
Effects of Lacosamide Treatment on Epileptogenesis, Neuronal Damage and Behavioral Comorbidities in a Rat Model of Temporal Lobe Epilepsy

 , ,
, ,  , ,
, , 
Abstract
:1. Introduction
2. Results
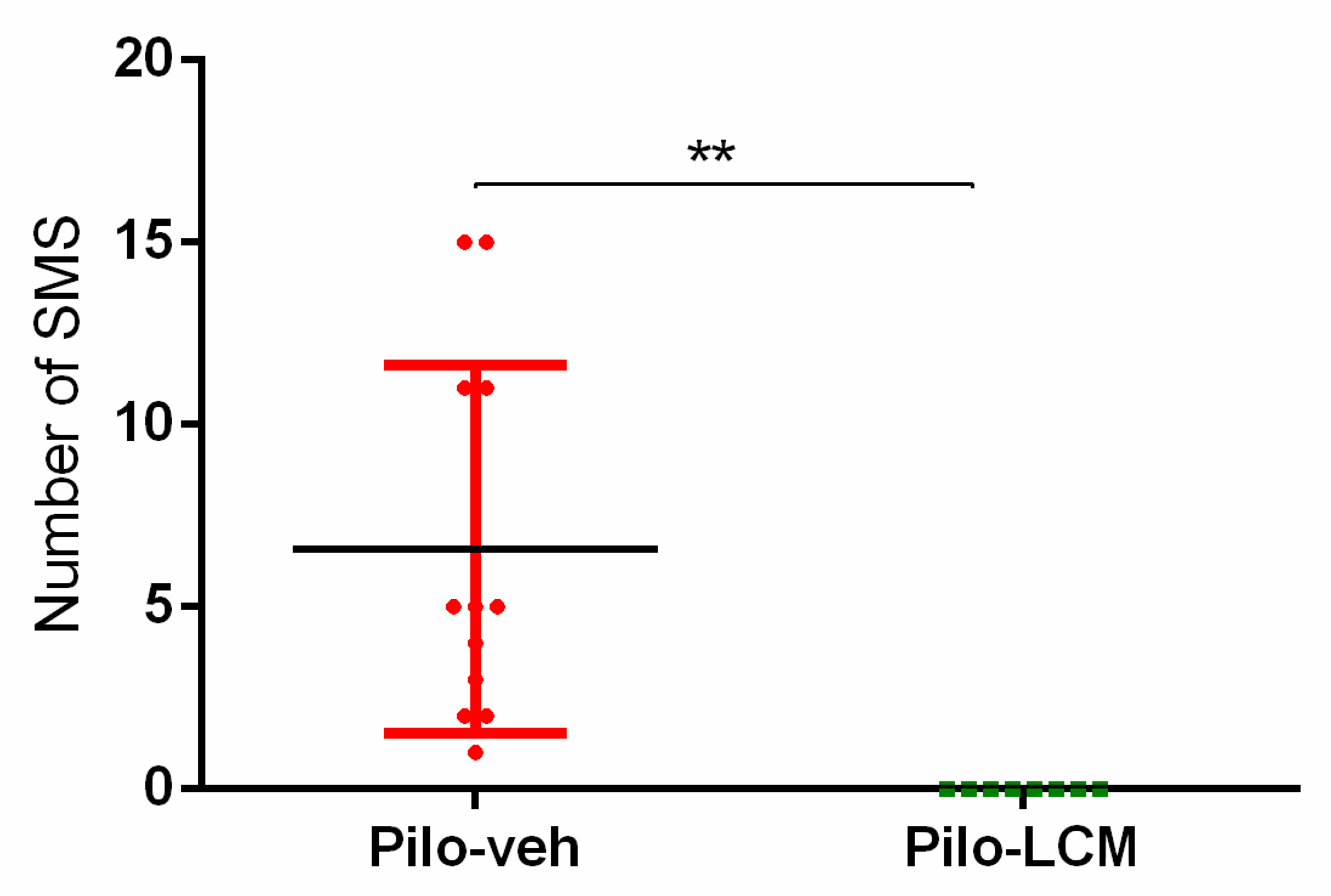
2.1. Seizure Activity in Rats Treated with Pilo
2.2. Behavioral Tests
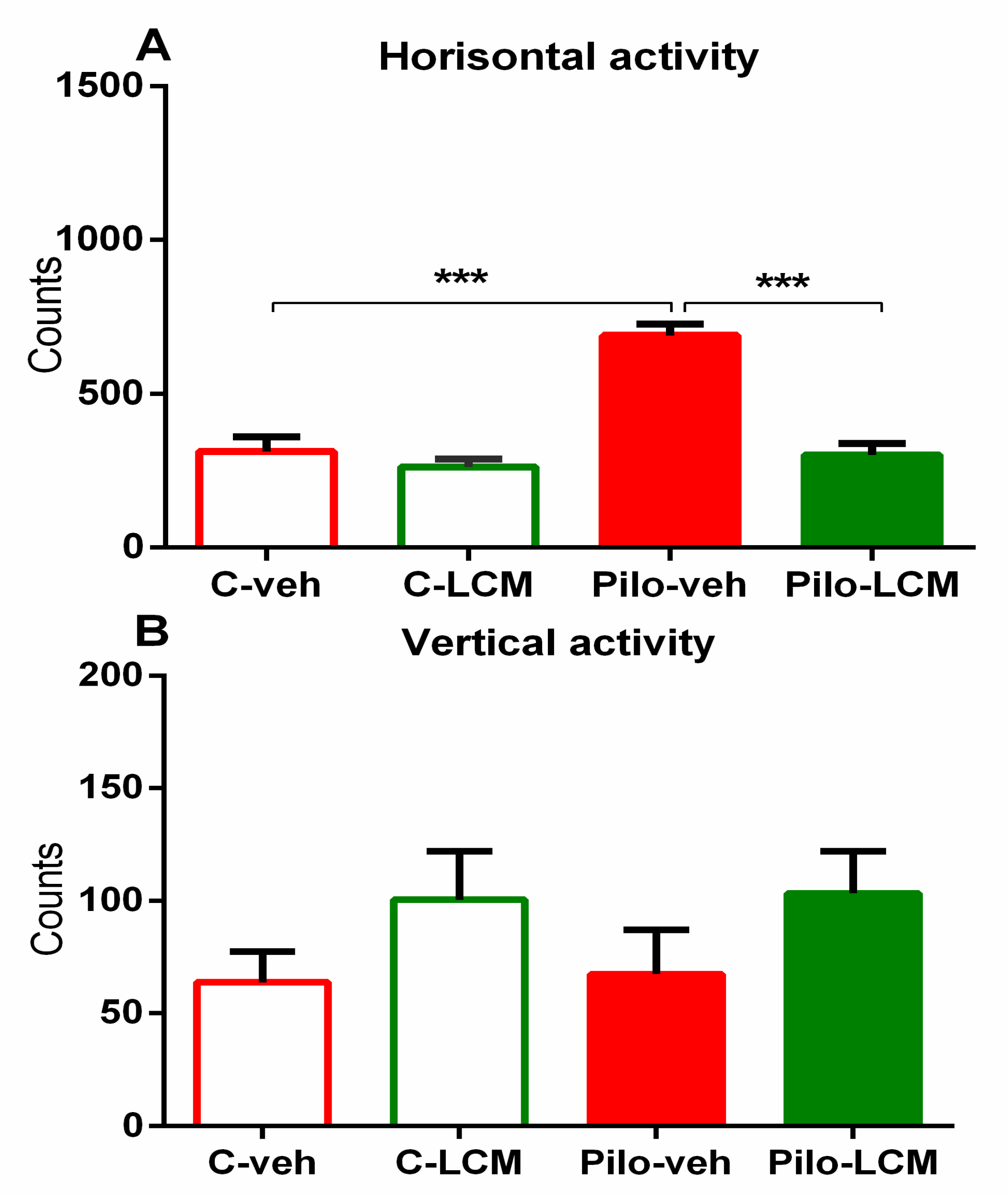
2.2.1. Activity Cage
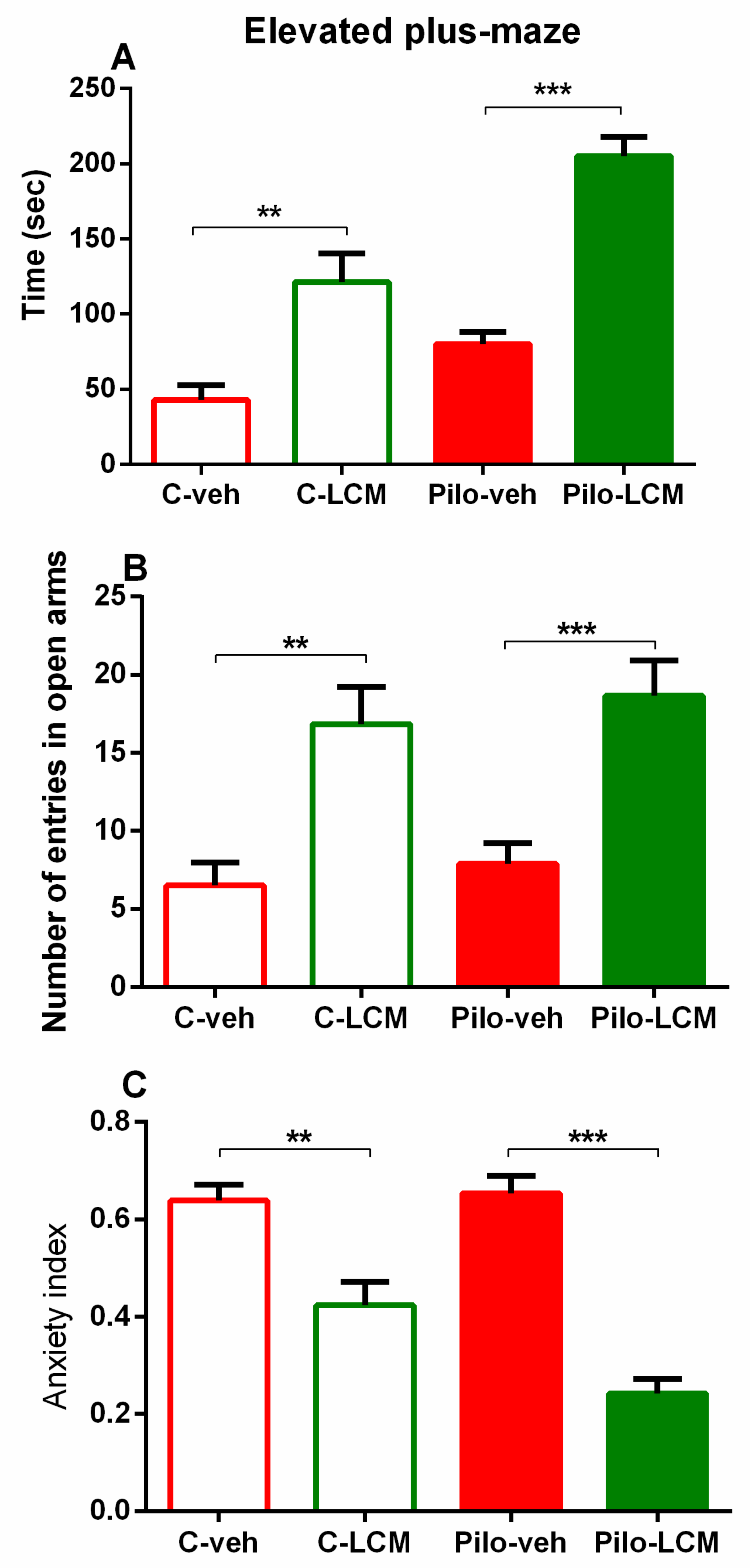
2.2.2. Elevated Plus Maze (EPM) Test
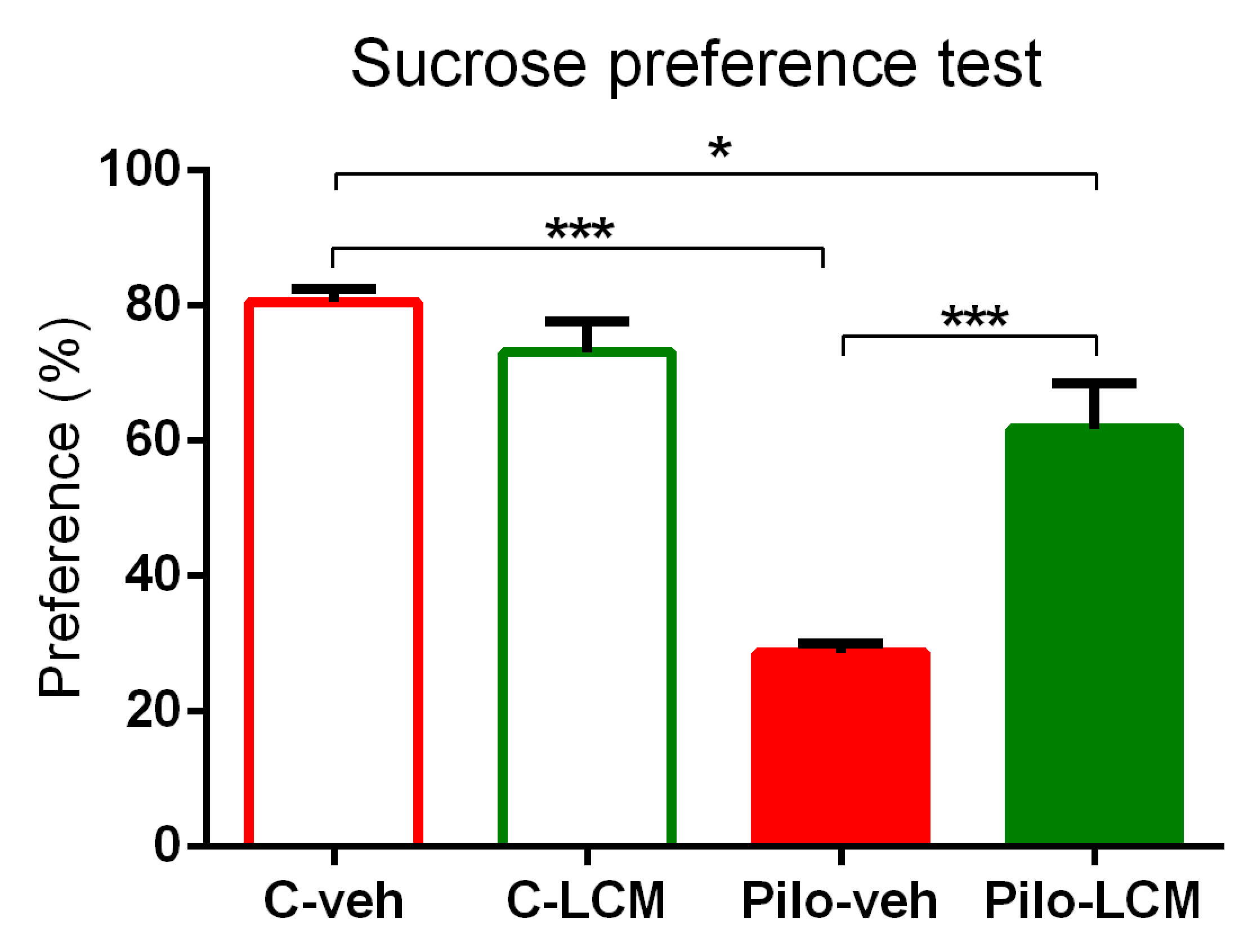
2.2.3. Sucrose Preference Test (SPT)
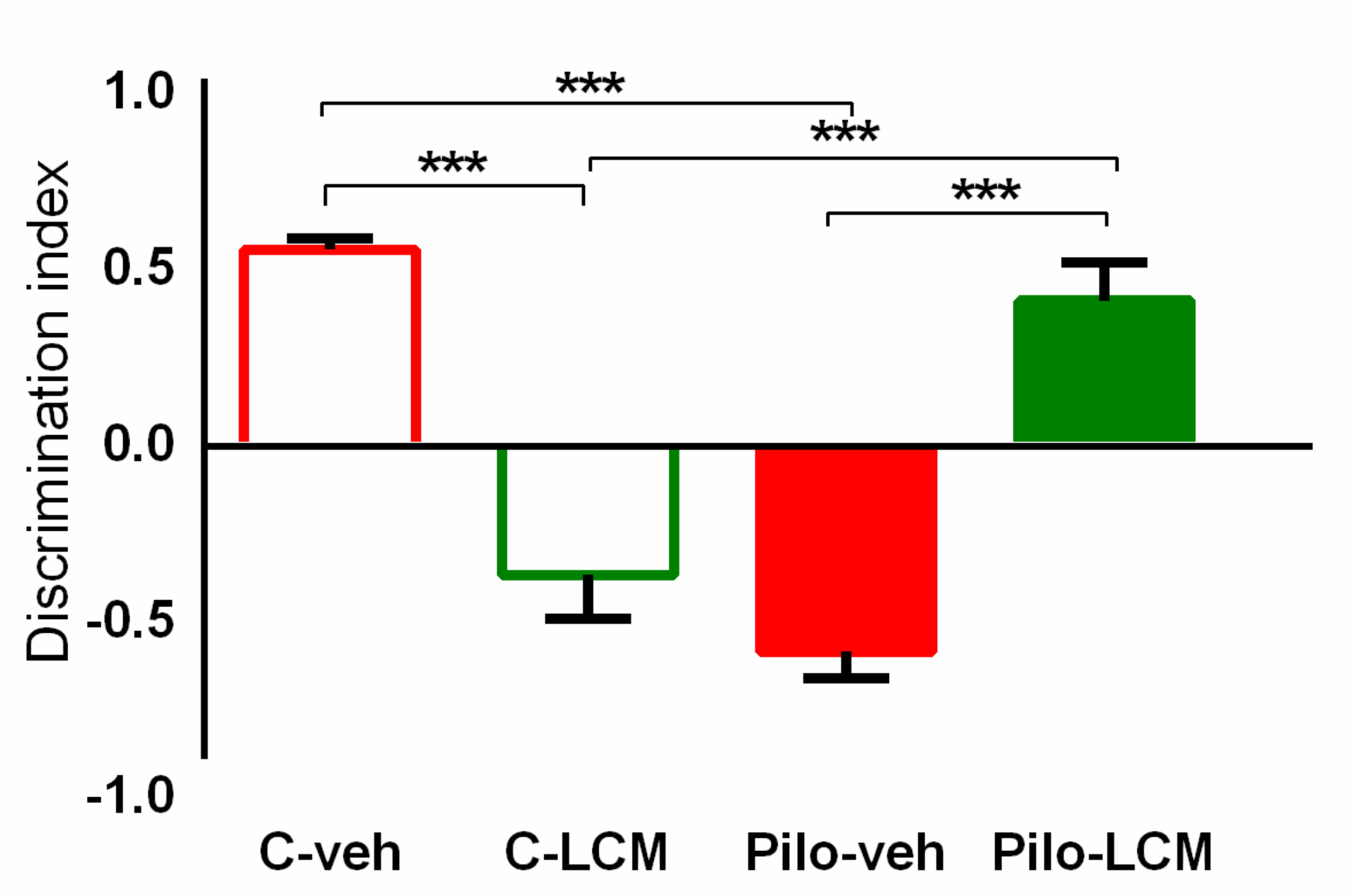
2.2.4. Object Recognition Test (ORT)
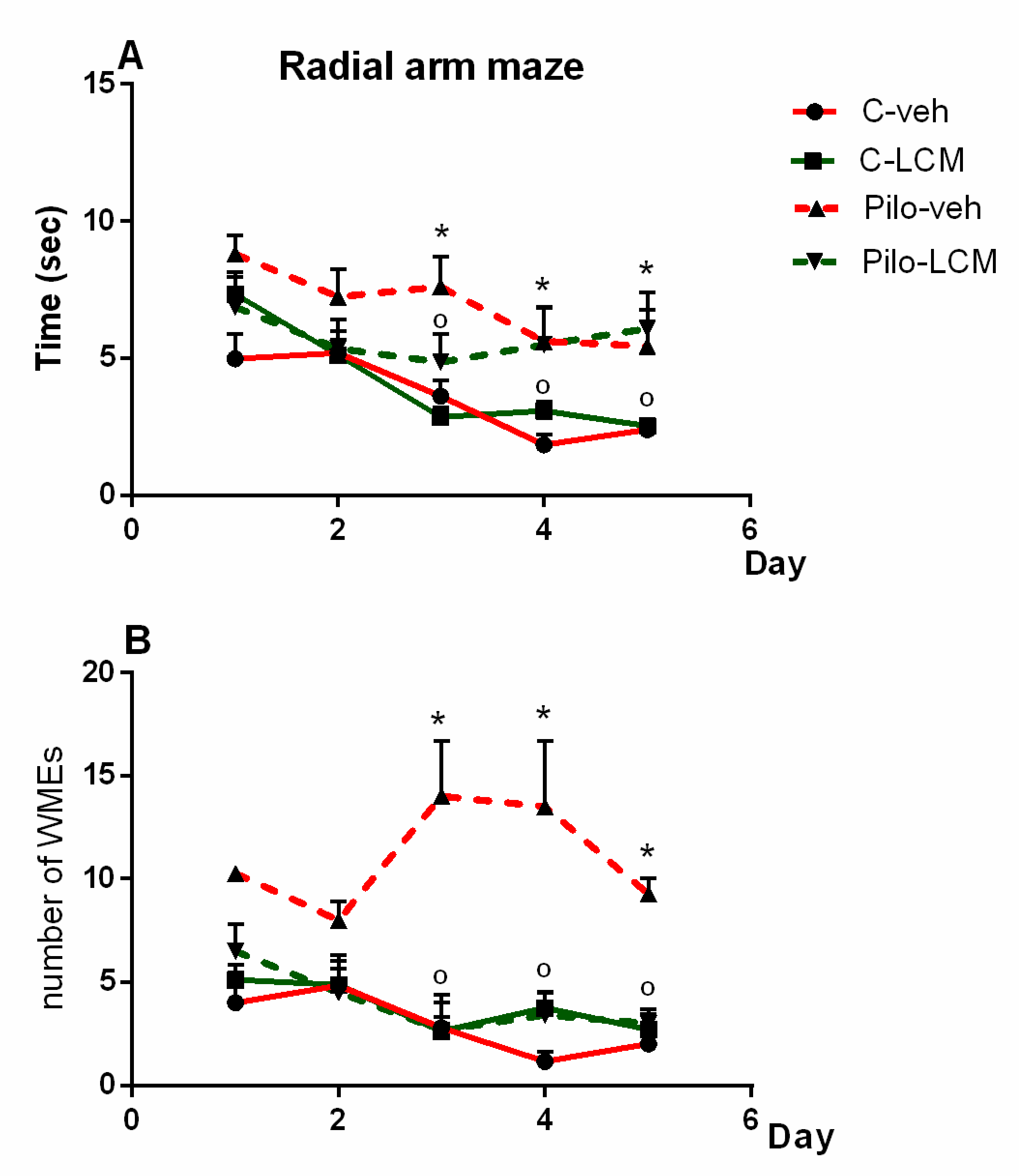
2.2.5. Radial Arm Maze Test (RAM)
2.3. Markers of Oxidative Stress
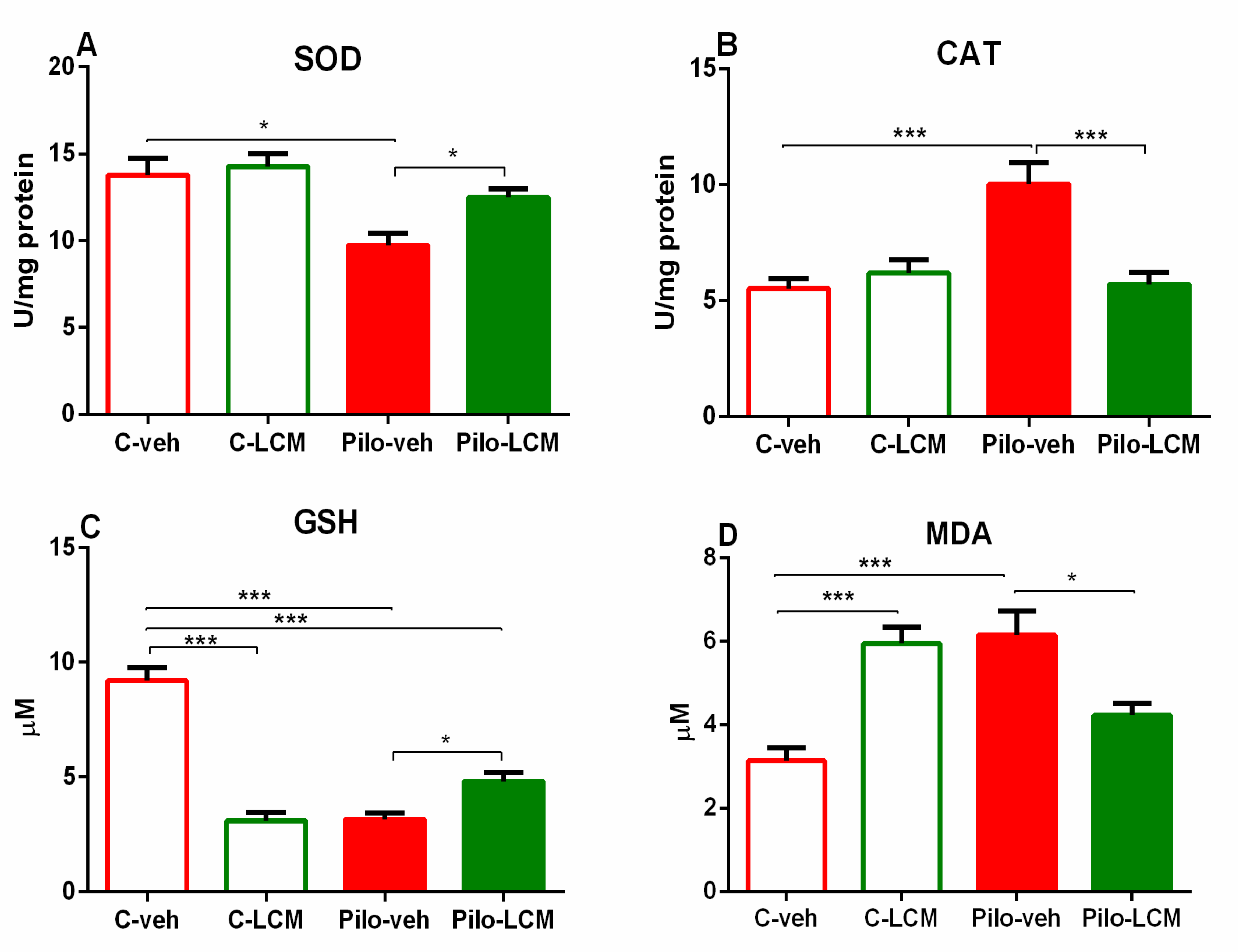
2.3.1. SOD Activity Assay
2.3.2. CAT Activity Assay
2.3.3. GSH Assay
2.3.4. MDA Levels
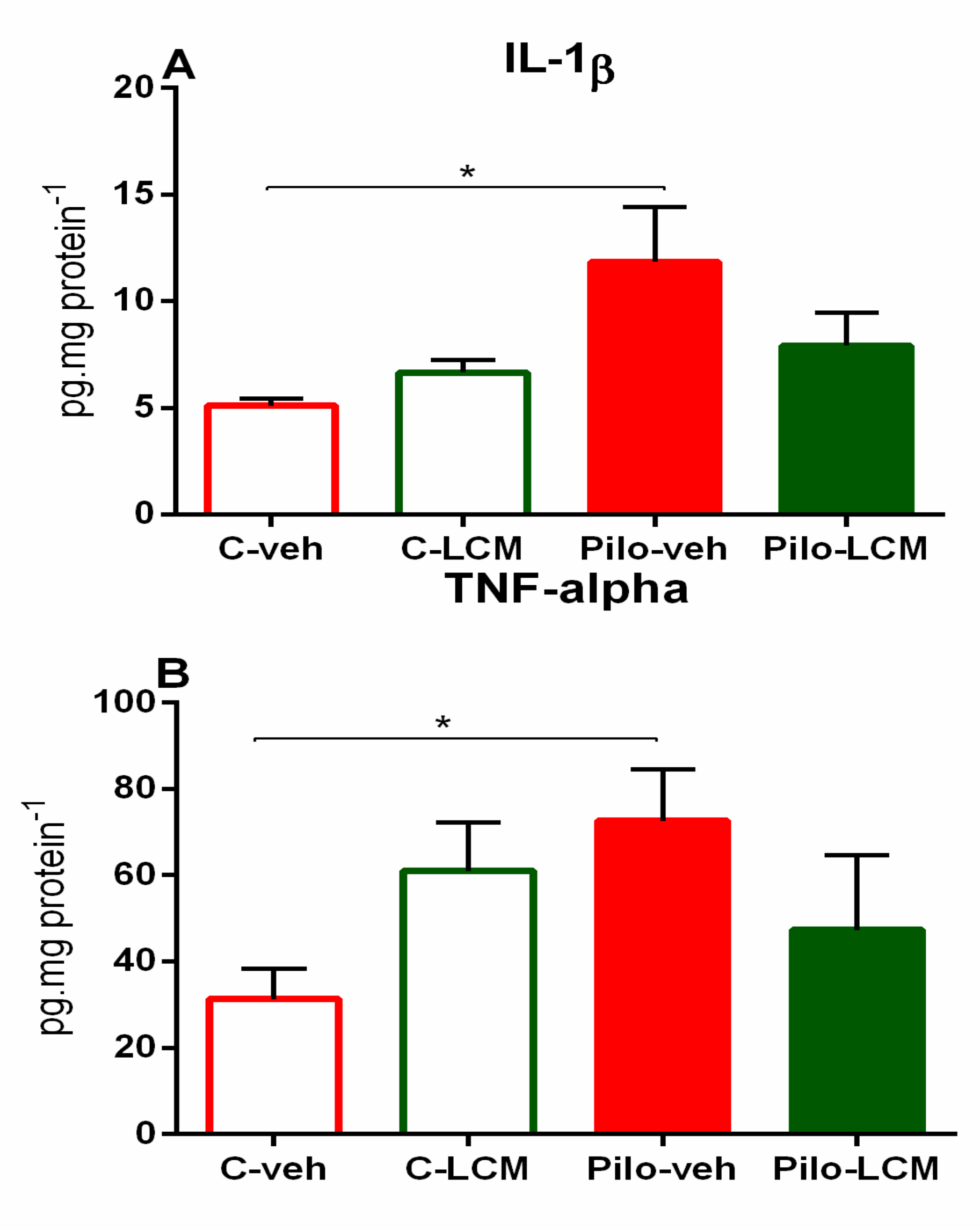
2.4. IL-1β Levels and TNF-α Levels
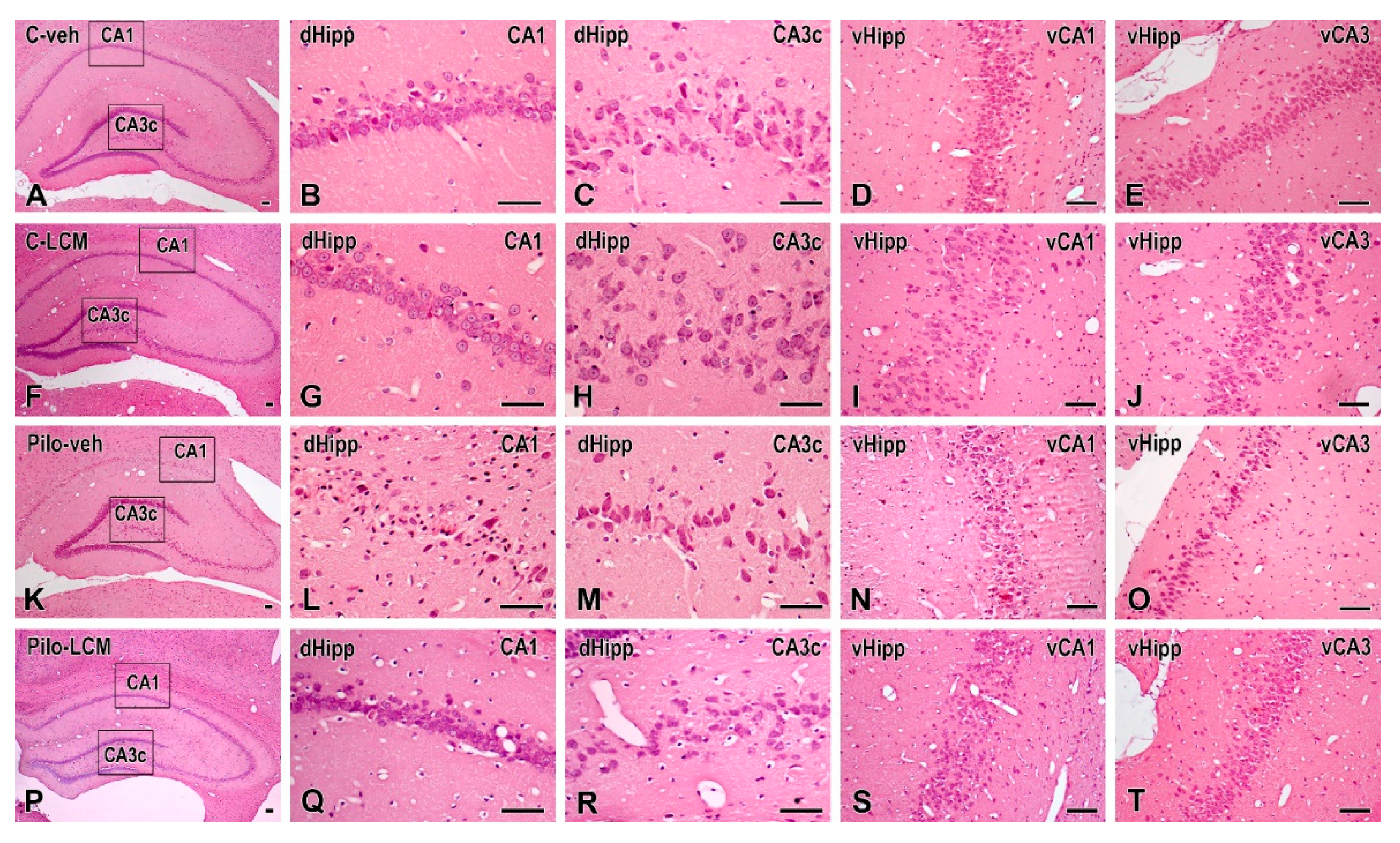
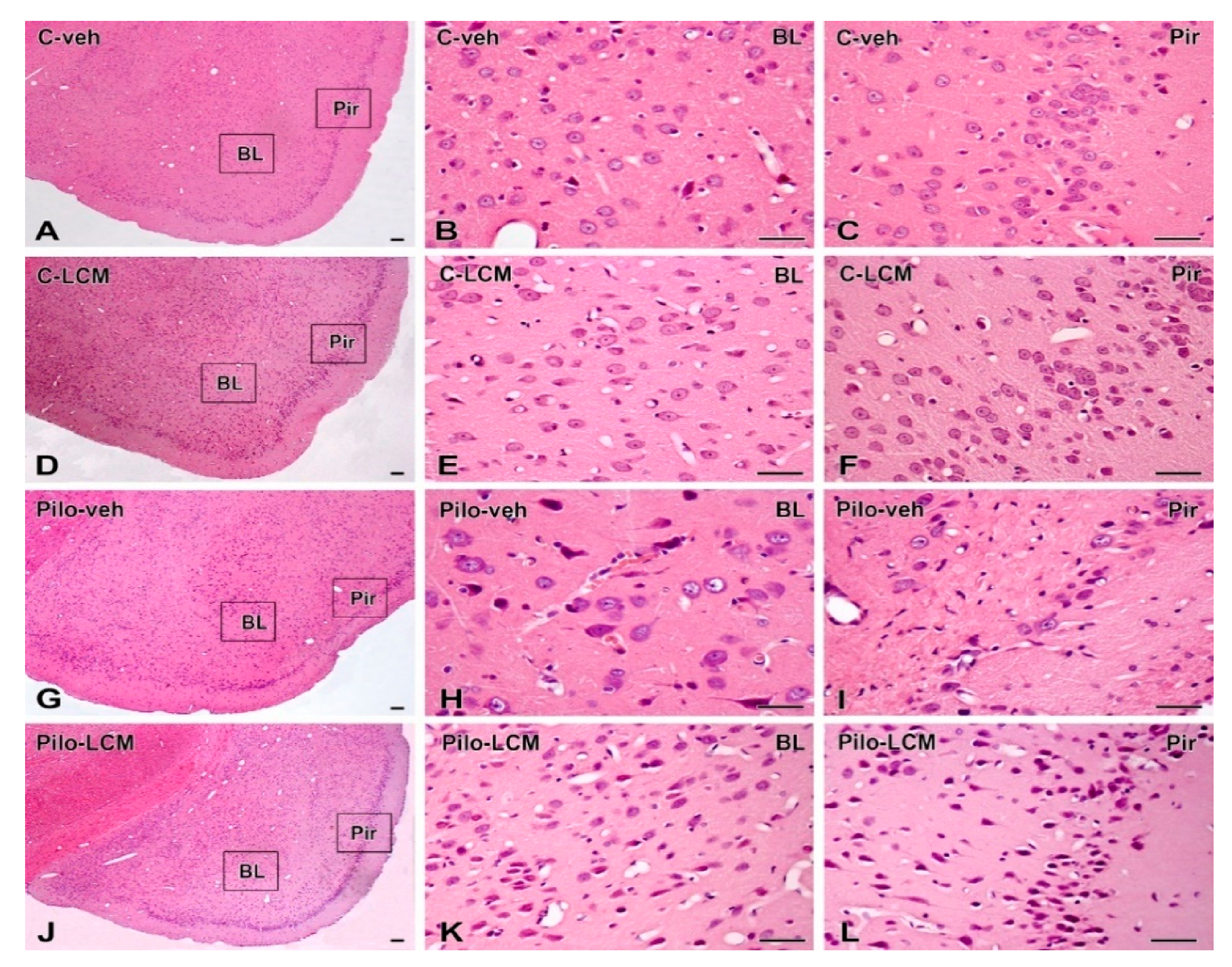
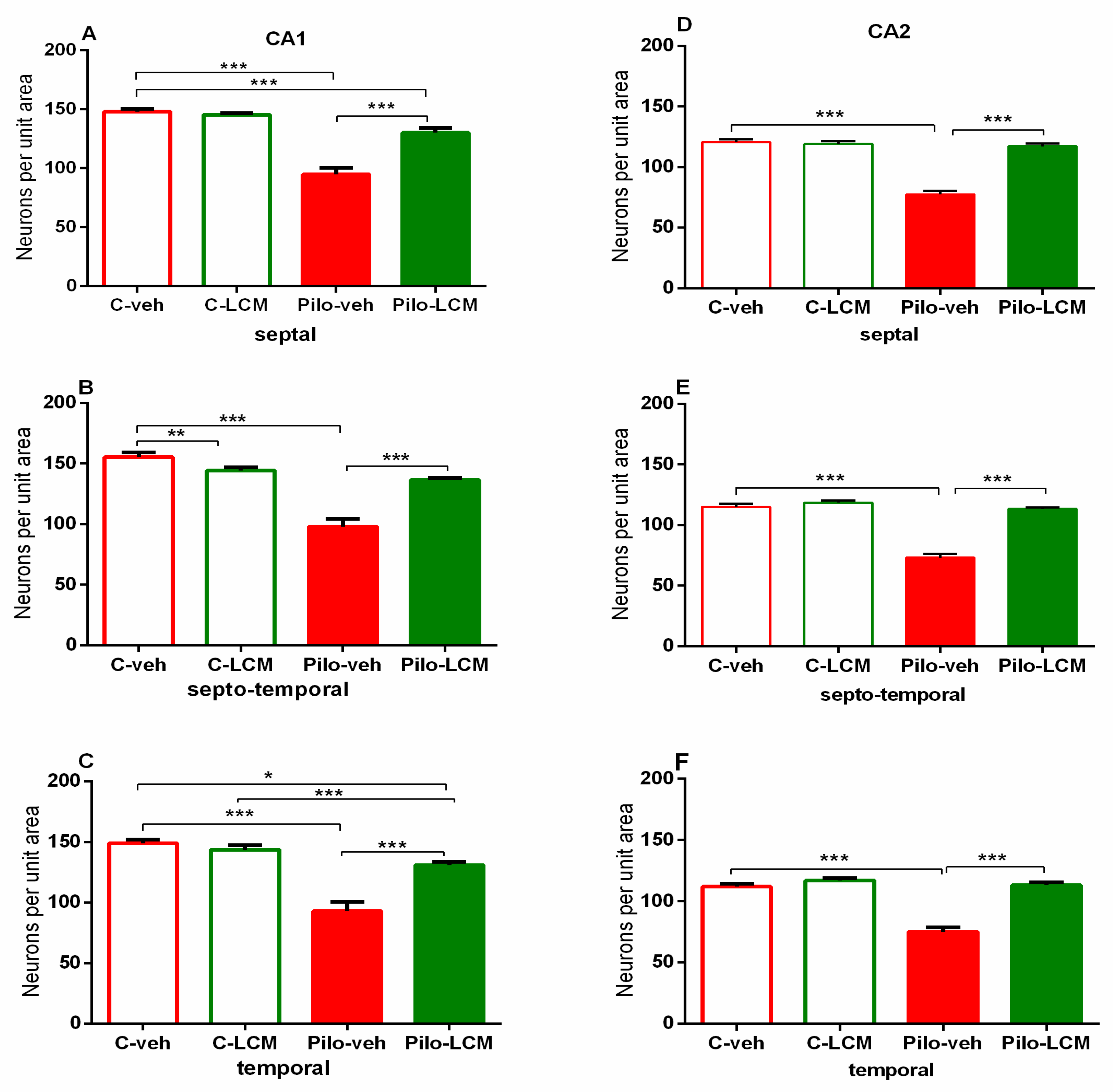
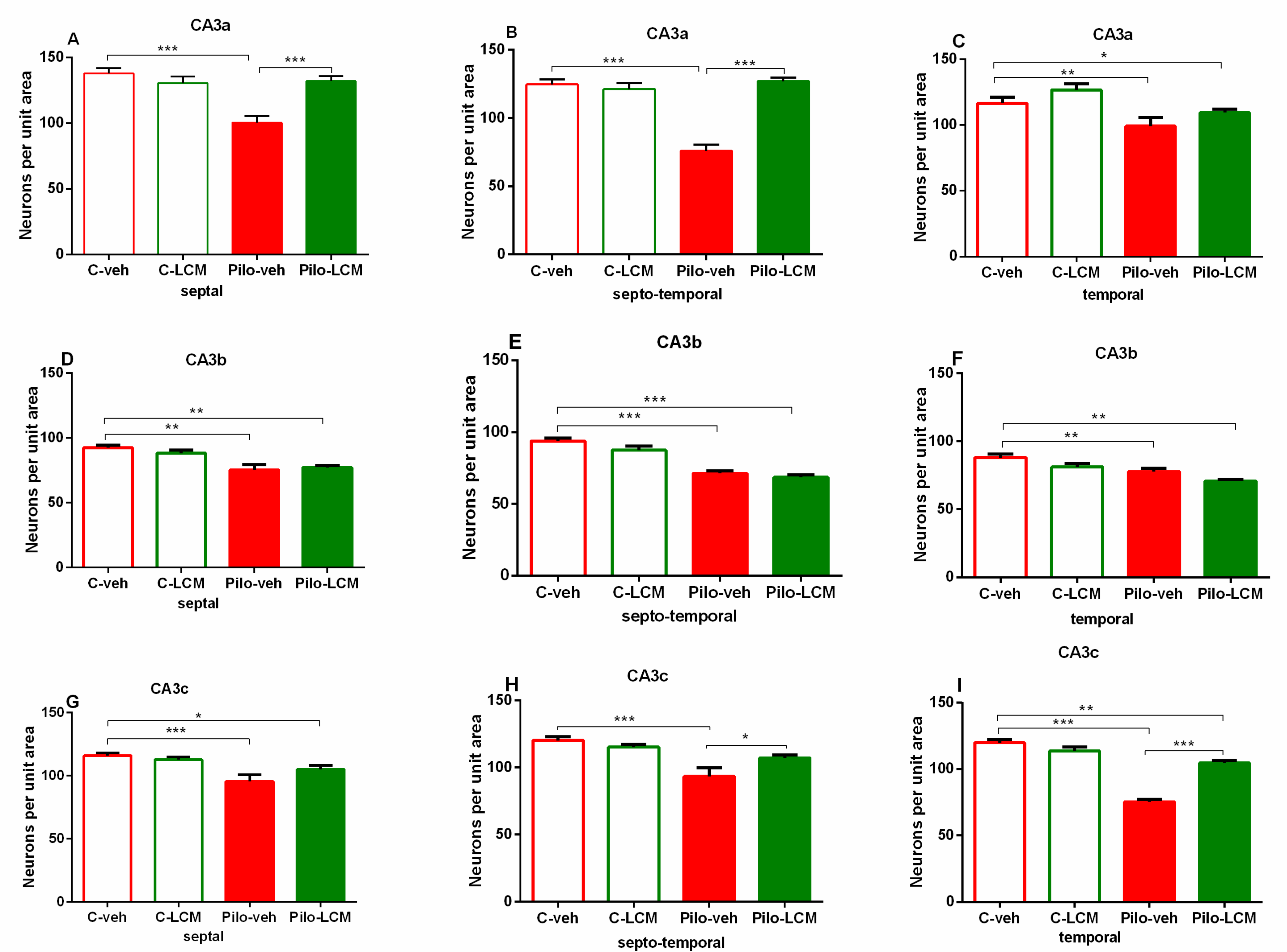
2.5. Histology
3. Discussion
4. Materials and Methods
4.1. Drugs and Reagents
4.2. Animals
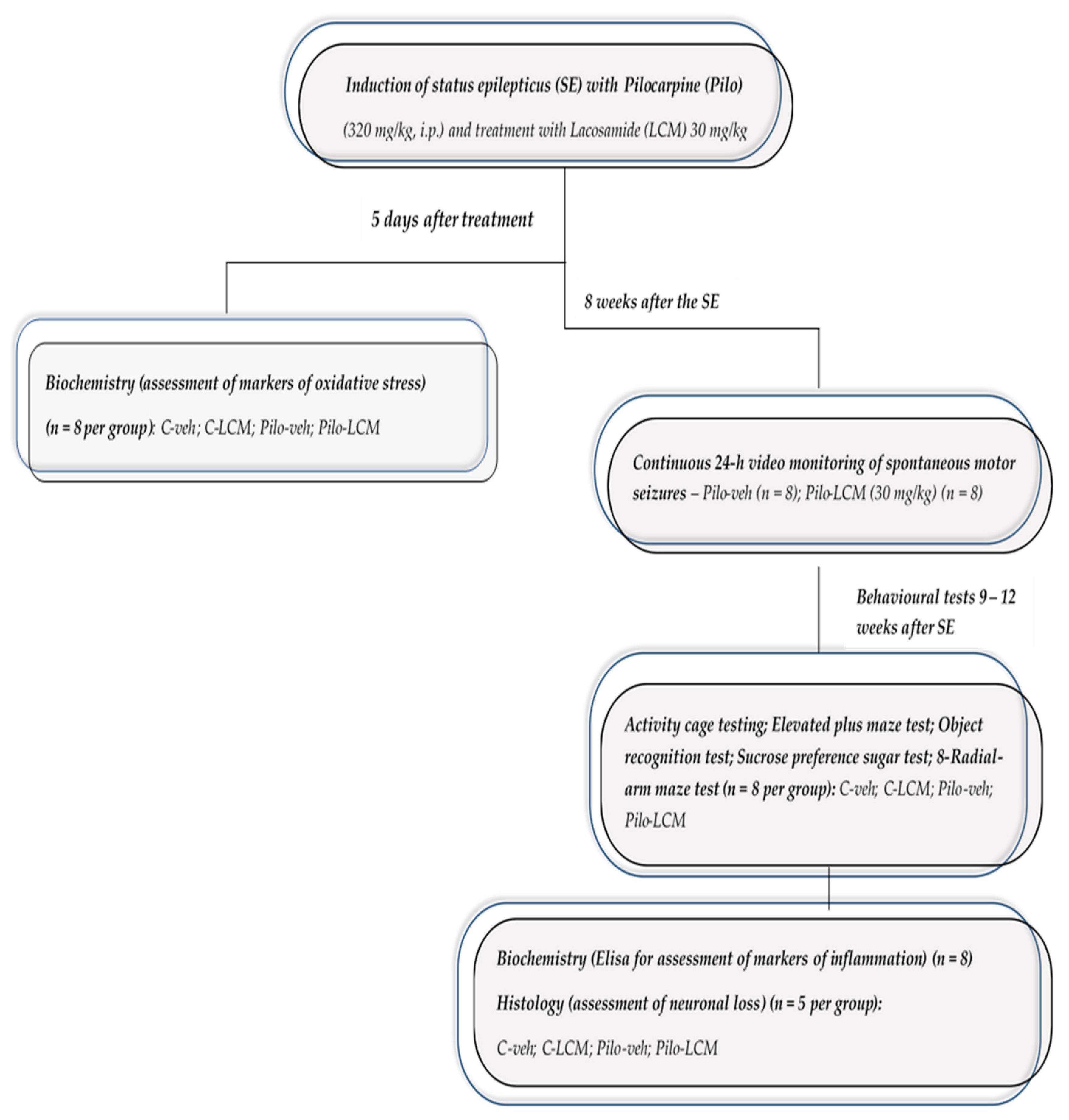
4.3. Study Design, Induction of Status Epilepticus and Treatment with LCM
4.4. Video Monitoring of Spontaneous Motor Seizures
4.5. Behavioral Tests
4.5.1. Activity Cage
4.5.2. Elevated Plus Maze Test
4.5.3. Object Recognition Test
4.5.4. Sucrose Preference Test
4.5.5. Radial Arm Maze Test
4.6. Markers of Oxidative Stress
4.6.1. Preparation of Tissue Homogenate
4.6.2. Protein Content
4.6.3. SOD Activity Assay
4.6.4. GSH Assay
4.6.5. CAT Activity Assay
4.6.6. MDA Assay
4.7. Measurement of IL-1 Beta and TNF-Alpha
4.8. Histology
4.9. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Perucca, P.; Bahlo, M.; Berkovic, S.F. The Genetics of Epilepsy. Annu. Rev. Genom. Hum. Genet. 2020. [Google Scholar] [CrossRef] [PubMed]
- Klee, R.; Töllner, K.; Rankovic, V.; Römermann, K.; Schidlitzki, A.; Bankstahl, M.; Löscher, W. Network pharmacology for antiepileptogenesis: Tolerability of multitargeted drug combinations in nonepileptic vs. post-status epilepticus mice. Epilepsy Res. 2015, 118, 34–48. [Google Scholar] [CrossRef] [PubMed]
- Mao, K.; Lei, D.; Zhang, H.; You, C. Anticonvulsant effect of piperine ameliorates memory impairment, inflammation and oxidative stress in a rat model of pilocarpine-induced epilepsy. Exp. Ther. Med. 2017, 13, 695–700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crans, R.A.; Daelemans, S.; Raedt, R.; Ciruela, F.; Stove, C.P. Kainic acid-induced status epilepticus decreases mGlu5 receptor and phase-specifically downregulates Homer1b/c expression. Brain Res. 2020, 1730, 146640. [Google Scholar] [CrossRef]
- Mohamed, H.K.; Eltony, S.A. Effect of acute pentylenetetrazol injection induced epileptic seizures on rat dentate gyrus at different postnatal ages. Anat. Cell Biol. 2020, 53, 84–94. [Google Scholar] [CrossRef] [Green Version]
- Watkins, L.V.; Pickrell, W.O.; Kerr, M.P. Treatment of psychiatric comorbidities in patients with epilepsy and intellectual disabilities: Is there a role for the neurologist? Epilepsy Behav. 2019, 98, 322–327. [Google Scholar] [CrossRef]
- Scorza, F.A.; Arida, R.M.; Naffah-Mazzacoratti, M.G.; Scerni, D.; Calderazzo, L.; Cavalheiro, E.A. The pilocarpine model of epilepsy: What have we learned? An. Acad. Bras. Cienc. 2009, 81, 345–365. [Google Scholar] [CrossRef] [Green Version]
- Clarkson, C.; Smeal, R.M.; Hasenoehrl, M.G.; White, J.A.; Rubio, M.E.; Wilcox, K.S. Ultrastructural and functional changes at the tripartite synapse during epileptogenesis in a model of temporal lobe epilepsy. Exp. Neurol. 2020, 326, 113196. [Google Scholar] [CrossRef]
- Cavalheiro, E.A. The pilocarpine model of epilepsy. Neurol. Sci. 1995, 16, 33–37. [Google Scholar] [CrossRef]
- Shetty, A.K. Hippocampal injury-induced cognitive and mood dysfunction, altered neurogenesis, and epilepsy: Can early neural stem cell grafting intervention provide protection? Epilepsy Behav. 2014, 38, 117–124. [Google Scholar] [CrossRef] [Green Version]
- Xie, T.; Wang, W.-P.; Mao, Z.-F.; Qu, Z.-Z.; Luan, S.-Q.; Jia, L.-J.; Kan, M.-C. Effects of epigallocatechin-3-gallate on pentylenetetrazole-induced kindling, cognitive impairment and oxidative stress in rats. Neurosci. Lett. 2012, 516, 237–241. [Google Scholar] [CrossRef]
- Folbergrová, J.; Ješina, P.; Kubová, H.; Otáhal, J. Effect of Resveratrol on Oxidative Stress and Mitochondrial Dysfunction in Immature Brain during Epileptogenesis. Mol. Neurobiol. 2018, 55, 7512–7522. [Google Scholar] [CrossRef]
- Waldbaum, S.; Patel, M. Mitochondria, oxidative stress, and temporal lobe epilepsy. Epilepsy Res. 2010, 88, 23–45. [Google Scholar] [CrossRef] [Green Version]
- Folbergrová, J. Oxidative Stress in Immature Brain Following Experimentally-Induced Seizures. Physiol. Res. 2013, 62, S39–S48. [Google Scholar] [CrossRef]
- Puttachary, S.; Sharma, S.; Stark, S.; Thippeswamy, T. Seizure-Induced Oxidative Stress in Temporal Lobe Epilepsy. BioMed Res. Int. 2015, 2015, 1–20. [Google Scholar] [CrossRef]
- Pauletti, A.; Terrone, G.; Shekh-Ahmad, T.; Salamone, A.; Ravizza, T.; Rizzi, M.; Pastore, A.; Pascente, R.; Liang, L.-P.; Villa, B.R.; et al. Targeting oxidative stress improves disease outcomes in a rat model of acquired epilepsy. Brain 2019, 142, e39. [Google Scholar] [CrossRef]
- Pearson, J.N.; Rowley, S.; Liang, L.-P.; White, A.M.; Day, B.J.; Patel, M. Reactive oxygen species mediate cognitive deficits in experimental temporal lobe epilepsy. Neurobiol. Dis. 2015, 82, 289–297. [Google Scholar] [CrossRef] [Green Version]
- Terrone, G.; Frigerio, F.; Balosso, S.; Ravizza, T.; Vezzani, A. Inflammation and reactive oxygen species in status epilepticus: Biomarkers and implications for therapy. Epilepsy Behav. 2019, 101, 106275. [Google Scholar] [CrossRef]
- Ravizza, T.; Gagliardi, B.; Noé, F.; Boer, K.; Aronica, E.; Vezzani, A. Innate and adaptive immunity during epileptogenesis and spontaneous seizures: Evidence from experimental models and human temporal lobe epilepsy. Neurobiol. Dis. 2008, 29, 142–160. [Google Scholar] [CrossRef]
- Vezzani, A.; Rüegg, S. Introduction. Epilepsia 2011, 52, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Pugh, C.; Johnson, J.D.; Martin, D.; Rudy, J.W.; Maier, S.F.; Watkins, L.R. Human immunodeficiency virus-1 coat protein gp120 impairs contextual fear conditioning: A potential role in AIDS related learning and memory impairments. Brain Res. 2000, 861, 8–15. [Google Scholar] [CrossRef]
- Pickering, M.; O’Connor, J.J. Pro-inflammatory cytokines and their effects in the dentate gyrus. Prog. Brain Res. 2007, 163, 339–354. [Google Scholar] [CrossRef] [PubMed]
- Vereyken, E.J.F.; Bajova, H.; Chow, S.; De Graan, P.N.E.; Gruol, N.L. Chronic interleukin-6 alters the level of synaptic proteins in hippocampus in culture and in vivo. Eur. J. Neurosci. 2007, 25, 3605–3616. [Google Scholar] [CrossRef] [PubMed]
- McAfoose, J.; Baune, B. Evidence for a cytokine model of cognitive function. Neurosci. Biobehav. Rev. 2009, 33, 355–366. [Google Scholar] [CrossRef] [Green Version]
- Yirmiya, R.; Goshen, I. Immune modulation of learning, memory, neural plasticity and neurogenesis. Brain Behav. Immun. 2011, 25, 181–213. [Google Scholar] [CrossRef]
- Vezzani, A.; Viviani, B. Neuromodulatory properties of inflammatory cytokines and their impact on neuronal excitability. Neuropharmacology 2015, 96, 70–82. [Google Scholar] [CrossRef]
- Alves, M.; Garcia, L.D.D.; Conte, G.; Jimenez-Mateos, E.M.; D’Orsi, B.; Sanz-Rodriguez, A.; Prehn, J.H.; Henshall, D.C.; Engel, T. Context-Specific Switch from Anti- to Pro-epileptogenic Function of the P2Y1 Receptor in Experimental Epilepsy. J. Neurosci. 2019, 39, 5377–5392. [Google Scholar] [CrossRef] [Green Version]
- Temkin, N.R. Antiepileptogenesis and Seizure Prevention Trials with Antiepileptic Drugs: Meta-Analysis of Controlled Trials. Epilepsia 2001, 42, 515–524. [Google Scholar] [CrossRef] [Green Version]
- Licko, T.; Seeger, N.; Zellinger, C.; Russmann, V.; Matagne, A.; Potschka, H. Lacosamide treatment following status epilepticus attenuates neuronal cell loss and alterations in hippocampal neurogenesis in a rat electrical status epilepticus model. Epilepsia 2013, 54, 1176–1185. [Google Scholar] [CrossRef]
- Nissinen, J.; Pitkänen, A. Effect of antiepileptic drugs on spontaneous seizures in epileptic rats. Epilepsy Res. 2007, 73, 181–191. [Google Scholar] [CrossRef]
- Grabenstatter, H.L.; Clark, S.; Dudek, F.E. Anticonvulsant Effects of Carbamazepine on Spontaneous Seizures in Rats with Kainate-induced Epilepsy: Comparison of Intraperitoneal Injections with Drug-in-food Protocols. Epilepsia 2007, 48, 2287–2295. [Google Scholar] [CrossRef]
- Miziak, B.; Konarzewska, A.; Ułamek-Kozioł, M.; Dudra-Jastrzębska, M.; Pluta, R.; Czuczwar, S.J. Anti-Epileptogenic Effects of Antiepileptic Drugs. Int. J. Mol. Sci. 2020, 21, 2340. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Yu, Y.; Ma, R.; Shao, N.; Meng, H. Lacosamide modulates collapsin response mediator protein 2 and inhibits mossy fiber sprouting after kainic acid-induced status epilepticus. NeuroReport 2018, 29, 1384–1390. [Google Scholar] [CrossRef]
- Tchekalarova, J.; Atanasova, D.; Kortenska, L.; Lazarov, N.; Shishmanova-Doseva, M.; Galchev, T.; Marinov, P. Agomelatine alleviates neuronal loss through BDNF signaling in the post-status epilepticus model induced by kainic acid in rat. Brain Res. Bull. 2019, 147, 22–35. [Google Scholar] [CrossRef]
- Racine, R.; Rose, P.A.; Burnham, W.M. Afterdischarge Thresholds and Kindling Rates in Dorsal and Ventral Hippocampus and Dentate Gyrus. Can. J. Neurol. Sci. J. Can. Sci. Neurol. 1977, 4, 273–278. [Google Scholar] [CrossRef] [Green Version]
- Akaike, K.; Tanaka, S.; Tojo, H.; Fukumoto, S.-I.; Imamura, S.-I.; Takigawa, M. Kainic acid-induced dorsal and ventral hippocampal seizures in rats. Brain Res. 2001, 900, 65–71. [Google Scholar] [CrossRef]
- Fanselow, M.S.; Dong, H.-W. Are the Dorsal and Ventral Hippocampus Functionally Distinct Structures? Neuron 2010, 65, 7–19. [Google Scholar] [CrossRef] [Green Version]
- Deng, X.; Wang, M.; Hu, S.; Feng, Y.; Shao, Y.; Xie, Y.; Wu, M.; Chen, Y.; Shi, X. The Neuroprotective Effect of Astaxanthin on Pilocarpine-Induced Status Epilepticus in Rats. Front. Cell. Neurosci. 2019, 13, 123. [Google Scholar] [CrossRef]
- Shannon, H.E.; Love, P.L. Effects of antiepileptic drugs on attention as assessed by a five-choice serial reaction time task in rats. Epilepsy Behav. 2005, 7, 620–628. [Google Scholar] [CrossRef]
- De Lima, M.N.M.; Presti-Torres, J.; Dornelles, A.; Bromberg, E.; Schröder, N. Differential effects of low and high doses of topiramate on consolidation and retrieval of novel object recognition memory in rats. Epilepsy Behav. 2007, 10, 32–37. [Google Scholar] [CrossRef]
- Barker-Haliski, M.L.; Vanegas, F.; Mau, M.J.; Underwood, T.K.; White, H.S. Acute cognitive impact of antiseizure drugs in naive rodents and corneal-kindled mice. Epilepsia 2016, 57, 1386–1397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Helmstaedter, C.; Witt, J.-A. The longer-term cognitive effects of adjunctive antiepileptic treatment with lacosamide in comparison with lamotrigine and topiramate in a naturalistic outpatient setting. Epilepsy Behav. 2013, 26, 182–187. [Google Scholar] [CrossRef] [PubMed]
- Liguori, C.; Izzi, F.; Manfredi, N.; Mercuri, N.B.; Placidi, F. Lacosamide may improve cognition in patients with focal epilepsy: EpiTrack to compare cognitive side effects of lacosamide and carbamazepine. Epilepsy Behav. Case Rep. 2018, 10, 35–37. [Google Scholar] [CrossRef] [PubMed]
- Higgins, G.A.; Breysse, N.; Undzys, E.; Derksen, D.R.; Jeffrey, M.; Scott, B.W.; Xin, T.; Roucard, C.; Bressand, K.; Depaulis, A.; et al. Comparative study of five antiepileptic drugs on a translational cognitive measure in the rat: Relationship to antiepileptic property. Psychopharmacology 2010, 207, 513–527. [Google Scholar] [CrossRef]
- Andres-Mach, M.; Szewczyk, A.; Zagaja, M.; Luszczki, J.; Maj, M.; Rola, R.; Abram, M.; Kaminski, K. Evaluation of the impact of compound C11 a new anticonvulsant candidate on cognitive functions and hippocampal neurogenesis in mouse brain. Neuropharmacology 2020, 163, 107849. [Google Scholar] [CrossRef]
- Shishmanova-Doseva, M.; Peychev, L.; Koeva, Y.; Terzieva, D.; Georgieva, K.; Peychev, Z. Chronic treatment with the new anticonvulsant drug lacosamide impairs learning and memory processes in rats: A possible role of BDNF/TrkB ligand receptor system. Pharmacol. Biochem. Behav. 2018, 169, 1–9. [Google Scholar] [CrossRef]
- Rocamora, R.; Ley, M.; Molins, A.; Toledo, M.; Sansa, G.; Bertol, V.; Becerra, J.-L.; Carreño, M.; Mauri, J.Á. Effect of lacosamide on depression and anxiety symptoms in patients with focal refractory epilepsy: A prospective multicenter study. Epilepsy Behav. 2018, 79, 87–92. [Google Scholar] [CrossRef]
- Moseley, B.D.; Cole, D.; Iwuora, O.; Strawn, J.R.; Privitera, M. The effects of lacosamide on depression and anxiety in patients with epilepsy. Epilepsy Res. 2015, 110, 115–118. [Google Scholar] [CrossRef]
- Alfaro, A.; Asensio, M.; García-Escrivá, A.; Medrano, V.; Salom, J.; Tortosa, D.; Palao, S.; Lezcano, M.; Berenguer, L.; Navarro, M.; et al. Estudio LAM: Conducta y calidad de vida en pacientes diagnosticados de epilepsia tratados con lacosamida. Neurología 2019, 34, 1–6. [Google Scholar] [CrossRef]
- Higgins, G.A.; Breysse, N.; Undzys, E.; Kuo, C.; Joharchi, N.; Derksen, D.R.; Xin, T.; Isaac, M.; Slassi, M. The anti-epileptic drug lacosamide (Vimpat®) has anxiolytic property in rodents. Eur. J. Pharmacol. 2009, 624, 1–9. [Google Scholar] [CrossRef]
- Cuomo, I.; Piacentino, D.; Kotzalidis, G.D.; Lionetto, L.; De Filippis, S. Lacosamide in bipolar disorder: A 30-day comparison to a retrospective control group treated with other antiepileptics. Psychiatry Clin. Neurosci. 2018, 72, 864–875. [Google Scholar] [CrossRef]
- Frantseva, M.; Velazquez, J.P.; Tsoraklidis, G.; Mendonca, A.; Adamchik, Y.; Mills, L.; Carlen, P.; Burnham, M. Oxidative stress is involved in seizure-induced neurodegeneration in the kindling model of epilepsy. Neuroscience 2000, 97, 431–435. [Google Scholar] [CrossRef]
- Stadtman, E.R. Protein Oxidation in Aging and Age-Related Diseases. Ann. N. Y. Acad. Sci. 2001, 928, 22–38. [Google Scholar] [CrossRef]
- Kovac, S.; Kostova, A.T.D.; Herrmann, A.M.; Melzer, N.; Meuth, S.G.; Gorji, A. Metabolic and Homeostatic Changes in Seizures and Acquired Epilepsy—Mitochondria, Calcium Dynamics and Reactive Oxygen Species. Int. J. Mol. Sci. 2017, 18, 1935. [Google Scholar] [CrossRef] [Green Version]
- Cevik, M.U.; Dönmezdil, N.; Ozdemir, H.H.; Taşin, M. Investigation of PON1 activity and MDA levels in patients with epilepsy not receiving antiepileptic treatment. Neuropsychiatr. Dis. Treat. 2016, 12, 1013–1017. [Google Scholar] [CrossRef]
- González-Reyes, S.; Santillán-Cigales, J.J.; Jiménez-Osorio, A.S.; Pedraza-Chaverri, J.; Guevara-Guzmán, R. Glycyrrhizin ameliorates oxidative stress and inflammation in hippocampus and olfactory bulb in lithium/pilocarpine-induced status epilepticus in rats. Epilepsy Res. 2016, 126, 126–133. [Google Scholar] [CrossRef]
- Ali, A.E.; Mahdy, H.M.; Elsherbiny, D.M.; Azab, S.S. Rifampicin ameliorates lithium-pilocarpine-induced seizures, consequent hippocampal damage and memory deficit in rats: Impact on oxidative, inflammatory and apoptotic machineries. Biochem. Pharmacol. 2018, 156, 431–443. [Google Scholar] [CrossRef]
- Ahmed, M.A.E. Neuroprotective Effects of Idebenone against Pilocarpine-Induced Seizures: Modulation of Antioxidant Status, DNA Damage and Na+, K+-ATPase Activity in Rat Hippocampus. Neurochem. Res. 2014, 39, 394–402. [Google Scholar] [CrossRef]
- Nirwan, N.; Siraj, F.; Vohora, D. Inverted-U response of lacosamide on pilocarpine-induced status epilepticus and oxidative stress in C57BL/6 mice is independent of hippocampal collapsin response mediator protein-2. Epilepsy Res. 2018, 145, 93–101. [Google Scholar] [CrossRef]
- Demiroz, S.; Ur, K.; Ulucan, A.; Bengu, A.S.; Ur, F.D.; Gergin, O.O.; Erdem, S. Neuroprotective Effects of Lacosamide in Experimental Traumatic Spinal Cord Injury in Rats. Turk. Neurosurg. 2019, 29, 718–723. [Google Scholar] [CrossRef]
- Savran, M.; Ozmen, O.; Erzurumlu, Y.; Savas, H.B.; Asci, S.; Kaynak, M. The Impact of Prophylactic Lacosamide on LPS-Induced Neuroinflammation in Aged Rats. Inflammation 2019, 42, 1913–1924. [Google Scholar] [CrossRef]
- Roseti, C.; van Vliet, E.A.; Cifelli, P.; Ruffolo, G.; Baayen, J.C.; Di Castro, M.A.; Bertollini, C.; Limatola, C.; Aronica, E.; Vezzani, A.; et al. GABAA currents are decreased by IL-1β in epileptogenic tissue of patients with temporal lobe epilepsy: Implications for ictogenesis. Neurobiol. Dis. 2015, 82, 311–320. [Google Scholar] [CrossRef]
- Frigerio, F.; Flynn, C.; Han, Y.; Lyman, K.; Lugo, J.N.; Ravizza, T.; Ghestem, A.; Pitsch, J.; Becker, A.; Anderson, A.E.; et al. Neuroinflammation Alters Integrative Properties of Rat Hippocampal Pyramidal Cells. Mol. Neurobiol. 2018, 55, 7500–7511. [Google Scholar] [CrossRef]
- Dantzer, R.; O’Connor, J.C.; Freund, G.G.; Johnson, R.W.; Kelley, K.W. From inflammation to sickness and depression: When the immune system subjugates the brain. Nat. Rev. Neurosci. 2008, 9, 46–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Avila, J.C.; Siqueira, L.D.; Mazeraud, A.; Azevedo, E.P.; Foguel, D.; Castro-Faria-Neto, H.C.; Sharshar, T.; Chrétien, F.; Bozza, F.A. Age-related cognitive impairment is associated with long-term neuroinflammation and oxidative stress in a mouse model of episodic systemic inflammation. J. Neuroinflamm. 2018, 15, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paudel, Y.N.; Shaikh, M.F.; Shah, S.; Kumari, Y.; Othman, I. Role of inflammation in epilepsy and neurobehavioral comorbidities: Implication for therapy. Eur. J. Pharmacol. 2018, 837, 145–155. [Google Scholar] [CrossRef] [PubMed]
- Kodali, M.; Hattiangady, B.; Shetty, G.; Bates, A.; Shuai, B.; Shetty, A. Curcumin treatment leads to better cognitive and mood function in a model of Gulf War Illness with enhanced neurogenesis, and alleviation of inflammation and mitochondrial dysfunction in the hippocampus. Brain Behav. Immun. 2018, 69, 499–514. [Google Scholar] [CrossRef] [PubMed]
- Voutsinosporche, B.; Koning, E.; Kaplan, H.; Ferrandon, A.; Guenounou, M.; Nehlig, A.; Motte, J. Temporal patterns of the cerebral inflammatory response in the rat lithium–pilocarpine model of temporal lobe epilepsy. Neurobiol. Dis. 2004, 17, 385–402. [Google Scholar] [CrossRef]
- Al-Massri, K.F.; Ahmed, L.A.; El-Abhar, H.S. Pregabalin and lacosamide ameliorate paclitaxel-induced peripheral neuropathy via inhibition of JAK/STAT signaling pathway and Notch-1 receptor. Neurochem. Int. 2018, 120, 164–171. [Google Scholar] [CrossRef]
- Racine, R.J. Modification of seizure activity by electrical stimulation: II. Motor seizure. Electroencephalogr. Clin. Neurophysiol. 1972, 32, 281–294. [Google Scholar] [CrossRef]
- Atanasova, D.; Tchekalarova, J.; Ivanova, N.; Nenchovska, Z.; Pavlova, E.; Atanassova, N.; Lazarov, N. Losartan suppresses the kainate-induced changes of angiotensin AT 1 receptor expression in a model of comorbid hypertension and epilepsy. Life Sci. 2018, 193, 40–46. [Google Scholar] [CrossRef]
- Tchekalarova, J.; Atanasova, D.; Kortenska, L.; Atanasova, M.; Lazarov, N. Chronic agomelatine treatment prevents comorbid depression in the post-status epilepticus model of acquired epilepsy through suppression of inflammatory signaling. Neurobiol. Dis. 2018, 115, 127–144. [Google Scholar] [CrossRef]
- Kakkar, P.; Das, B.; Viswanathan, P.N. A modified spectrophotometric assay of superoxide dismutase. Indian J. Biochem. Biophys. 1984, 21, 130–132. [Google Scholar]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Tchekalarova, J.; Petkova, Z.; Pechlivanova, D.; Moyanova, S.; Kortenska, L.; Mitreva, R.; Lozanov, V.; Atanasova, D.; Lazarov, N.; Stoynev, A.; et al. Prophylactic treatment with melatonin after status epilepticus: Effects on epileptogenesis, neuronal damage, and behavioral changes in a kainate model of temporal lobe epilepsy. Epilepsy Behav. 2013, 27, 174–187. [Google Scholar] [CrossRef]
- Tchekalarova, J.; Atanasova, D.; Nenchovska, Z.; Atanasova, M.; Kortenska, L.; Gesheva, R.; Lazarov, N. Agomelatine protects against neuronal damage without preventing epileptogenesis in the kainate model of temporal lobe epilepsy. Neurobiol. Dis. 2017, 104, 1–14. [Google Scholar] [CrossRef]


















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
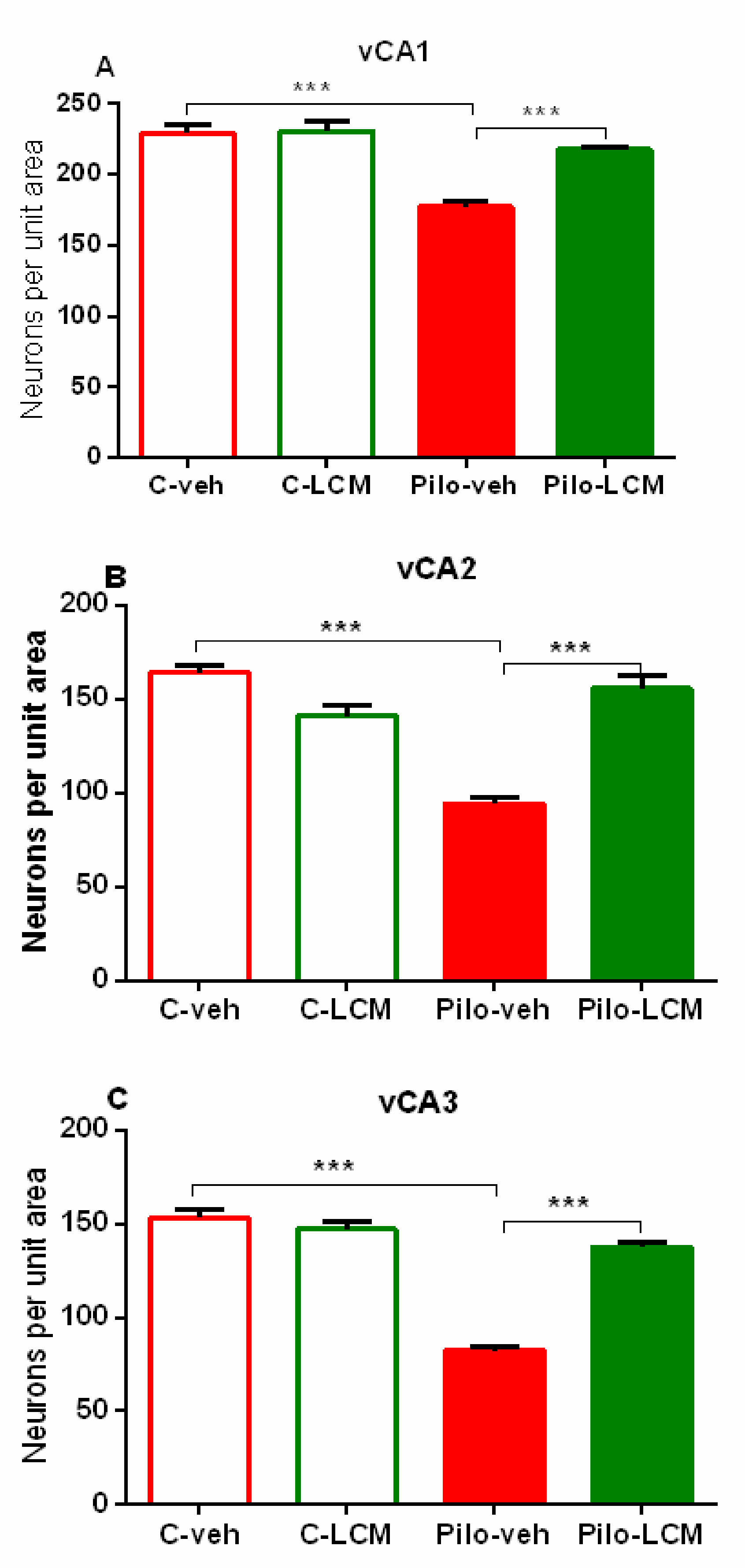
| CA1. Septal | Epilepsy [F1,88 = 72.742, p < 0.001] Treatment [F1,87 = 31.036, p < 0.001] Epilepsy x Treatment [F1,88 = 17.776, p < 0.001] |
| Septo-temporal | Epilepsy [F1,88 = 72.050, p < 0.001] Treatment [F1,88 = 13.228, p < 0.001] Epilepsy x Treatment [F1,88 = 42.946, p < 0.001] |
| Temporal | Epilepsy [F1,96 = 59.882, p < 0.001] Treatment [F1,96 = 13.564. p < 0.001] Epilepsy x Treatment [F1,96 = 23.695, p < 0.001] |
| CA2 Septal | Epilepsy [F1,59 = 49.985, p < 0.001] Treatment [F1,59 = 71.588 p < 0.001] Epilepsy x Treatment [F1,59 = 59.156, p < 0.001] |
| Septo-temporal | Epilepsy [F1,81 = 70.550, p < 0.001] Treatment [F1,81 = 70.550, p < 0.001] Epilepsy x Treatment [F1,81 = 47.346, p < 0.001] |
| Temporal | Epilepsy [F1,65 = 3.531, p = 0.065] Treatment [F1,65 = 2.637, p = 0.109] Epilepsy x Treatment [F1,65 = 17.273, p < 0.001] |
| CA3a Septal | Epilepsy [F1,63 = 7.273, p = 0.009] Treatment [F1,63 = 8.951, p = 0.004] Epilepsy x Treatment [F1,63 = 8.762, p = 0.004] |
| Septo-temporal | Epilepsy [F1,63 = 23.515, p < 0.001] Treatment [F1,63 = 41.089, p < 0.001] Epilepsy x Treatment [F1,63 = 26.684, p < 0.001] |
| Temporal | Epilepsy [F1,63 = 11.451, p < 0.001] Treatment [F1,63 = 3.851, p =0.053] Epilepsy x Treatment [F1,63 = 0.0019, p = 0.965] |
| CA3b Septal | Epilepsy [F1,90 = 0.330, p = 0.567] Treatment [F1,90 = 0.330, p = 0.567] Epilepsy x Treatment [F1,90 = 1.356, p = 0.247] |
| Septo-temporal | Epilepsy [F1,93 = 4.408, p = 0.039] Treatment [F1,93 = 93.980, p < 0.001] Epilepsy x Treatment [F1,93 = 0.798, p = 0.374] |
| Temporal | Epilepsy [F1,76 = 8.365, p = 0.005] Treatment [F1,76 = 18.971, p < 0.001] Epilepsy x Treatment [F1,76 = 0.0003, p = 0.995] |
| CA3c Septal | Epilepsy [F1,90 = 70.424, p < 0.001] Treatment [F1,90 = 2.969, p = 0.0889] Epilepsy x Treatment [F1,90 = 4.358, p = 0.039] |
| Septo-temporal | Epilepsy [F1,92 = 47.971, p < 0.001] Treatment [F1,92 = 1.371, p = 0.245] Epilepsy x Treatment [F1,92 = 0.0000566, p = 0.994] |
| Temporal | Epilepsy [F1,92 = 265,837; p < 0,001] Treatment [F2,92 = 17,913; p < 0,001] Epilepsy x Treatment [F2,92 = 34,335; p < 0,001] |
| Ventral hippocampus CA1 | Epilepsy [F1,74 = 41.293, p < 0.001] Treatment [F1,74 = 14.954, p < 0.001] Epilepsy x Treatment [F1,74 = 15.396, p < 0.001] |
| Ventral hippocampus CA2 | Epilepsy [F1,93 = 81.391, p < 0.001] Treatment [F1,93 = 19.146, p < 0.001] Epilepsy x Treatment [F2,93 = 42.975, p < 0.001] |
| Ventral hippocampus CA3 | Epilepsy [F1,74 = 42.658, p < 0.001] Treatment [F1,74 = 42.658, p < 0.001] Epilepsy x Treatment [F1,74 = 64.381, p < 0,001] |
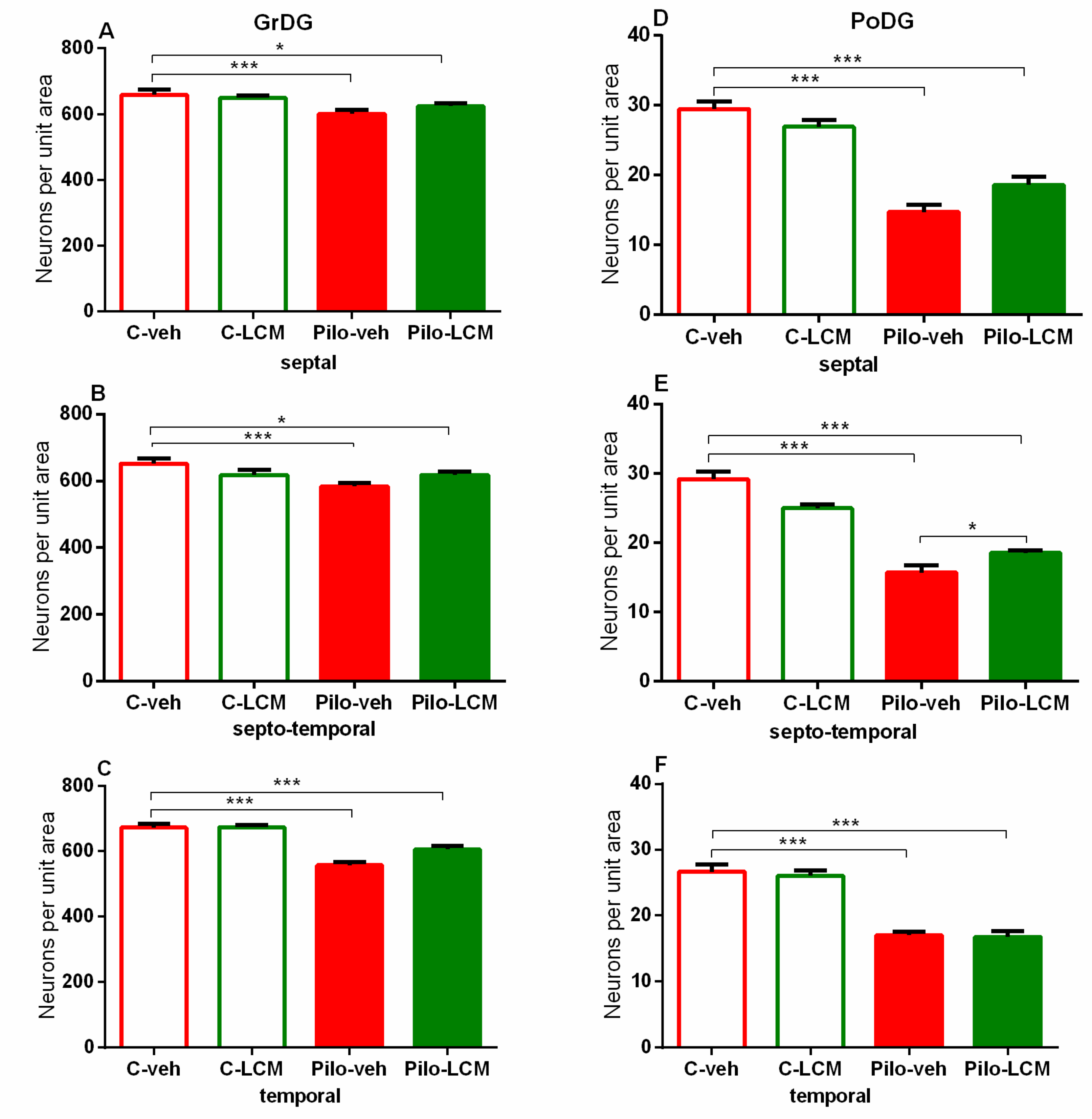
| Hilus of the dentate gyrus Septal | Epilepsy [F1,87 = 237.156, p < 0.001] Treatment [F1,87 = 0.0386, p = 0.845] Epilepsy x Treatment [F1,87 = 0.109, p = 0.742] |
| Septo-temporal | Epilepsy [F1,89 = 199.646, p < 0.001] Treatment [F1,89 = 0.709, p = 0.402] Epilepsy x Treatment [F1,89 = 1.117, p = 0.293] |
| Temporal | Epilepsy [F1,99 = 72.320, p < 0.001] Treatment [F1,99 = 0.0920, p = 0.762] Epilepsy x Treatment [F1,99 = 1.727, p = 0.192] |
| Dentate gyrus Septal | Epilepsy [F1,91 = 14.480, p < 0.001] Treatment [F1,91 = 0.173, p = 0.679] Epilepsy x Treatment [F1,9 1 = 0.173, p = 0.679] |
| Septo-temporal | Epilepsy [F1,91 = 23.872, p < 0.001] Treatment [F1,91= 0.374, p = 0.542] Epilepsy x Treatment [F1,91 = 3.047, p = 0.084] |
| Temporal | Epilepsy [F1,93 = 74,314; p < 0,001] Treatment [F1,93 = 1.502, p = 0.223] Epilepsy x Treatment [F1,93 = 4.562, p = 0.035] |
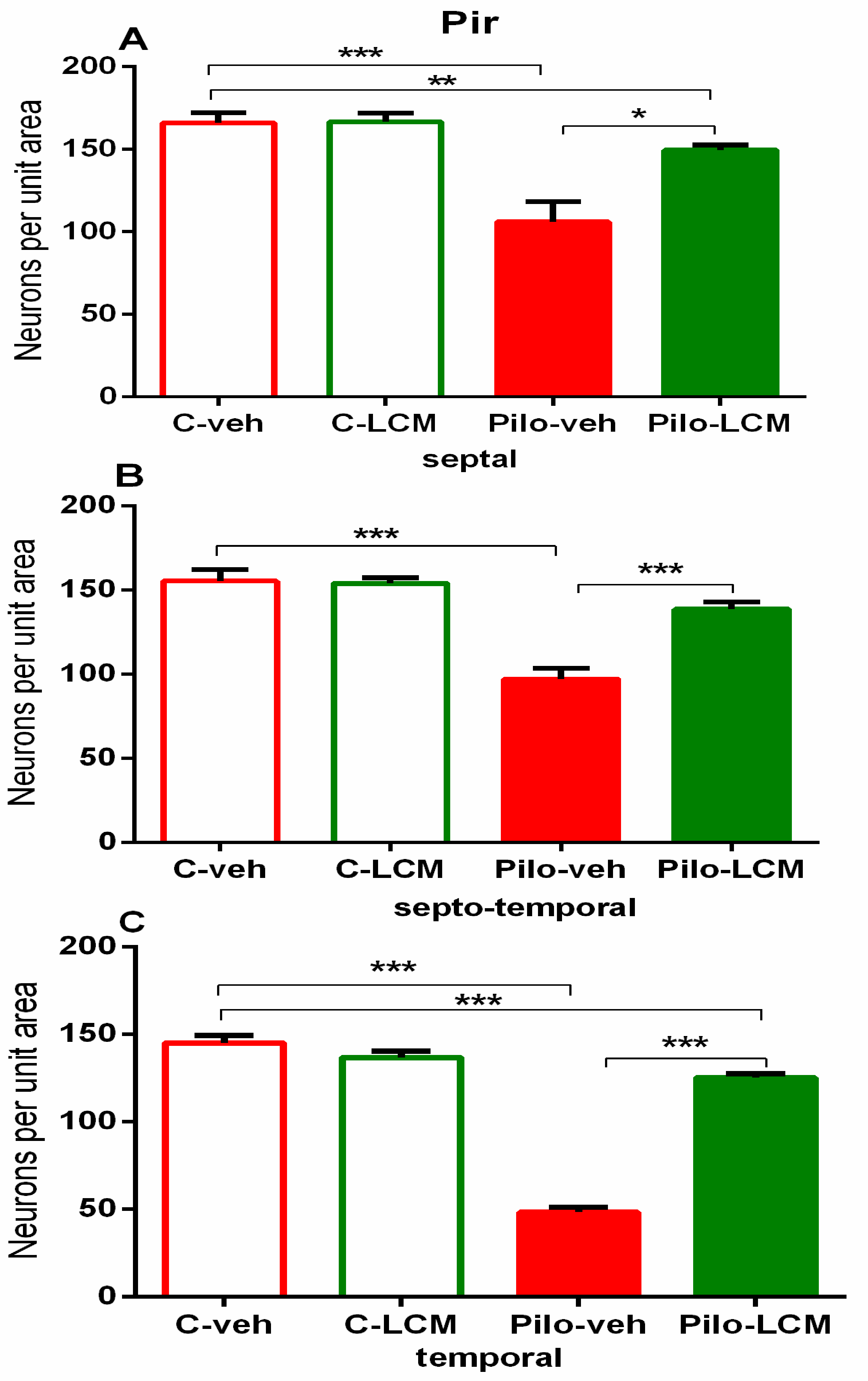
| Piriform cortex Septal | Epilepsy [F1,76 = 43.071, p < 0.001] Treatment [F2,76 = 2.328, p = 0.131] Epilepsy x Treatment [F2, 76 = 8.202, p = 0.005] |
| Septo-temporal | Epilepsy [F1,81 = 44.779, p < 0.001] Treatment [F2,81 = 2.859, p = 0.095] Epilepsy x Treatment [F2, 81 = 2.195, p = 0.143] |
| Temporal | Epilepsy [F1,81 = 178.178, p < 0.001] Treatment [F2,81= 9.193, p = 0.004] Epilepsy x Treatment [F1,81 = 24.629, p < 0.001] |
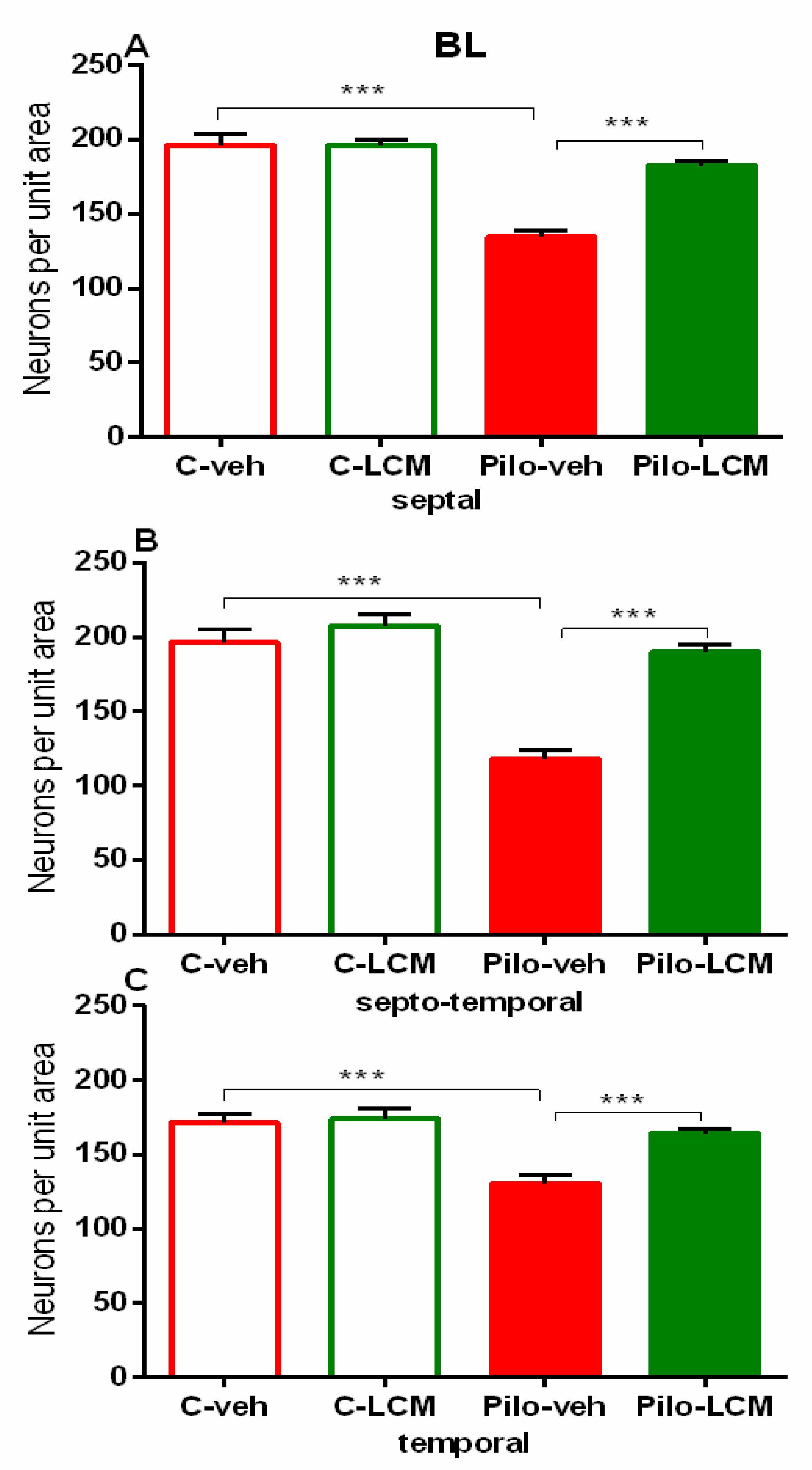
| Amygdala (basolateral) Septal | Epilepsy [F1,97 = 30.348, p < 0,001] Treatment [F1,97 = 9.565, p = 0.003] Epilepsy x Treatment [F2,97 = 17.838, p < 0.001] |
| Septo-temporal | Epilepsy [F1,96 = 43,690; p < 0,001] Treatment [F2,96 = 5.030, p = 0.0271] Epilepsy x Treatment [F2,96 = 28.454, p < 0.001] |
| Temporal | Epilepsy [F1,82 = 14.945, p < 0.001] Treatment [F1,82= 2.994, p = 0.087] Epilepsy x Treatment [F2,82 = 8.023, p = 0.006] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shishmanova-Doseva, M.; Atanasova, D.; Uzunova, Y.; Yoanidu, L.; Peychev, L.; Marinov, P.; Tchekalarova, J. Effects of Lacosamide Treatment on Epileptogenesis, Neuronal Damage and Behavioral Comorbidities in a Rat Model of Temporal Lobe Epilepsy. Int. J. Mol. Sci. 2021, 22, 4667. https://doi.org/10.3390/ijms22094667
Shishmanova-Doseva M, Atanasova D, Uzunova Y, Yoanidu L, Peychev L, Marinov P, Tchekalarova J. Effects of Lacosamide Treatment on Epileptogenesis, Neuronal Damage and Behavioral Comorbidities in a Rat Model of Temporal Lobe Epilepsy. International Journal of Molecular Sciences. 2021; 22(9):4667. https://doi.org/10.3390/ijms22094667
Chicago/Turabian StyleShishmanova-Doseva, Michaela, Dimitrinka Atanasova, Yordanka Uzunova, Lyubka Yoanidu, Lyudmil Peychev, Pencho Marinov, and Jana Tchekalarova. 2021. "Effects of Lacosamide Treatment on Epileptogenesis, Neuronal Damage and Behavioral Comorbidities in a Rat Model of Temporal Lobe Epilepsy" International Journal of Molecular Sciences 22, no. 9: 4667. https://doi.org/10.3390/ijms22094667
APA StyleShishmanova-Doseva, M., Atanasova, D., Uzunova, Y., Yoanidu, L., Peychev, L., Marinov, P., & Tchekalarova, J. (2021). Effects of Lacosamide Treatment on Epileptogenesis, Neuronal Damage and Behavioral Comorbidities in a Rat Model of Temporal Lobe Epilepsy. International Journal of Molecular Sciences, 22(9), 4667. https://doi.org/10.3390/ijms22094667