene expression in the rat amygdala complex. J. Mol. Neurosci. 2013, 49, 312–319. [Google Scholar] [CrossRef] [PubMed]
- D’Addario, C.; Caputi, F.F.; Rimondini, R.; Gandolfi, O.; Del Borrello, E.; Candeletti, S.; Romualdi, P. Different alcohol exposures induce selective alterations on the expression of dynorphin and nociceptin systems related genes in rat brain. Addict. Biol. 2013, 18, 425–433. [Google Scholar] [CrossRef]
- Stragier, E.; Massart, R.; Salery, M.; Hamon, M.; Geny, D.; Martin, V.; Boulle, F.; Lanfumey, L. Ethanol-induced epigenetic regulations at the Bdnf gene in C57BL/6J mice. Mol. Psychiatry 2015, 20, 405–412. [Google Scholar] [CrossRef]
- Qiang, M.; Denny, A.; Lieu, M.; Carreon, S.; Li, J. Histone H3K9 modifications are a local chromatin event involved in ethanol-induced neuroadaptation of the NR2B gene. Epigenetics 2011, 6, 1095–1104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pascual, M.; Couto, B.R.D.; Alfonso-Loeches, S.; Aguilar, M.A.; Rodriguez-Arias, M.; Guerri, C. Changes in histone acetylation in the prefrontal cortex of ethanol-exposed adolescent rats are associated with ethanol-induced place conditioning. Neuropharmacology 2012, 62, 2309–2319. [Google Scholar] [CrossRef]
- Kyzar, E.J.; Zhang, H.; Sakharkar, A.J.; Pandey, S.C. Adolescent alcohol exposure alters lysine demethylase 1 (LSD1) expression and histone methylation in the amygdala during adulthood. Addict. Biol. 2017, 22, 1191–1204. [Google Scholar] [CrossRef] [PubMed]
- Barbier, E.; Johnstone, A.L.; Khomtchouk, B.B.; Tapocik, J.D.; Pitcairn, C.; Rehman, F.; Augier, E.; Borich, A.; Schank, J.R.; Rienas, C.A.; et al. Dependence-induced increase of alcohol self-administration and compulsive drinking mediated by the histone methyltransferase PRDM2. Mol. Psychiatry 2017, 22, 1746–1758. [Google Scholar] [CrossRef] [PubMed]
- Wolstenholme, J.T.; Mahmood, T.; Harris, G.M.; Abbas, S.; Miles, M.F. Intermittent Ethanol during Adolescence Leads to Lasting Behavioral Changes in Adulthood and Alters Gene Expression and Histone Methylation in the PFC. Front. Mol. Neurosci. 2017, 10, 307. [Google Scholar] [CrossRef]
- Gavin, D.P.; Hashimoto, J.G.; Lazar, N.H.; Carbone, L.; Crabbe, J.C.; Guizzetti, M. Stable Histone Methylation Changes at Proteoglycan Network Genes Following Ethanol Exposure. Front. Genet. 2018, 9, 346. [Google Scholar] [CrossRef] [PubMed]
- Kyzar, E.J.; Zhang, H.; Pandey, S.C. Adolescent Alcohol Exposure Epigenetically Suppresses Amygdala Arc Enhancer RNA Expression to Confer Adult Anxiety Susceptibility. Biol. Psychiatry 2019, 85, 904–914. [Google Scholar] [CrossRef]
- Hashimoto, J.G.; Gavin, D.P.; Wiren, K.M.; Crabbe, J.C.; Guizzetti, M. Prefrontal cortex expression of chromatin modifier genes in male WSP and WSR mice changes across ethanol dependence, withdrawal, and abstinence. Alcohol 2017, 60, 83–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Veazey, K.J.; Parnell, S.E.; Miranda, R.C.; Golding, M.C. Dose-dependent alcohol-induced alterations in chromatin structure persist beyond the window of exposure and correlate with fetal alcohol syndrome birth defects. Epigenetics Chromatin 2015, 8, 39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Govorko, D.; Bekdash, R.A.; Zhang, C.; Sarkar, D.K. Male germline transmits fetal alcohol adverse effect on hypothalamic proopiomelanocortin gene across generations. Biol. Psychiatry 2012, 72, 378–388. [Google Scholar] [CrossRef] [Green Version]
- Bekdash, R.A.; Zhang, C.; Sarkar, D.K. Gestational Choline Supplementation Normalized Fetal Alcohol-Induced Alterations in Histone Modifications, DNA Methylation, and Proopiomelanocortin (POMC) Gene Expression in beta-Endorphin-Producing POMC Neurons of the Hypothalamus. Alcohol Clin. Exp. Res. 2013, 37, 1133–1142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Subbanna, S.; Nagre, N.N.; Shivakumar, M.; Umapathy, N.S.; Psychoyos, D.; Basavarajappa, B.S. Ethanol induced acetylation of histone at G9a exon1 and G9a-mediated histone H3 dimethylation leads to neurodegeneration in neonatal mice. Neuroscience 2014, 258, 422–432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Subbanna, S.; Basavarajappa, B.S. Pre-administration of G9a/GLP inhibitor during Synaptogenesis Prevents Postnatal Ethanol-induced LTP Deficits and Neurobehavioral Abnormalities in Adult Mice. Exp. Neurol. 2014, 261, 34–43. [Google Scholar] [CrossRef] [Green Version]
- Subbanna, S.; Joshi, V.; Basavarajappa, B.S. Activity-dependent Signaling and Epigenetic Abnormalities in Mice Exposed to Postnatal Ethanol. Neuroscience 2018, 392, 230–240. [Google Scholar] [CrossRef]
- Joshi, V.; Subbanna, S.; Shivakumar, M.; Basavarajappa, B.S. CB1R regulates CDK5 signaling and epigenetically controls Rac1 expression contributing to neurobehavioral abnormalities in mice postnatally exposed to ethanol. Neuropsychopharmacology 2019, 44, 514–525. [Google Scholar] [CrossRef] [Green Version]
- Subbanna, S.; Nagre, N.N.; Umapathy, N.S.; Pace, B.S.; Basavarajappa, B.S. Ethanol exposure induces neonatal neurodegeneration by enhancing CB1R Exon1 histone H4K8 acetylation and up-regulating CB1R function causing neurobehavioral abnormalities in adult mice. Int. J. Neuropsychopharmacol. 2015, 18, 1–15. [Google Scholar] [CrossRef]
- Zhang, C.R.; Ho, M.-F.; Vega, M.C.S.; Burne, T.H.J.; Chong, S. Prenatal ethanol exposure alters adult hippocampal VGLUT2 expression with concomitant changes in promoter DNA methylation, H3K4 trimethylation and miR-467b-5p levels. Epigenetics Chromatin 2015, 8, 40. [Google Scholar] [CrossRef] [Green Version]
- Jarmasz, J.S.; Stirton, H.; Basalah, D.; Davie, J.R.; Clarren, S.K.; Astley, S.J.; Del Bigio, M.R. Global DNA Methylation and Histone Posttranslational Modifications in Human and Nonhuman Primate Brain in Association with Prenatal Alcohol Exposure. Alcohol Clin. Exp. Res. 2019, 43, 1145–1162. [Google Scholar] [CrossRef] [PubMed]
- Schaffner, S.L.; Lussier, A.A.; Baker, J.A.; Goldowitz, D.; Hamre, K.M.; Kobor, M.S. Neonatal Alcohol Exposure in Mice Induces Select Differentiation- and Apoptosis-Related Chromatin Changes Both Independent of and Dependent on Sex. Front. Genet. 2020, 11, 35. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Diseases | Enzyme | Histones | Model | Genetic Background | Genes | Brain Region | Effect | References |
|---|---|---|---|---|---|---|---|---|
| AD | H2BK108me | Human | PFC | ↓ | [91] | |||
| H4R55me | Human | PFC | ↓ | [91] | ||||
| H3K9me2 | Human | OC | ↑ | [92] | ||||
| H3K9me2/ me3 | Human | CA1 | ↓ | [99] | ||||
| H3K9me2 | FAD mice | C57BL/6J | PFC HP | ↑ | [94] | |||
| H3K9me2 | Human | PFC | ↑ | [94] | ||||
| EMT1 (GLP) EMT2 (G9a) | FAD mice | C57BL/6J | PFC HP | ↑ | [94] | |||
| G9a/GLP | Human | PFC | ↑ | [94] | ||||
| H3K9me2 | FAD mice | C57BL/6J | Gria2/ GluA2 | PFC HC | ↑ | [94] | ||
| H3K9me2 | FAD mice | C57BL/6J | Grin2b/ NR2B | PFC HC | ↑ | [94] | ||
| H3K9me2 | FAD mice | C57BL/6J | Shank2 | PFC HC | ↑ | [94] | ||
| G9a | Aβ-treated human stem cell-derived neurons. | Cell Cultures | ↑ | [95] | ||||
| H3K9me2/ me3 | Human | PFC | ↓ | [96] | ||||
| H3K4me3 | CK-p25 AD | lncRNA | ↑ | [100] | ||||
| H3K27me3 | CK-p25 AD | lncRNA | ↓ | [100] | ||||
| H3K4me3 | Human | ANK1 | [101] | |||||
| H3K4me3 | Human | Nucleus | ↓ | [103] | ||||
| H3K4me3 | Human | Cytoplasm | ↑ | [103] | ||||
| H3K4me3 | 3×Tg mice | Nucleus | ↓ | [103] | ||||
| H3K4me3 | Human | PFC-Nucleus | ↑ | [104] | ||||
| H3K4me3 | PS19 mice | PFC-Nucleus | ↑ | [104] | ||||
| KMT2A-D | Human | PFC-Nucleus | ↑ | [104] | ||||
| Kmt2a-d | PS19 | PFC-Nucleus | ↑ | [104] | ||||
| HD | H3K9me2 | R6/2 mice | C57BL/6J | ST | ↑ | [110,111,112] | ||
| SETDB1 | H3K9me3 | Human R6/2 mice | C57BL/6J | NC, ST, CuN | ↑ | [113] | ||
| H3K9me3 | STHdh Q7/7 and STHdh Q111/111 cells | ↑ | [117] | |||||
| H3K9me3 | Human, R6/7 mice, HD cell lines | Chrm1, Pdgfb, Inpp5j, Hrh1, Irf6, Eya1, and Kif5c | ↑ | [117] | ||||
| PRMT5 | H2A/H4 sDMA of R3 | mutantHtt fragment | In vitro activity | ↓ | [119] | |||
| H2A/H4 sDMA of R3 | mutantHtt fragment | Transfected primary neurons | ↓ | [119] | ||||
| PRC2 | Hdhex4/5 Embryos | ↑ | [120] | |||||
| H3K27me3 | Hdhex4/5 Embryos | ↓ | [120] | |||||
| H3K27me3 | Hdhex4/5/ex4/5 ESC and NPC | Bivalent loci | ↓ | [123] | ||||
| H3K4me3 | HD and R6/2 mouse | Bdnf, Penk1, Drd2 | ↓ | [131] | ||||
| H3K4me3 | HD and R6/2 mouse | REST/NRSF | ↓ | [131] | ||||
| Jarid1c | - | HD and R6/2 mouse | ↑ | [131] | ||||
| Jarid1c | - | Htt (Q150) knockin mice | ↑ | [134] | ||||
| PD | H3K9me1 H3K9me2 | Transgenic Drosophila and inducible SH-SY5Y neuroblastoma cells | ↑ | [144] | ||||
| G9a | H3K9me2 | αS-induced SH-SY5Y cells | L1cam, Snap25 | ↑ | [144] | |||
| H3K4me3 | Human PD | Snca | ↑ | [145] | ||||
| H3K27me3 | Human PD | - | ↑ | [145] | ||||
| H3K27me3 | SH-SY5Y cells+ 6-OHDA | - | ↓ | [146] | ||||
| H3K4me3 | SH-SY5Y cells+ 6-OHDA | - | ↓ | [146] | ||||
| H3K27me3 | SH-SY5Y cells+ 6-OHDA | - | ↓ | [146] | ||||
| H3K4me3 | SH-SY5Y cells+ 6-OHDA | - | ↓ | [146] | ||||
| H3K27me3 | SH-SY5Y cells+ 6-OHDA +GSK-J4 | - | ↑ | [146] | ||||
| H3K4me3 | SH-SY5Y cells+ 6-OHDA++GSK-J4 | - | ↑ | [146] | ||||
| H3K27me3 | SH-SY5Y cells+ 6-OHDA++GSK-J4 | - | ↑ | [146] | ||||
| H3K4me3 | SH-SY5Y cells+ 6-OHDA++GSK-J4 | - | ↑ | [146] | ||||
| ALS | H3K9me3, H3K27me3 H4K20me3 | Human ALS (c9FTD/ALS) | C9orf72 | ↑ | [160] | |||
| H4R3me2asym | yeast models of ALS (over expression of FUS) | - | ↓ | [160] | ||||
| H3K36me3 | yeast models of ALS (over expression of TDP-43) | - | ↓ | [160] | ||||
| PMRT1 | FUSR521C ALS model (Overexpression of PMRT1) | - | ↓ | [160] | ||||
| H4R3me2asym | FUSR521C ALS model (loss of PMRT1 function) | - | ↓ | [160] | ||||
| H3K9me3 | C9ALS/FTD BAC mice | C9 or f72 | ↑ | [167] | ||||
| H3K9me3 | C9ALS/FTD BAC mice | SC, NC, HP | ↑ | [167] | ||||
| H3K9me3 | C9ALS/FTD BAC mice | Cultured Astrocytes and neurons | ↓ | [168] |
| Alcohol Exposure | Tissue Examined | Effects |
|---|---|---|
| Postmortem Human alcoholic brain | PFC | Increased Global H3K4me3 at GIPC1, BCL2L1, and UBE1 genes [180]. |
| Postmortem Human alcoholic brain | HP | Increased H3K4me3 at expressed, non-expressed, and non-genic gene regions [179]. |
| Postmortem Human alcoholic brain | Amygdala | Increased recruitment of Ezh2, which regulates H3K27me3 levels, at BDNF and ARC gene locus in early-onset AUD group [181]. |
| Postmortem Human alcoholic brain | ACC | Increased KDM6b [182] that regulates H3K27me3 levels. |
| Acute alcohol in mice | CCx and HP | Decreased H3K27me3 at Mt1 gene promoter; Increased H3K4me3 at Mt2 gene promoter [183]. |
| Acute alcohol in rats | Amygdala | Decreased H3K27me3 levels; Increased H3K9me2 [184,185]. |
| Chronic ethanol (free choice paradigm) in mice | HP | Increased H3K27me3 at Bdnf PII and PIII. Decreased H3K4me3 at Bdnf PVIII [186]. |
| Chronic intermittent ethanol (CIE) in mice cortical neurons | Cortical | Decreased H3K9me2; Decreased Setd1a, Setd1b, Setdb2, Suv39 h1, Setd4, Setd6, Setdb1, Prmt6, and G9a; Decreased G9a, Suv39h1 at the NR2B gene promoter [187]. |
| Binge-like ethanol in adolescent rats | PFC | Increased H3K4me2 at cFos, Cdk5, and FosB genes promoters [188] |
| Intermittent alcohol exposure in adolescent rats | Amygdala | Decreased LSD1 and LSD1+8A in CeA and MeA; Increased H3K9me2 in CeA and MeA [189]. |
| CIA in rats | dmPFC | Decreased PRDM2 expession; Inhibition of PRDM2 in dmPFC via shRNA increased alcohol self-administration [190]. |
| Intermittent alcohol exposure in adolescent mice | PFC | Reduced H3K36m1, me2 and me3 levels [191]. |
| Alcohol vapor exposure in WSR mice | PFC | Increased H3K27me3 Reduced H3K4me3 |
| Alcohol vapor exposure for 72h in WP and WSP mice | PFC | Reduced Smyd3, which di- and trimethylates H3K4. Reduced Setdb1, which trimethylates H3K9. Reduced Setd6, which mono-methylates the lysine 8 on the histone variant H2AZ (H2AZK8me1). Increased Setd7, which mono-methylates H3K4; Increased Setd3, which methylates H3K4 and H3K36; Increased Ash1l, which methylates H3K36 [192]. |
| Intermittent alcohol exposure in adolescent rats | Amygdala | Decreased Kdm6b, increased H3K27me3 at Arc SARE site; Decreased Arc expression; Kdm6b siRNA in CeA causes anxiety and AIE phenotype [193]. |
| Intermittent alcohol vapor exposure in rats | mPFC | Increased KDM6B protein and decreased H3K27me3 [182]. |
| Gestational Day (GD) 7 | GD 17 cortex | Increased H3K9me2, and decreased H3K27me3; Altered G9a, Setdb1, Kdm1a, Kdm4c, Uhrf1 and Ezh2 mRNA levels [195]. |
| GD 7–21 | Embryonic days (ED) 7.0–14.5 | Decreased H3K4me2, H3K4me3; Decreased mRNA levels of Set7/9; Increased G9a mRNA and H3K9me2 [196]. |
| PD 60–80 from F1-F3 generation ED14.5–PD 7 | Decreased H3K4me2, H3K4me3, and Set7/9 mRNA; Increased G9a and setdb1 mRNA, H3K9me2 [197] | |
| PD7 mice | PD7 HP and NC | Increased G9a [198]; Increased degradation of H3K9me2 and H3K27me2 by activated caspase 3 [34] Inhibition of G9a before ethanol treatment rescued degradation of H3K9me2 and LTP and spatial memory deficits [199] Enhanced H3K9me2 and G9a at Arc [200] and Rac1 [201] gene promoters; Reduced H3K9me2 and G9a at Cnr1 gene exon-1 [202] |
| GD 0–8 days mice | PD87 HP | Increased H3K4me3 at Slcl7a6 gene; Increased Slc17a6 gene and reduced VGLUT2 protein levels [203] |
| Macaca nemestrina (GD 33–46) | DG Ependyma | Increased H3K4me3 at Slcl7a6 gene; Increased Slc17a6 gene and reduced VGLUT2 protein levels [203]. |
| Human prenatal ethanol | Fetal brain ependyma cells | Reduced H3K4me3 [204] |
| PD7 | PD7 7 h after ethanol exposure (NC and Cerebellum) | PD7 7 h after ethanol exposure (NC and Cerebellum) [205]. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Basavarajappa, B.S.; Subbanna, S. Histone Methylation Regulation in Neurodegenerative Disorders. Int. J. Mol. Sci. 2021, 22, 4654. https://doi.org/10.3390/ijms22094654
Basavarajappa BS, Subbanna S. Histone Methylation Regulation in Neurodegenerative Disorders. International Journal of Molecular Sciences. 2021; 22(9):4654. https://doi.org/10.3390/ijms22094654
Chicago/Turabian StyleBasavarajappa, Balapal S., and Shivakumar Subbanna. 2021. "Histone Methylation Regulation in Neurodegenerative Disorders" International Journal of Molecular Sciences 22, no. 9: 4654. https://doi.org/10.3390/ijms22094654
APA StyleBasavarajappa, B. S., & Subbanna, S. (2021). Histone Methylation Regulation in Neurodegenerative Disorders. International Journal of Molecular Sciences, 22(9), 4654. https://doi.org/10.3390/ijms22094654