
Suppression of PGC-1α Drives Metabolic Dysfunction in TGFβ2-Induced EMT of Retinal Pigment Epithelial Cells




Abstract
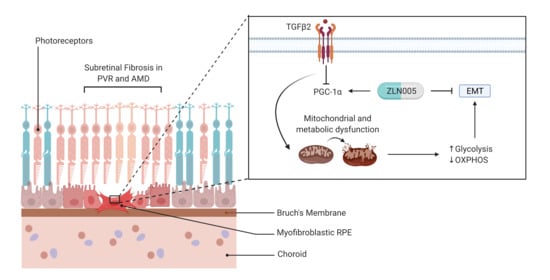
:
1. Introduction
2. Results
2.1. TGFβ2-Induced EMT of ARPE-19 Is Accompanied by Mitochondrial Dysfunction
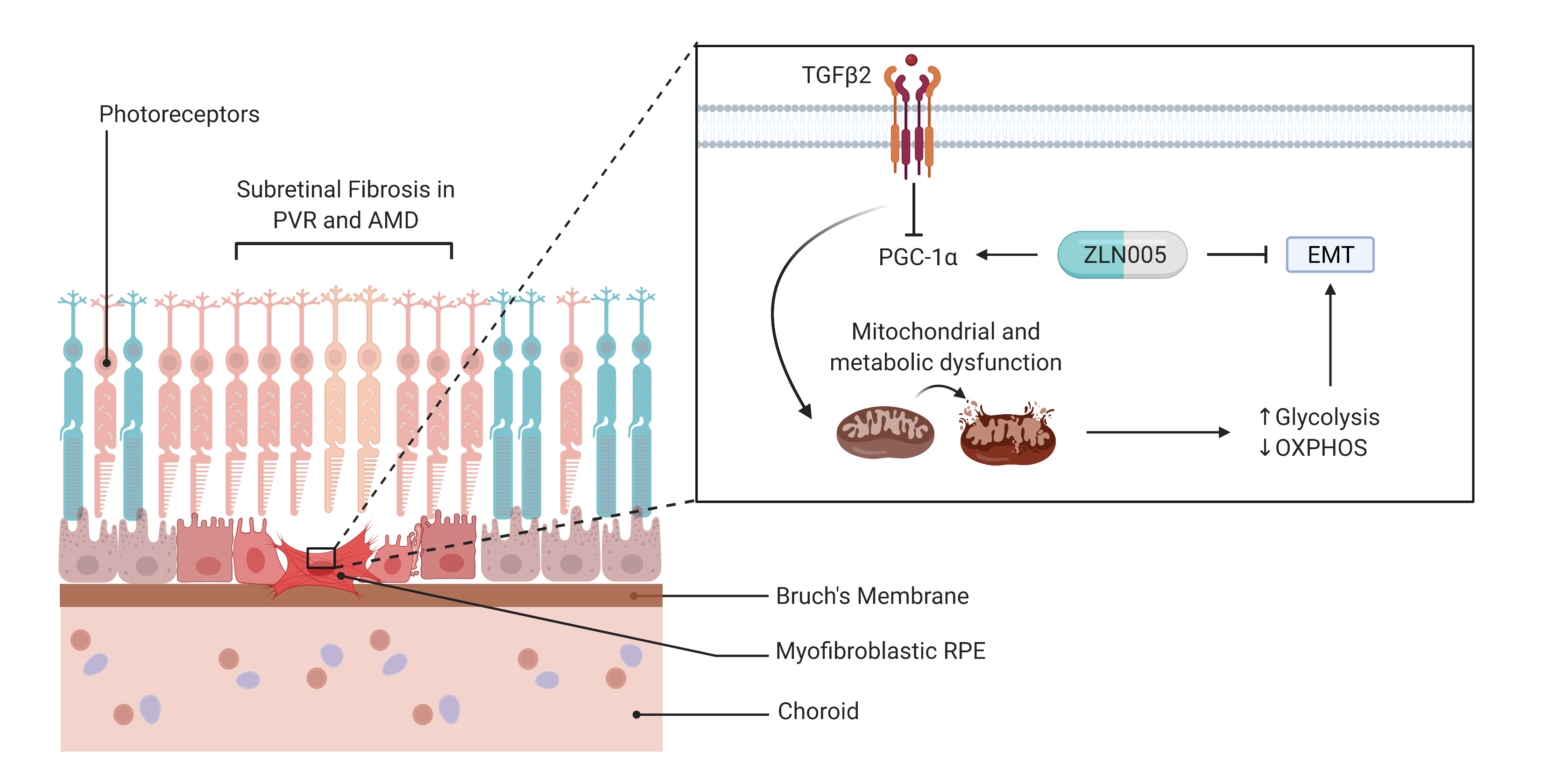
2.2. TGFβ2 Reduces Mitochondrial Respiration in RPE
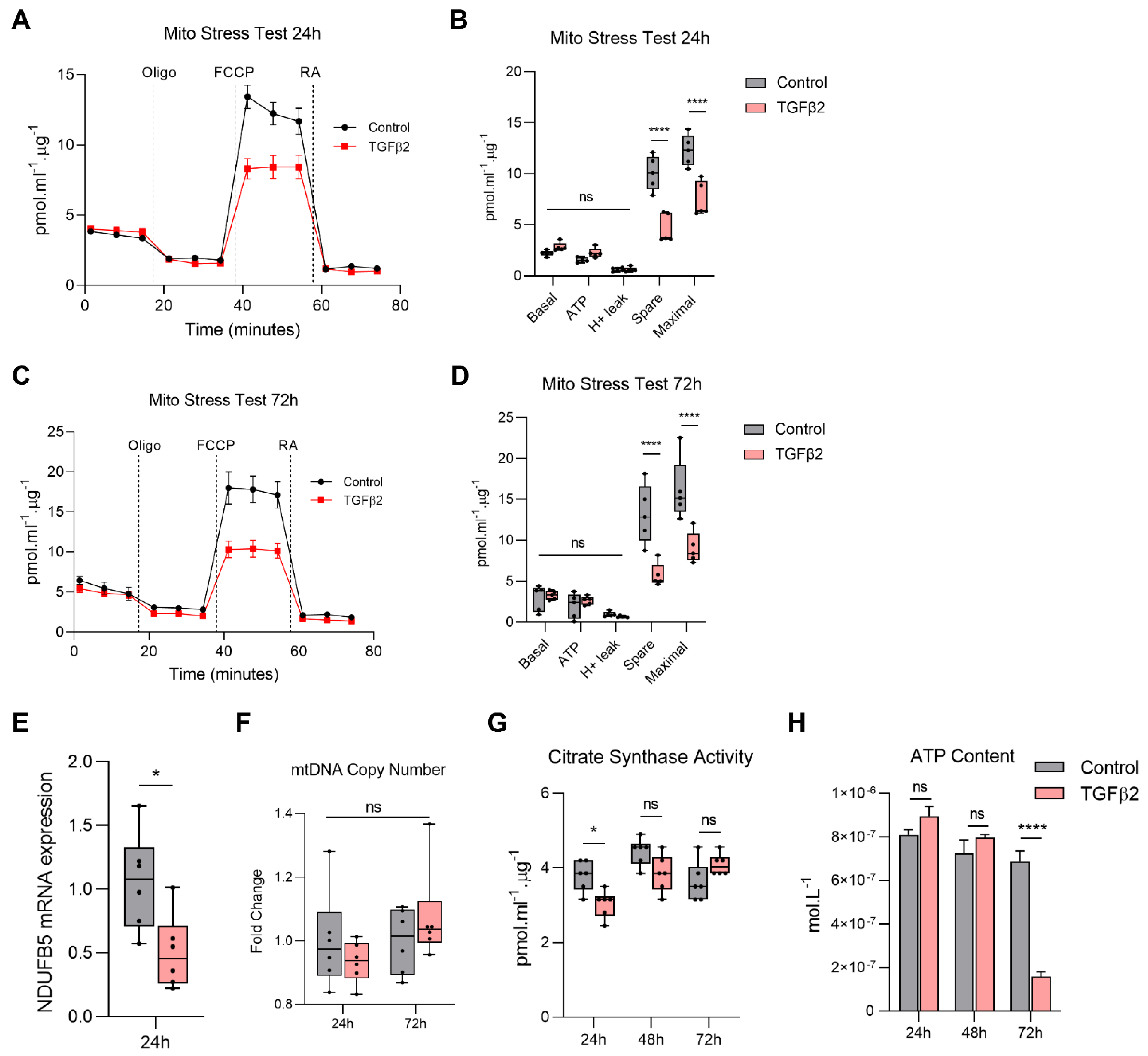
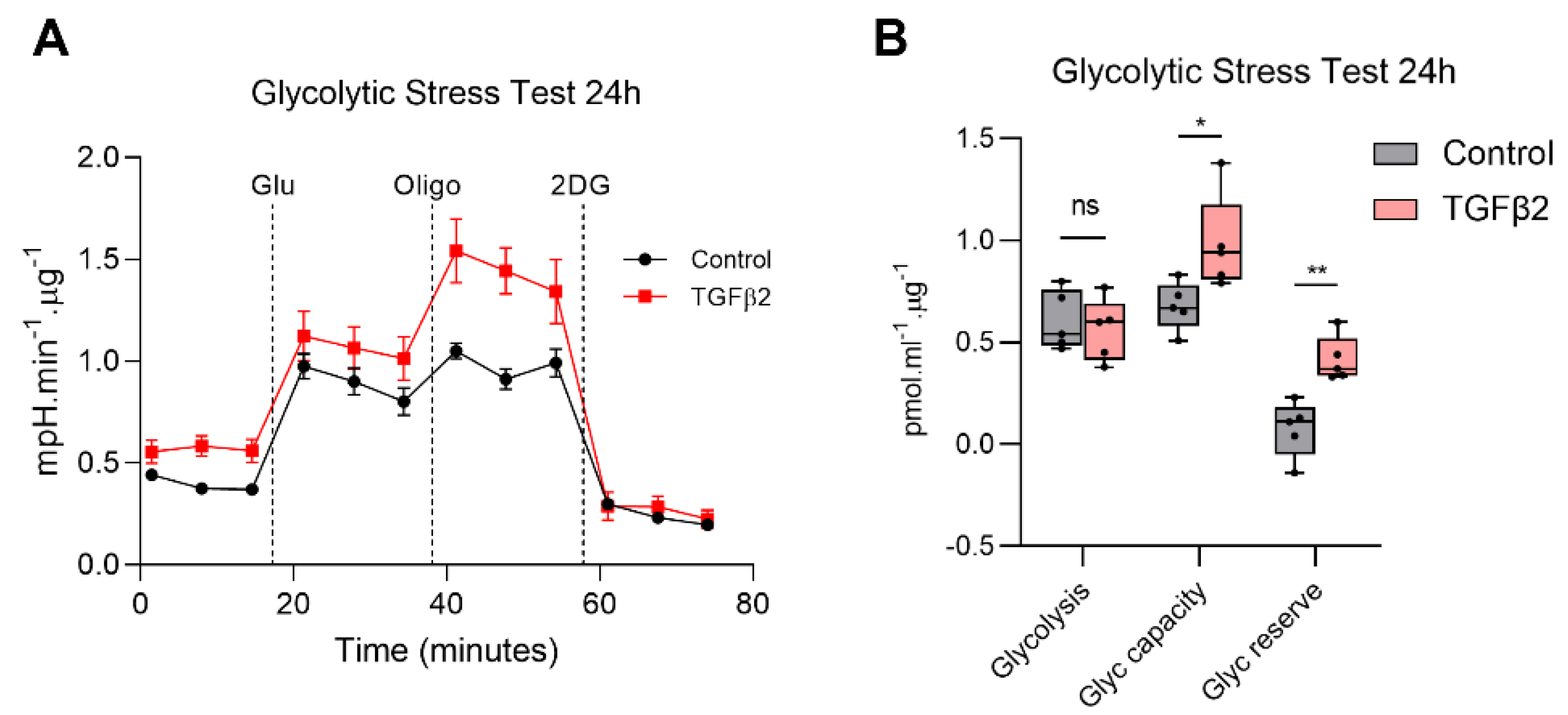
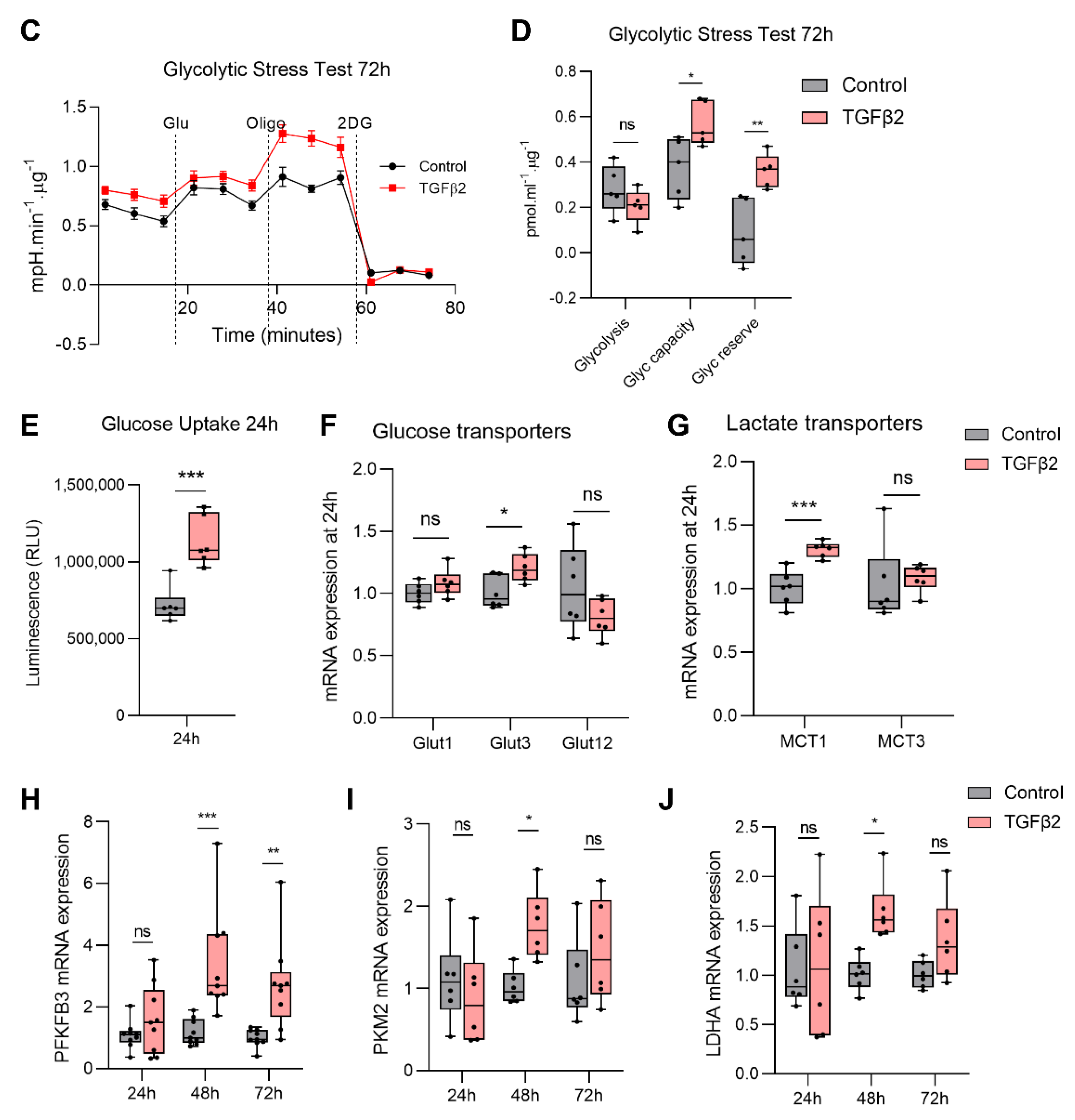
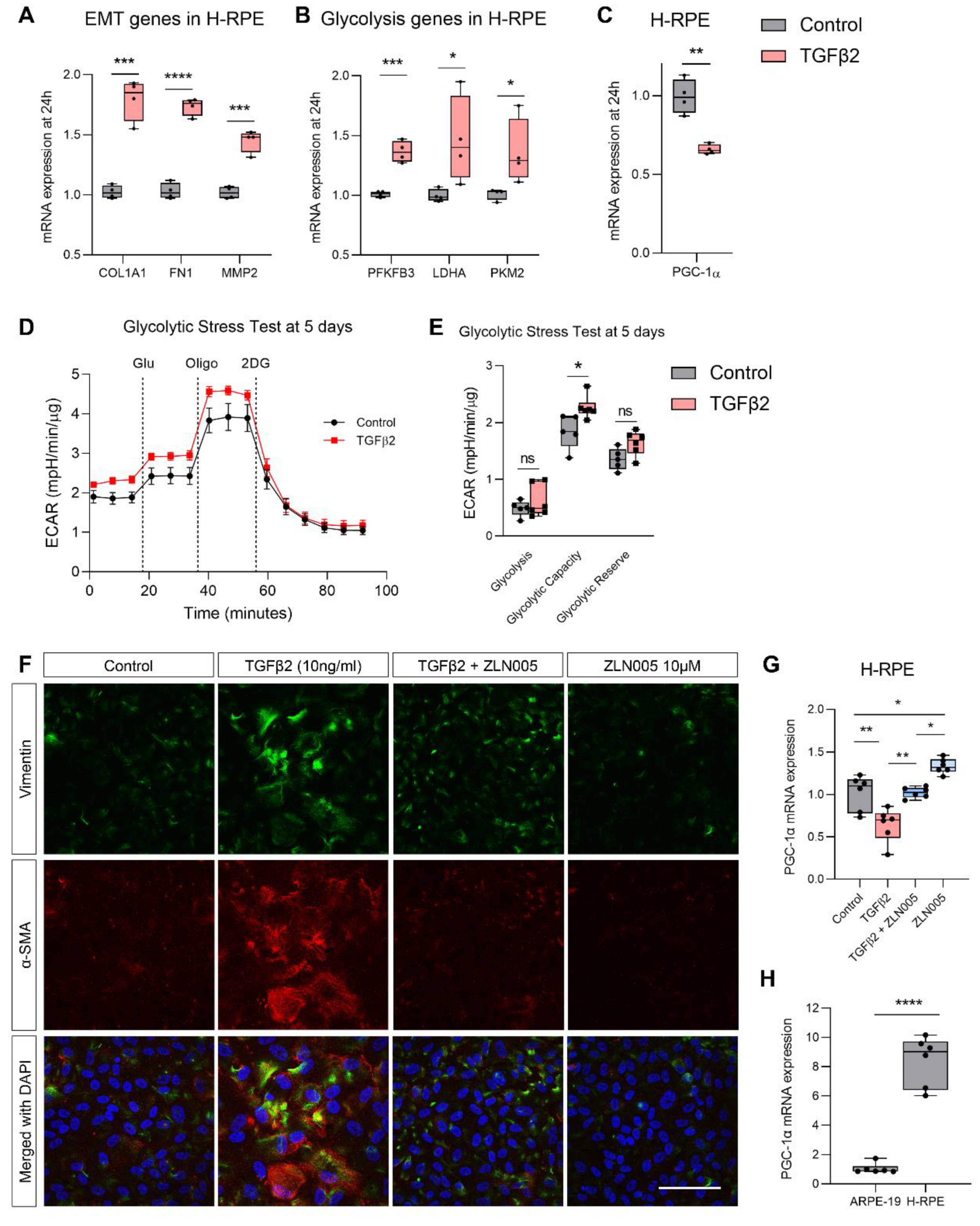
2.3. TGFβ2 Enhances Glycolytic Reserve in RPE and Increases Glycolysis Gene Expression
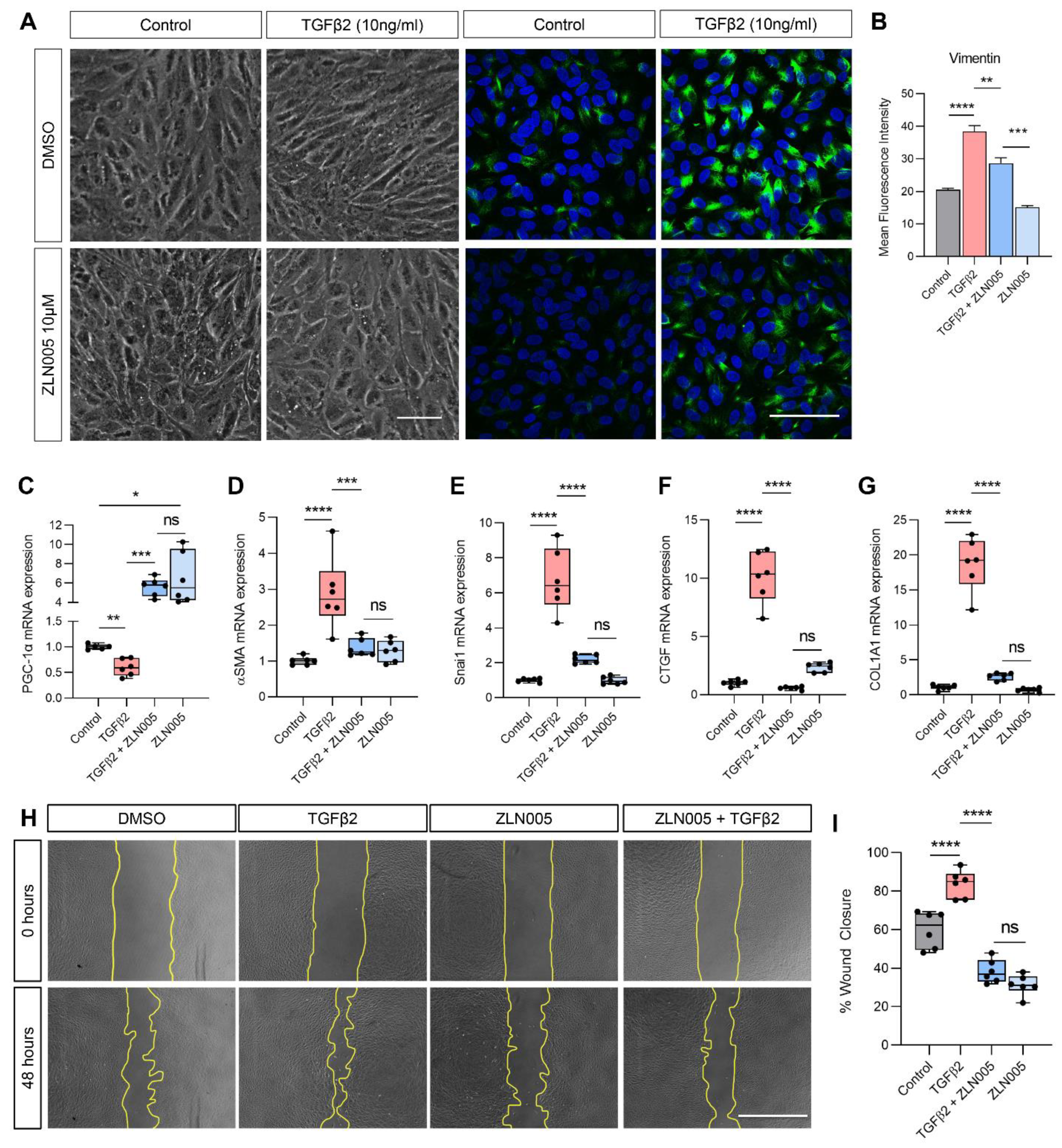
2.4. TGFβ2 Suppresses PGC-1α Gene Expression and ZLN005 Blocks TGFβ2-Induced EMT in ARPE-19 and Primary Human RPE
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Immunofluorescence Confocal Microscopy
4.3. Quantitative PCR (qPCR)
4.4. Mitochondrial Morphology Imaging
4.5. Automated Processing of Mitochondrial Network Morphology
4.6. High-Resolution Respirometry
4.7. Quantification of Intracellular ATP Content
4.8. Citrate Synthase Assay
4.9. Mitochondrial Copy Number
4.10. Glucose Uptake Assay
4.11. Scratch Wound Migration Assay
4.12. Statistical Analysis
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Guerra, F.; Guaragnella, N.; Arbini, A.A.; Bucci, C.; Giannattasio, S.; Moro, L. Mitochondrial Dysfunction: A Novel Potential Driver of Epithelial-to-Mesenchymal Transition in Cancer. Front. Oncol. 2017, 7, 295. [Google Scholar] [CrossRef] [Green Version]
- Georgakopoulos-Soares, I.; Chartoumpekis, D.V.; Kyriazopoulou, V.; Zaravinos, A. EMT Factors and Metabolic Pathways in Cancer. Front. Oncol. 2020, 10, 499. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Chen, Y.; Zhang, P.; Huang, S.; Zhu, C.; Ding, G.; Liu, B.; Yang, T.; Zhang, A. Mitochondrial dysfunction accounts for aldosterone-induced epithelial-to-mesenchymal transition of renal proximal tubular epithelial cells. Free Radic. Biol. Med. 2012, 53, 30–43. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Zhang, Y.; Chang, X.; Zhang, X. Imbalance in mitochondrial dynamics induced by low PGC-1alpha expression contributes to hepatocyte EMT and liver fibrosis. Cell Death Dis. 2020, 11, 226. [Google Scholar] [CrossRef] [Green Version]
- Shu, D.Y.; Lovicu, F.J. Myofibroblast transdifferentiation: The dark force in ocular wound healing and fibrosis. Prog. Retin. Eye Res. 2017, 60, 44–65. [Google Scholar] [CrossRef] [PubMed]
- Shu, D.Y.; Butcher, E.; Saint-Geniez, M. EMT and EndMT: Emerging Roles in Age-Related Macular Degeneration. Int. J. Mol. Sci. 2020, 21, 4271. [Google Scholar] [CrossRef] [PubMed]
- Connor, T.B., Jr.; Roberts, A.B.; Sporn, M.B.; Danielpour, D.; Dart, L.L.; Michels, R.G.; de Bustros, S.; Enger, C.; Kato, H.; Lansing, M.; et al. Correlation of fibrosis and transforming growth factor-beta type 2 levels in the eye. J. Clin. Investig. 1989, 83, 1661–1666. [Google Scholar] [CrossRef]
- Esser, P.; Heimann, K.; Bartz-schmidt, K.U.; Fontana, A.; Schraermeyer, U.; Thumann, G.; Weller, M. Apoptosis in proliferative vitreoretinal disorders: Possible involvement of TGF-beta-induced RPE cell apoptosis. Exp. Eye Res. 1997, 65, 365–378. [Google Scholar] [CrossRef] [PubMed]
- Kon, C.H.; Occleston, N.L.; Aylward, G.W.; Khaw, P.T. Expression of vitreous cytokines in proliferative vitreoretinopathy: A prospective study. Investig. Ophthalmol. Vis. Sci. 1999, 40, 705–712. [Google Scholar]
- Gonzalez-Avila, G.; Lozano, D.; Manjarrez, M.E.; Ruiz, V.M.; Teran, L.; Vadillo-Ortega, F.; Selman, M. Influence on collagen metabolism of vitreous from eyes with proliferative vitreoretinopathy. Ophthalmology 1995, 102, 1400–1405. [Google Scholar] [CrossRef]
- Bochaton-Piallat, M.L.; Kapetanios, A.D.; Donati, G.; Redard, M.; Gabbiani, G.; Pournaras, C.J. TGF-beta1, TGF-beta receptor II and ED-A fibronectin expression in myofibroblast of vitreoretinopathy. Investig. Ophthalmol. Vis. Sci. 2000, 41, 2336–2342. [Google Scholar]
- Rosales, M.A.B.; Shu, D.Y.; Iacovelli, J.; Saint-Geniez, M. Loss of PGC-1alpha in RPE induces mesenchymal transition and promotes retinal degeneration. Life Sci. Alliance 2019, 2. [Google Scholar] [CrossRef]
- Jiang, L.; Xiao, L.; Sugiura, H.; Huang, X.; Ali, A.; Kuro-o, M.; Deberardinis, R.J.; Boothman, D.A. Metabolic reprogramming during TGFbeta1-induced epithelial-to-mesenchymal transition. Oncogene 2015, 34, 3908–3916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liberti, M.V.; Locasale, J.W. The Warburg Effect: How Does it Benefit Cancer Cells? Trends Biochem. Sci. 2016, 41, 211–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Z.; Lee, Y.F.; Yeo, X.H.; Loo, S.Y.; Tam, W.L. Shifting the Gears of Metabolic Plasticity to Drive Cell State Transitions in Cancer. Cancers 2021, 13, 1316. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, T.; Aggarwal, K.; Pattnaik, B.; Mukherjee, S.; Sethi, T.; Tiwari, B.K.; Kumar, M.; Micheal, A.; Mabalirajan, U.; Ghosh, B.; et al. Computational classification of mitochondrial shapes reflects stress and redox state. Cell Death Dis. 2013, 4, e461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nikolaisen, J.; Nilsson, L.I.; Pettersen, I.K.; Willems, P.H.; Lorens, J.B.; Koopman, W.J.; Tronstad, K.J. Automated quantification and integrative analysis of 2D and 3D mitochondrial shape and network properties. PLoS ONE 2014, 9, e101365. [Google Scholar] [CrossRef]
- Berman, S.B.; Pineda, F.J.; Hardwick, J.M. Mitochondrial fission and fusion dynamics: The long and short of it. Cell Death Differ. 2008, 15, 1147–1152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larsen, S.; Nielsen, J.; Hansen, C.N.; Nielsen, L.B.; Wibrand, F.; Stride, N.; Schroder, H.D.; Boushel, R.; Helge, J.W.; Dela, F.; et al. Biomarkers of mitochondrial content in skeletal muscle of healthy young human subjects. J. Physiol. 2012, 590, 3349–3360. [Google Scholar] [CrossRef]
- Sprowl-Tanio, S.; Habowski, A.N.; Pate, K.T.; McQuade, M.M.; Wang, K.; Edwards, R.A.; Grun, F.; Lyou, Y.; Waterman, M.L. Lactate/pyruvate transporter MCT-1 is a direct Wnt target that confers sensitivity to 3-bromopyruvate in colon cancer. Cancer Metab. 2016, 4, 20. [Google Scholar] [CrossRef] [Green Version]
- Philp, N.J.; Yoon, H.; Lombardi, L. Mouse MCT3 gene is expressed preferentially in retinal pigment and choroid plexus epithelia. Am J. Physiol. Cell Physiol. 2001, 280, C1319–C1326. [Google Scholar] [CrossRef] [PubMed]
- Satish, S.; Philipose, H.; Rosales, M.A.B.; Saint-Geniez, M. Pharmaceutical Induction of PGC-1alpha Promotes Retinal Pigment Epithelial Cell Metabolism and Protects against Oxidative Damage. Oxid. Med. Cell. Longev. 2018, 2018, 9248640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iacovelli, J.; Rowe, G.C.; Khadka, A.; Diaz-Aguilar, D.; Spencer, C.; Arany, Z.; Saint-Geniez, M. PGC-1alpha Induces Human RPE Oxidative Metabolism and Antioxidant Capacity. Investig. Ophthalmol. Vis. Sci. 2016, 57, 1038–1051. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Youle, R.J.; van der Bliek, A.M. Mitochondrial fission, fusion, and stress. Science 2012, 337, 1062–1065. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Das, R.; Chakrabarti, O. Mitochondrial hyperfusion: A friend or a foe. Biochem. Soc. Trans. 2020, 48, 631–644. [Google Scholar] [CrossRef]
- Bach, D.; Pich, S.; Soriano, F.X.; Vega, N.; Baumgartner, B.; Oriola, J.; Daugaard, J.R.; Lloberas, J.; Camps, M.; Zierath, J.R.; et al. Mitofusin-2 determines mitochondrial network architecture and mitochondrial metabolism. A novel regulatory mechanism altered in obesity. J. Biol. Chem. 2003, 278, 17190–17197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shah, S.I.; Paine, J.G.; Perez, C.; Ullah, G. Mitochondrial fragmentation and network architecture in degenerative diseases. PLoS ONE 2019, 14, e0223014. [Google Scholar] [CrossRef]
- Benard, G.; Bellance, N.; James, D.; Parrone, P.; Fernandez, H.; Letellier, T.; Rossignol, R. Mitochondrial bioenergetics and structural network organization. J. Cell Sci. 2007, 120, 838–848. [Google Scholar] [CrossRef] [Green Version]
- Westermann, B. Bioenergetic role of mitochondrial fusion and fission. Biochim. Biophys. Acta 2012, 1817, 1833–1838. [Google Scholar] [CrossRef] [Green Version]
- Detmer, S.A.; Chan, D.C. Functions and dysfunctions of mitochondrial dynamics. Nat. Rev. Mol. Cell Biol. 2007, 8, 870–879. [Google Scholar] [CrossRef]
- Cheung, E.C.; McBride, H.M.; Slack, R.S. Mitochondrial dynamics in the regulation of neuronal cell death. Apoptosis 2007, 12, 979–992. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vesce, S.; Jekabsons, M.B.; Johnson-Cadwell, L.I.; Nicholls, D.G. Acute glutathione depletion restricts mitochondrial ATP export in cerebellar granule neurons. J. Biol. Chem. 2005, 280, 38720–38728. [Google Scholar] [CrossRef] [Green Version]
- Yadava, N.; Nicholls, D.G. Spare respiratory capacity rather than oxidative stress regulates glutamate excitotoxicity after partial respiratory inhibition of mitochondrial complex I with rotenone. J. Neurosci. 2007, 27, 7310–7317. [Google Scholar] [CrossRef] [Green Version]
- Feng, J.; Li, J.; Wu, L.; Yu, Q.; Ji, J.; Wu, J.; Dai, W.; Guo, C. Emerging roles and the regulation of aerobic glycolysis in hepatocellular carcinoma. J. Exp. Clin. Cancer Res. 2020, 39, 126. [Google Scholar] [CrossRef] [PubMed]
- Hua, W.; Ten Dijke, P.; Kostidis, S.; Giera, M.; Hornsveld, M. TGFbeta-induced metabolic reprogramming during epithelial-to-mesenchymal transition in cancer. Cell. Mol. Life Sci. 2020, 77, 2103–2123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yalcin, A.; Solakoglu, T.H.; Ozcan, S.C.; Guzel, S.; Peker, S.; Celikler, S.; Balaban, B.D.; Sevinc, E.; Gurpinar, Y.; Chesney, J.A. 6-phosphofructo-2-kinase/fructose 2,6-bisphosphatase-3 is required for transforming growth factor β1-enhanced invasion of Panc1 cells in vitro. Biochem. Biophys. Res. Commun. 2017, 484, 687–693. [Google Scholar] [CrossRef]
- Shi, L.; Pan, H.; Liu, Z.; Xie, J.; Han, W. Roles of PFKFB3 in cancer. Signal Transduct. Target. Ther. 2017, 2, 17044. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, F.; Yoshimoto, S.; Okamura, K.; Ikebe, T.; Hashimoto, S. Nuclear PKM2 promotes the progression of oral squamous cell carcinoma by inducing EMT and post-translationally repressing TGIF2. Oncotarget 2018, 9, 33745–33761. [Google Scholar] [CrossRef] [Green Version]
- Spencer, N.Y.; Stanton, R.C. The Warburg Effect, Lactate, and Nearly a Century of Trying to Cure Cancer. Semin. Nephrol. 2019, 39, 380–393. [Google Scholar] [CrossRef]
- Yalamanchi, N.; Klein, M.B.; Pham, H.M.; Longaker, M.T.; Chang, J. Flexor tendon wound healing in vitro: Lactate up-regulation of TGF-beta expression and functional activity. Plast. Reconstr. Surg. 2004, 113, 625–632. [Google Scholar] [CrossRef]
- Li, X.; Zhang, Z.; Zhang, Y.; Cao, Y.; Wei, H.; Wu, Z. Upregulation of lactate-inducible snail protein suppresses oncogene-mediated senescence through p16(INK4a) inactivation. J. Exp. Clin. Cancer Res. CR 2018, 37, 39. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.B.; Viji, R.I.; Kiran, M.S.; Sudhakaran, P.R. Endothelial cell response to lactate: Implication of PAR modification of VEGF. J. Cell. Physiol. 2007, 211, 477–485. [Google Scholar] [CrossRef]
- Seliger, C.; Leukel, P.; Moeckel, S.; Jachnik, B.; Lottaz, C.; Kreutz, M.; Brawanski, A.; Proescholdt, M.; Bogdahn, U.; Bosserhoff, A.K.; et al. Lactate-modulated induction of THBS-1 activates transforming growth factor (TGF)-beta2 and migration of glioma cells in vitro. PLoS ONE 2013, 8, e78935. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Du, J.; Justus, S.; Hsu, C.W.; Bonet-Ponce, L.; Wu, W.H.; Tsai, Y.T.; Wu, W.P.; Jia, Y.; Duong, J.K.; et al. Reprogramming metabolism by targeting sirtuin 6 attenuates retinal degeneration. J. Clin. Investig. 2016, 126, 4659–4673. [Google Scholar] [CrossRef] [Green Version]
- Venkatesh, A.; Ma, S.; Le, Y.Z.; Hall, M.N.; Ruegg, M.A.; Punzo, C. Activated mTORC1 promotes long-term cone survival in retinitis pigmentosa mice. J. Clin. Investig. 2015, 125, 1446–1458. [Google Scholar] [CrossRef] [Green Version]
- Kurihara, T.; Westenskow, P.D.; Gantner, M.L.; Usui, Y.; Schultz, A.; Bravo, S.; Aguilar, E.; Wittgrove, C.; Friedlander, M.; Paris, L.P.; et al. Hypoxia-induced metabolic stress in retinal pigment epithelial cells is sufficient to induce photoreceptor degeneration. Elife 2016, 5, e14319. [Google Scholar] [CrossRef]
- Zhao, C.; Yasumura, D.; Li, X.; Matthes, M.; Lloyd, M.; Nielsen, G.; Ahern, K.; Snyder, M.; Bok, D.; Dunaief, J.L.; et al. mTOR-mediated dedifferentiation of the retinal pigment epithelium initiates photoreceptor degeneration in mice. J. Clin. Investig. 2011, 121, 369–383. [Google Scholar] [CrossRef] [Green Version]
- Brown, E.E.; DeWeerd, A.J.; Ildefonso, C.J.; Lewin, A.S.; Ash, J.D. Mitochondrial oxidative stress in the retinal pigment epithelium (RPE) led to metabolic dysfunction in both the RPE and retinal photoreceptors. Redox Biol. 2019, 24, 101201. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Yanagida, A.; Knight, K.; Engel, A.L.; Vo, A.H.; Jankowski, C.; Sadilek, M.; Tran, V.T.; Manson, M.A.; Ramakrishnan, A.; et al. Reductive carboxylation is a major metabolic pathway in the retinal pigment epithelium. Proc. Natl. Acad. Sci. USA 2016, 113, 14710–14715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, H.; Kim, H.; Lee, S.; Youn, H.; Youn, B. Role of Metabolic Reprogramming in Epithelial⁻Mesenchymal Transition (EMT). Int. J. Mol. Sci. 2019, 20, 2042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masin, M.; Vazquez, J.; Rossi, S.; Groeneveld, S.; Samson, N.; Schwalie, P.C.; Deplancke, B.; Frawley, L.E.; Gouttenoire, J.; Moradpour, D.; et al. GLUT3 is induced during epithelial-mesenchymal transition and promotes tumor cell proliferation in non-small cell lung cancer. Cancer Metab. 2014, 2, 11. [Google Scholar] [CrossRef] [Green Version]
- Dhup, S.; Dadhich, R.K.; Porporato, P.E.; Sonveaux, P. Multiple biological activities of lactic acid in cancer: Influences on tumor growth, angiogenesis and metastasis. Curr. Pharm. Des. 2012, 18, 1319–1330. [Google Scholar] [CrossRef] [Green Version]
- Zeuthen, T.; Hamann, S.; La Cour, M. Cotransport of H+, lactate and H2O by membrane proteins in retinal pigment epithelium of bullfrog. J. Physiol. 1996, 497 Pt 1, 3–17. [Google Scholar] [CrossRef] [Green Version]
- Adijanto, J.; Philp, N.J. The SLC16A family of monocarboxylate transporters (MCTs)--physiology and function in cellular metabolism, pH homeostasis, and fluid transport. Curr. Top. Membr. 2012, 70, 275–311. [Google Scholar] [CrossRef]
- Daniele, L.L.; Sauer, B.; Gallagher, S.M.; Pugh, E.N., Jr.; Philp, N.J. Altered visual function in monocarboxylate transporter 3 (Slc16a8) knockout mice. Am J. Physiol. Cell Physiol. 2008, 295, C451–C457. [Google Scholar] [CrossRef] [Green Version]
- Yu, J.; Auwerx, J. The role of sirtuins in the control of metabolic homeostasis. Ann. N. Y. Acad. Sci. 2009, 1173 (Suppl. S1), E10–E19. [Google Scholar] [CrossRef] [PubMed]
- Lagouge, M.; Argmann, C.; Gerhart-Hines, Z.; Meziane, H.; Lerin, C.; Daussin, F.; Messadeq, N.; Milne, J.; Lambert, P.; Elliott, P.; et al. Resveratrol improves mitochondrial function and protects against metabolic disease by activating SIRT1 and PGC-1alpha. Cell 2006, 127, 1109–1122. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, K.; He, S.; Terasaki, H.; Nazari, H.; Zhang, H.; Spee, C.; Kannan, R.; Hinton, D.R. Resveratrol inhibits epithelial-mesenchymal transition of retinal pigment epithelium and development of proliferative vitreoretinopathy. Sci. Rep. 2015, 5, 16386. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Li, X.; Wang, B.; Chen, Y.; Xiao, A.; Zeng, D.; Ou, D.; Yan, S.; Li, W.; Zheng, Q. ZLN005 protects cardiomyocytes against high glucose-induced cytotoxicity by promoting SIRT1 expression and autophagy. Exp. Cell Res. 2016, 345, 25–36. [Google Scholar] [CrossRef] [PubMed]
- Simic, P.; Williams, E.O.; Bell, E.L.; Gong, J.J.; Bonkowski, M.; Guarente, L. SIRT1 suppresses the epithelial-to-mesenchymal transition in cancer metastasis and organ fibrosis. Cell Rep. 2013, 3, 1175–1186. [Google Scholar] [CrossRef] [Green Version]
- Bugyei-Twum, A.; Ford, C.; Civitarese, R.; Seegobin, J.; Advani, S.L.; Desjardins, J.F.; Kabir, G.; Zhang, Y.; Mitchell, M.; Switzer, J.; et al. Sirtuin 1 activation attenuates cardiac fibrosis in a rodent pressure overload model by modifying Smad2/3 transactivation. Cardiovasc. Res. 2018, 114, 1629–1641. [Google Scholar] [CrossRef] [PubMed]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [PubMed] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Symbol | Gene Name | Forward Sequence (5′-3′) | Reverse Sequence (5′-3′) |
|---|---|---|---|
| A-SMA | Alpha-smooth muscle actin | AAAAGACAGCTACGTGGGTGA | GCCATGTTCTATCGGGTACTTC |
| B2M | Beta-2 microglobulin | TTCTGGTGCTTGTCTCACTGA | CAGTATGTTCGGCTTCCCATTC |
| COL1A1 | Collagen, type I, alpha 1 | GTGCGATGACGTGATCTGTGA | CGGTGGTTTCTTGGTCGGT |
| CTGF | Connective tissue growth factor | CAGCATGGACGTTCGTCTG | AACCACGGTTTGGTCCTTGG |
| FIS1 | Fission 1 | TGACATCCGTAAAGGCATCG | CTTCTCGTATTCCTTGAGCCG |
| FN1 | Fibronectin 1 | CGGTGGCTGTCAGTCAAAG | AAACCTCGGCTTCCTCCATAA |
| GLUT1 | Glucose Transporter 1 (SLC2A1) | GGCCAAGAGTGTGCTAAAGAA | ACAGCGTTGATGCCAGACAG |
| GLUT12 | Glucose Transporter 12 (SLC2A12) | GAGGCTGCGGCATGTTTAC | CCAAGTTCATAACCCACCAGG |
| GLUT3 | Glucose Transporter 3 (SLC2A3) | GCTGGGCATCGTTGTTGGA | GCACTTTGTAGGATAGCAGGAAG |
| LDHA | Lactate dehydrogenase A | GGCCTGTGCCATCAGTATCT | GGAGATCCATCATCTCTCCC |
| MCT1 | Monocarboxylic acid transporter 1 (SLC16A1) | AGGTCCAGTTGGATACACCCC | GCATAAGAGAAGCCGATGGAAAT |
| MCT3 | Monocarboxylate transporter, member 3 (SLC16A3) | CCATGCTCTACGGGACAGG | GCTTGCTGAAGTAGCGGTT |
| MFN1 | Mitofusin 1 | TGCCCTTCACATGGACAAAG | CTCTGTAGTGACATCTGTGCC |
| MFN2 | Mitofusin 2 | ATGTGGCCCAACTCTAAGTG | CACAAACACATCAGCATCCAG |
| MMP2 | Matrix metalloproteinase-2 | CTTCCAAGTCTGGAGCGATGT | TACCGTCAAAGGGGTATCCAT |
| NDUFB5 | NADH dehydrogenase [ubiquinone] 1 beta subcomplex, 5, 16 kDa | CACTCGCCTCGGATTTGG | CGCCTGTCATAGAATCTAGAAGG |
| PFKFB3 | 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 3 | CAGTTGTGCCTCCAATATC | GGCTTCATAGCAACTGATCC |
| PGC-1A | Peroxisome proliferator-activated receptor gamma, coactivator 1 alpha (PPARGC1A) | GTCACCACCCAAATCCTTAT | ATCTACTGCCTGGAGACCTT |
| PKM2 | Pyruvate kinase muscle isozyme M2 | CAAAGGACCTCAGCAGCCATGTC | GGGAAGCTGGGCCAATGGTACAGA |
| PPIH | Peptidylprolyl isomerase H (cyclophilin H) | CCCCAACAATAAGCCCAAG | CACCACCAAGAAGAAGGGAA |
| SNAI1 | Snail 1 | TCGGAAGCCTAACTACAGCGA | AGATGAGCATTGGCAGCGAG |
| TBP | TATA-binding protein | TGCACAGGAGCCAAGAGTGAA | CACATCACAGCTCCCCACCA |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shu, D.Y.; Butcher, E.R.; Saint-Geniez, M. Suppression of PGC-1α Drives Metabolic Dysfunction in TGFβ2-Induced EMT of Retinal Pigment Epithelial Cells. Int. J. Mol. Sci. 2021, 22, 4701. https://doi.org/10.3390/ijms22094701
Shu DY, Butcher ER, Saint-Geniez M. Suppression of PGC-1α Drives Metabolic Dysfunction in TGFβ2-Induced EMT of Retinal Pigment Epithelial Cells. International Journal of Molecular Sciences. 2021; 22(9):4701. https://doi.org/10.3390/ijms22094701
Chicago/Turabian StyleShu, Daisy Y., Erik R. Butcher, and Magali Saint-Geniez. 2021. "Suppression of PGC-1α Drives Metabolic Dysfunction in TGFβ2-Induced EMT of Retinal Pigment Epithelial Cells" International Journal of Molecular Sciences 22, no. 9: 4701. https://doi.org/10.3390/ijms22094701
APA StyleShu, D. Y., Butcher, E. R., & Saint-Geniez, M. (2021). Suppression of PGC-1α Drives Metabolic Dysfunction in TGFβ2-Induced EMT of Retinal Pigment Epithelial Cells. International Journal of Molecular Sciences, 22(9), 4701. https://doi.org/10.3390/ijms22094701