
Genomic Tackling of Human Satellite DNA: Breaking Barriers through Time


Abstract
:1. Introduction
2. Human Satellite DNA Families
2.1. α Satellite DNA
2.2. Satellite DNA I
2.3. Satellite DNA II/III
2.4. β Satellite DNA
2.5. γ Satellite DNA
3. Satellite DNA: Repetitively Challenging
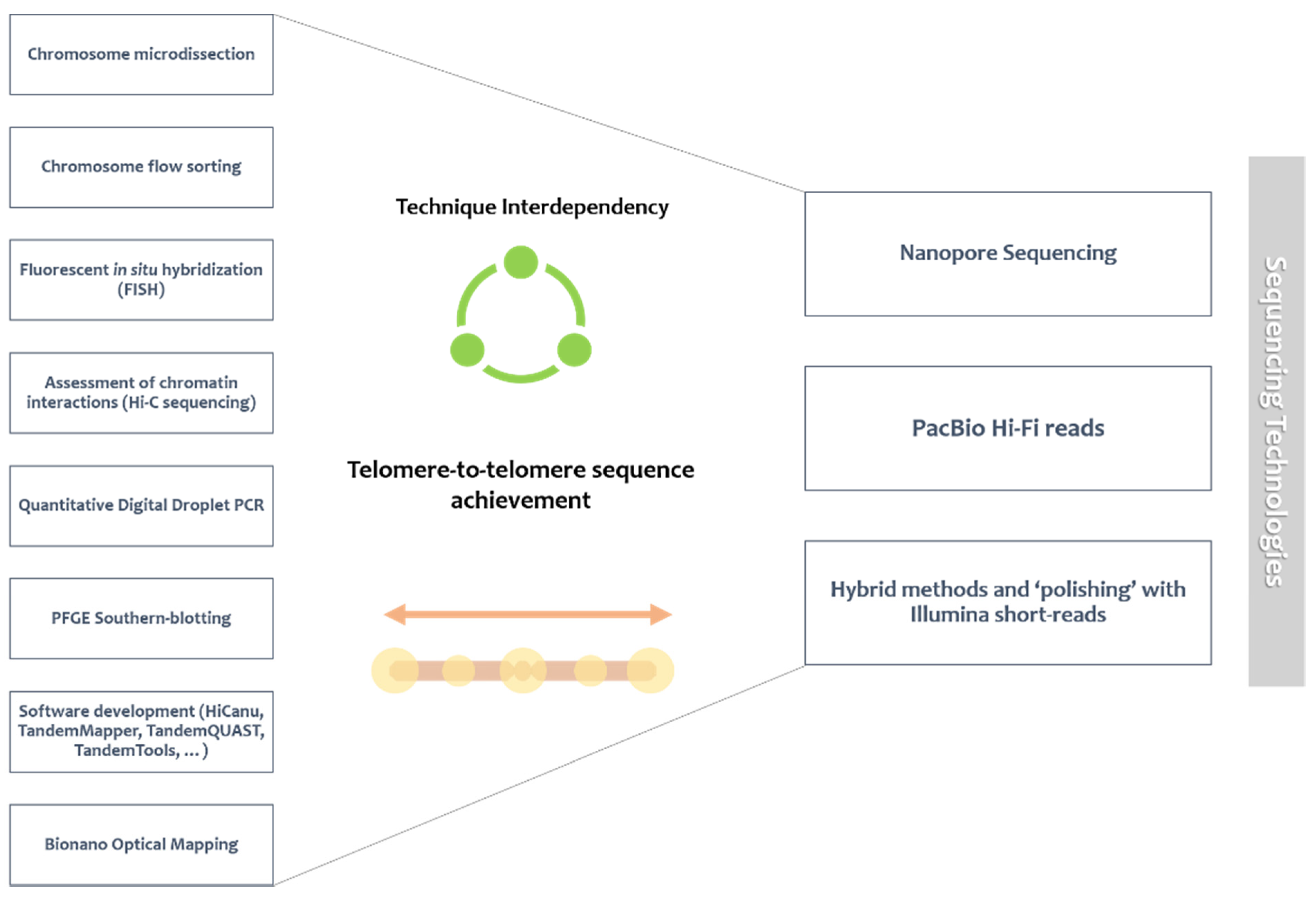
4. Satellite DNA Analysis: The Promise behind Long Reads and Technique Interdependency
4.1. Sequencing Methodologies over Time
4.2. Technique Interdependency
4.3. The Achieved Vs. the Achievable

5. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Britten, R.J.; Kohne, D.E. Repeated sequences in DNA. Science 1968, 161, 529–540. [Google Scholar] [CrossRef] [PubMed]
- Ohno, S. So much’junk’DNA in our genome. In Proceedings of Evolution of Genetic Systems. Brookhaven Symp. Biol. 1972, 23, 366–370. [Google Scholar] [PubMed]
- Palazzo, A.F.; Gregory, T.R. The case for junk DNA. PLoS Genet. 2014, 10, e1004351. [Google Scholar] [CrossRef] [Green Version]
- López-Flores, I.; Garrido-Ramos, M. The repetitive DNA content of eukaryotic genomes. In Repetitive DNA; Karger Publishers: Basel, Switzerland, 2012; Volume 7, pp. 1–28. [Google Scholar]
- McNulty, S.M.; Sullivan, B.A. Alpha satellite DNA biology: Finding function in the recesses of the genome. Chromosome Res. Int. J. Mol. Supramol. Evol. Asp. Chromosome Biol. 2018, 26, 115–138. [Google Scholar] [CrossRef]
- Tørresen, O.K.; Star, B.; Mier, P.; Andrade-Navarro, M.A.; Bateman, A.; Jarnot, P.; Gruca, A.; Grynberg, M.; Kajava, A.V.; Promponas, V.J. Tandem repeats lead to sequence assembly errors and impose multi-level challenges for genome and protein databases. Nucleic Acids Res. 2019, 47, 10994–11006. [Google Scholar] [CrossRef]
- Heitz, E. Das Heterochromatin der Moose; Bornträger: Berlin, Germany, 1928. [Google Scholar]
- Podgornaya, O.I.; Ostromyshenskii, D.I.; Enukashvily, N.I. Who Needs This Junk, or Genomic Dark Matter. Biochem. Biokhimiia 2018, 83, 450–466. [Google Scholar] [CrossRef] [PubMed]
- Kit, S. Equilibrium sedimentation in density gradients of DNA preparations from animal tissues. J. Mol. Biol. 1961, 3, 711–IN712. [Google Scholar] [CrossRef]
- Sueoka, N. Compositional correlation between deoxyribonucleic acid and protein. Cold Spring Harb. Symp. Quant. Biol. 1961, 26, 35–43. [Google Scholar] [CrossRef]
- Plohl, M.; Luchetti, A.; Mestrovic, N.; Mantovani, B. Satellite DNAs between selfishness and functionality: Structure, genomics and evolution of tandem repeats in centromeric (hetero)chromatin. Gene 2008, 409, 72–82. [Google Scholar] [CrossRef] [PubMed]
- Yasmineh, W.; Yunis, J. Localization of repeated DNA sequences in CsCl gradients by hybridization with complementary RNA. Exp. Cell Res. 1974, 88, 340–344. [Google Scholar] [CrossRef]
- Hartley, G.; O’Neill, R.J. Centromere repeats: Hidden gems of the genome. Genes 2019, 10, 223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jagannathan, M.; Yamashita, Y.M. Function of Junk: Pericentromeric Satellite DNA in Chromosome Maintenance. Cold Spring Harb. Symp. Quant. Biol. 2017, 82, 319–327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaves, R.; Ferreira, D.; Mendes-da-Silva, A.; Meles, S.; Adega, F. FA-SAT Is an Old Satellite DNA Frozen in Several Bilateria Genomes. Genome Biol. Evol. 2017, 9, 3073–3087. [Google Scholar] [CrossRef] [Green Version]
- Henikoff, S.; Dalal, Y. Centromeric chromatin: What makes it unique? Curr. Opin. Genet. Dev. 2005, 15, 177–184. [Google Scholar] [CrossRef] [PubMed]
- Plohl, M.; Meštrović, N.; Mravinac, B. Satellite DNA evolution. In Repetitive DNA; Karger Publishers: Basel, Switzerland, 2012; Volume 7, pp. 126–152. [Google Scholar]
- Biscotti, M.A.; Canapa, A.; Forconi, M.; Olmo, E.; Barucca, M. Transcription of tandemly repetitive DNA: Functional roles. Chromosome Res. Int. J. Mol. Supramol. Evol. Asp. Chromosome Biol. 2015, 23, 463–477. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, D.; Escudeiro, A.; Adega, F.; Anjo, S.I.; Manadas, B.; Chaves, R. FA-SAT ncRNA interacts with PKM2 protein: Depletion of this complex induces a switch from cell proliferation to apoptosis. Cell. Mol. Life Sci. 2019. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, D.; Meles, S.; Escudeiro, A.; Mendes-da-Silva, A.; Adega, F.; Chaves, R. Satellite non-coding RNAs: The emerging players in cells, cellular pathways and cancer. Chromosome Res. Int. J. Mol. Supramol. Evol. Asp. Chromosome Biol. 2015, 23, 479–493. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.-J.; Lee, J.; Han, K. Transposable elements: No more ‘Junk DNA’. Genom. Inform. 2012, 10, 226. [Google Scholar] [CrossRef]
- Makałowski, W. Genomic scrap yard: How genomes utilize all that junk. Gene 2000, 259, 61–67. [Google Scholar] [CrossRef]
- Puppo, I.; Saifitdinova, A.; Tonyan, Z. The Role of Satellite DNA in Causing Structural Rearrangements in Human Karyotype. Russ. J. Genet. 2020, 56, 41–47. [Google Scholar] [CrossRef]
- Veiko, N.N.; Ershova, E.S.; Malinovskaya, E.M.; Konkova, M.S.; Veiko, R.V.; Umriukhin, P.E.; Martynov, A.V.; Kutsev, S.I.; Kostyuk, S.V. Copy number variation of human satellite III (1q12) with Aging. Front. Genet. 2019, 10, 704. [Google Scholar]
- Yandım, C.; Karakülah, G. Expression dynamics of repetitive DNA in early human embryonic development. BMC Genom. 2019, 20, 439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shatskikh, A.S.; Kotov, A.A.; Adashev, V.E.; Bazylev, S.S.; Olenina, L.V. Functional Significance of Satellite DNAs: Insights from Drosophila. Front. Cell Dev. Biol. 2020, 8, 312. [Google Scholar] [CrossRef] [PubMed]
- Aldrup-MacDonald, M.; Sullivan, B. The Past, Present, and Future of Human Centromere Genomics. Genes 2014, 5, 33–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glunčić, M.; Vlahović, I.; Paar, V. Discovery of 33mer in chromosome 21–the largest alpha satellite higher order repeat unit among all human somatic chromosomes. Sci. Rep. 2019, 9, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.; Wevrick, R.; Fisher, R.B.; Ferguson-Smith, M.A.; Lin, C.C. Human centromeric DNAs. Hum. Genet. 1997, 100, 291–304. [Google Scholar] [CrossRef] [PubMed]
- Prosser, J.; Frommer, M.; Paul, C.; Vincent, P.C. Sequence relationships of three human satellite DNAs. J. Mol. Biol. 1986, 187, 145–155. [Google Scholar] [CrossRef]
- Vissel, B.; Nagy, A.; Choo, K. A satellite III sequence shared by human chromosomes 13, 14, and 21 that is contiguous with α satellite DNA. Cytogenet. Genome Res. 1992, 61, 81–86. [Google Scholar] [CrossRef]
- Plohl, M.; Mestrovic, N.; Mravinac, B. Centromere identity from the DNA point of view. Chromosoma 2014, 123, 313–325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hall, L.L.; Byron, M.; Carone, D.M.; Whitfield, T.W.; Pouliot, G.P.; Fischer, A.; Jones, P.; Lawrence, J.B. Demethylated HSATII DNA and HSATII RNA foci sequester PRC1 and MeCP2 into cancer-specific nuclear bodies. Cell Rep. 2017, 18, 2943–2956. [Google Scholar] [CrossRef]
- McNulty, S.M.; Sullivan, L.L.; Sullivan, B.A. Human Centromeres Produce Chromosome-Specific and Array-Specific Alpha Satellite Transcripts that Are Complexed with CENP-A and CENP-C. Dev. Cell, 2017; 42, 226–240.e6. [Google Scholar] [CrossRef]
- Bersani, F.; Lee, E.; Kharchenko, P.V.; Xu, A.W.; Liu, M.; Xega, K.; MacKenzie, O.C.; Brannigan, B.W.; Wittner, B.S.; Jung, H. Pericentromeric satellite repeat expansions through RNA-derived DNA intermediates in cancer. Proc. Natl. Acad. Sci. USA 2015, 112, 15148–15153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delpu, Y.; McNamara, T.; Griffin, P.; Kaleem, S.; Narayan, S.; Schildkraut, C.; Miga, K.; Tahiliani, M. Chromosomal rearrangements at hypomethylated Satellite 2 sequences are associated with impaired replication efficiency and increased fork stalling. bioRxiv 2019, 554410. [Google Scholar] [CrossRef]
- Erliandri, I.; Fu, H.; Nakano, M.; Kim, J.-H.; Miga, K.H.; Liskovykh, M.; Earnshaw, W.C.; Masumoto, H.; Kouprina, N.; Aladjem, M.I. Replication of alpha-satellite DNA arrays in endogenous human centromeric regions and in human artificial chromosome. Nucleic Acids Res. 2014, 42, 11502–11516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vlahović, I.; Glunčić, M.; Rosandić, M.; Ugarković, Đ.; Paar, V. Regular higher order repeat structures in beetle Tribolium castaneum genome. Genome Biol. Evol. 2017, 9, 2668–2680. [Google Scholar] [PubMed] [Green Version]
- Miga, K.H. Centromeric Satellite DNAs: Hidden Sequence Variation in the Human Population. Genes 2019, 10, 352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Warburton, P.E.; Hasson, D.; Guillem, F.; Lescale, C.; Jin, X.; Abrusan, G. Analysis of the largest tandemly repeated DNA families in the human genome. BMC Genom. 2008, 9, 533. [Google Scholar] [CrossRef] [Green Version]
- Altemose, N.; Miga, K.H.; Maggioni, M.; Willard, H.F. Genomic characterization of large heterochromatic gaps in the human genome assembly. PLoS Comput. Biol. 2014, 10, e1003628. [Google Scholar] [CrossRef] [Green Version]
- Fowler, C.; Drinkwater, R.; Burgoyne, L.; Skinner, J. Hypervariable lengths of human DNA associated with a human satellite III sequence found in the 3.4 kb Y-specific fragment. Nucleic Acids Res. 1987, 15, 3929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsu, L.Y.; Benn, P.A.; Tannenbaum, H.L.; Perlis, T.E.; Carlson, A.D.; Opitz, J.M.; Reynolds, J.F. Chromosomal polymorphisms of 1, 9, 16, and Y in 4 major ethnic groups: A large prenatal study. Am. J. Med Genet. 1987, 26, 95–101. [Google Scholar] [CrossRef]
- Podugolnikova, O.; Korostelev, A. The quantitative analysis of polymorphism on human chromosomes 1, 9, 16, and Y. IV. Heterogeneity of a normal population. Hum. Genet. 1980, 54, 163. [Google Scholar] [CrossRef] [PubMed]
- Tagarro, I.; Fernández-Peralta, A.M.; González-Aguilera, J.J. Chromosomal localization of human satellites 2 and 3 by a FISH method using oligonucleotides as probes. Hum. Genet. 1994, 93, 383–388. [Google Scholar] [CrossRef] [PubMed]
- Trowell, H.E.; Nagy, A.; Vissel, B.; Choo, K.A. Long-range analyses of the centromeric regions of human chromosomes 13, 14 and 21: Identification of a narrow domain containing two key centromeric DNA elements. Hum. Mol. Genet. 1993, 2, 1639–1649. [Google Scholar] [CrossRef] [PubMed]
- Agresti, A.; Rainaldi, G.; Lobbiani, A.; Magnani, I.; Di Lernia, R.; Meneveri, R.; Siccardi, A.; Ginelli, E. Chromosomal location by in situ hybridization of the human Sau3A family of DNA repeats. Hum. Genet. 1987, 75, 326–332. [Google Scholar] [CrossRef] [PubMed]
- Cooke, H.J.; Hindley, J. Cloning of human satellite III DNA: Different components are on different chromosomes. Nucleic Acids Res. 1979, 6, 3177–3198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gosden, J.; Mitchell, A.; Buckland, R.; Clayton, R.; Evans, H. The location of four human satellite DNAs on human chromosomes. Exp. Cell Res. 1975, 92, 148–158. [Google Scholar] [CrossRef]
- Jørgensen, A.L.; Kølvraa, S.; Jones, C.; Bak, A.L. A subfamily of alphoid repetitive DNA shared by the NOR-bearing human chromosomes 14 and 22. Genomics 1988, 3, 100–109. [Google Scholar] [CrossRef]
- Tagarro, I.; Wiegant, J.; Raap, A.K.; González-Aguilera, J.J.; Fernández-Peralta, A.M. Assignment of human satellite 1 DNA as revealed by fluorescent in situ hybridization with oligonucleotides. Hum. Genet. 1994, 93, 125–128. [Google Scholar] [CrossRef]
- Garrido-Ramos, M.A. Satellite DNA: An Evolving Topic. Genes 2017, 8, 230. [Google Scholar] [CrossRef]
- Maio, J.J. DNA strand reassociation and polyribonucleotide binding in the African green monkey, Cercopithecus aethiops. J. Mol. Biol. 1971, 56, 579–595. [Google Scholar] [CrossRef]
- Manuelidis, L. Chromosomal localization of complex and simple repeated human DNAs. Chromosoma 1978, 66, 23–32. [Google Scholar] [CrossRef] [PubMed]
- Rudd, M.; Schueler, M.; Willard, H. Sequence organization and functional annotation of human centromeres. Cold Spring Harb. Symp. Quant. Biol. 2003, 68, 141–149. [Google Scholar] [CrossRef]
- Willard, H.F. Chromosome-specific organization of human alpha satellite DNA. Am. J. Hum. Genet. 1985, 37, 524. [Google Scholar] [PubMed]
- Willard, H.F.; Waye, J.S. Chromosome-specific subsets of human alpha satellite DNA: Analysis of sequence divergence within and between chromosomal subsets and evidence for an ancestral pentameric repeat. J. Mol. Evol. 1987, 25, 207–214. [Google Scholar] [CrossRef] [PubMed]
- AIexandrov, I.; Medvedev, L.; Mashkova, T.; Kisselev, L.; Romanova, L.; Yurov, Y. Definition of a new alpha satellite suprachromosomal family characterized by monomeric organization. Nucleic Acids Res. 1993, 21, 2209–2215. [Google Scholar] [CrossRef]
- Shepelev, V.; Uralsky, L.; Alexandrov, A.; Yurov, Y.; Rogaev, E.I.; Alexandrov, I. Annotation of suprachromosomal families reveals uncommon types of alpha satellite organization in pericentromeric regions of hg38 human genome assembly. Genom. Data 2015, 5, 139–146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muro, Y.; Masumoto, H.; Yoda, K.; Nozaki, N.; Ohashi, M.; Okazaki, T. Centromere protein B assembles human centromeric alpha-satellite DNA at the 17-bp sequence, CENP-B box. J. Cell Biol. 1992, 116, 585–596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sullivan, K.F.; Glass, C.A. CENP-B is a highly conserved mammalian centromere protein with homology to the helix-loop-helix family of proteins. Chromosoma 1991, 100, 360–370. [Google Scholar] [CrossRef] [PubMed]
- Fachinetti, D.; Han, J.S.; McMahon, M.A.; Ly, P.; Abdullah, A.; Wong, A.J.; Cleveland, D.W. DNA sequence-specific binding of CENP-B enhances the fidelity of human centromere function. Dev. Cell 2015, 33, 314–327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Devilee, P.; Cremer, T.; Slagboom, P.; Bakker, E.; Scholl, H.P.; Hager, H.; Stevenson, A.; Cornelisse, C.; Pearson, P. Two subsets of human alphoid repetitive DNA show distinct preferential localization in the pericentric regions of chromosomes 13, 18, and 21. Cytogenet. Genome Res. 1986, 41, 193–201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choo, K.; Vissel, B.; Nagy, A.; Earle, E.; Kalitsis, P. A survey of the genomic distribution of alpha satellite DNA on all the human chromosomes, and derivation of a new consensus sequence. Nucleic Acids Res. 1991, 19, 1179. [Google Scholar] [CrossRef] [Green Version]
- Sullivan, L.L.; Chew, K.; Sullivan, B.A. α satellite DNA variation and function of the human centromere. Nucleus 2017, 8, 331–339. [Google Scholar] [CrossRef] [Green Version]
- Sullivan, L.L.; Sullivan, B.A. Genomic and functional variation of human centromeres. Exp. Cell Res. 2020, 111896. [Google Scholar] [CrossRef] [PubMed]
- Hayden, K.E.; Strome, E.D.; Merrett, S.L.; Lee, H.-R.; Rudd, M.K.; Willard, H.F. Sequences associated with centromere competency in the human genome. Mol. Cell. Biol. 2013, 33, 763–772. [Google Scholar] [CrossRef] [Green Version]
- Henikoff, J.G.; Thakur, J.; Kasinathan, S.; Henikoff, S. A unique chromatin complex occupies young α-satellite arrays of human centromeres. Sci. Adv. 2015, 1, e1400234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waye, J.S.; Willard, H.F. Nucleotide sequence heterogeneity of alpha satellite repetitive DNA: A survey of alphoid sequences from different human chromosomes. Nucleic Acids Res. 1987, 15, 7549–7569. [Google Scholar] [CrossRef] [Green Version]
- Schueler, M.G.; Dunn, J.M.; Bird, C.P.; Ross, M.T.; Viggiano, L.; Rocchi, M.; Willard, H.F.; Green, E.D.; Program, N.C.S. Progressive proximal expansion of the primate X chromosome centromere. Proc. Natl. Acad. Sci. USA 2005, 102, 10563–10568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Logsdon, G.A.; Vollger, M.R.; Hsieh, P.; Mao, Y.; Liskovykh, M.A.; Koren, S.; Nurk, S.; Mercuri, L.; Dishuck, P.C.; Rhie, A. The structure, function and evolution of a complete human chromosome 8. Nature 2021, 1–7. [Google Scholar] [CrossRef]
- Shepelev, V.A.; Alexandrov, A.A.; Yurov, Y.B.; Alexandrov, I.A. The evolutionary origin of man can be traced in the layers of defunct ancestral alpha satellites flanking the active centromeres of human chromosomes. PLoS Genet 2009, 5, e1000641. [Google Scholar] [CrossRef]
- Meyne, J.; Goodwin, E.H.; Moyzis, R.K. Chromosome localization and orientation of the simple sequence repeat of human satellite I DNA. Chromosoma 1994, 103, 99–103. [Google Scholar] [CrossRef] [PubMed]
- Kalitsis, P.; Earle, E.; Vissel, B.; Shaffer, L.G.; Choo, K.A. A chromosome 13-specific human satellite I DNA subfamily with minor presence on chromosome 21: Further studies on Robertsonian translocations. Genomics 1993, 16, 104–112. [Google Scholar] [CrossRef]
- Jeanpierre, M. Human satellites 2 and 3. Ann. De Genet. 1994, 37, 163–171. [Google Scholar]
- Moyzis, R.K.; Torney, D.C.; Meyne, J.; Buckingham, J.M.; Wu, J.-R.; Burks, C.; Sirotkin, K.M.; Goad, W.B. The distribution of interspersed repetitive DNA sequences in the human genome. Genomics 1989, 4, 273–289. [Google Scholar] [CrossRef]
- Schwarzacher-Robinson, T.; Cram, L.; Meyne, J.; Moyzis, R. Characterization of human heterochromatin by in situ hybridization with satellite DNA clones. Cytogenet. Cell Genet. 1988, 47, 192–196. [Google Scholar] [CrossRef] [PubMed]
- Nakahori, Y.; Mitani, K.; Yamada, M.; Nakagome, Y. A human Y-chromosome specific repeated DNA family (DYZ1) consists of a tandem array of pentanucleotides. Nucleic Acids Res. 1986, 14, 7569–7580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choo, K.A. The Centromere; Oxford University Press: Oxford, UK, 1997; Volume 320. [Google Scholar]
- Cooke, H. Repeated sequence specific to human males. Nature 1976, 262, 182–186. [Google Scholar] [CrossRef] [PubMed]
- Kunkel, L.M.; Smith, K.D.; Boyer, S.H. Human Y-chromosome-specific reiterated DNA. Science 1976, 191, 1189–1190. [Google Scholar] [CrossRef] [PubMed]
- Choo, K.; Earle, E.; McQuillan, C. A homologous subfamily of satellite III DNA on human chromosomes 14 and 22. Nucleic Acids Res. 1990, 18, 5641–5648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choo, K.; Earle, E.; Vissel, B.; Kalitsis, P. A chromosome 14-specific human satellite III DNA subfamily that shows variable presence on different chromosomes 14. Am. J. Hum. Genet. 1992, 50, 706. [Google Scholar] [PubMed]
- Bandyopadhyay, R.; McQuillan, C.; Page, S.; Choo, K.; Shaffer, L. Identification and characterization of satellite III subfamilies to the acrocentric chromosomes. Chromosome Res. 2001, 9, 223–233. [Google Scholar] [CrossRef] [PubMed]
- Earle, E.; Shaffer, L.; Kalitsis, P.; McQuillan, C.; Dale, S.; Choo, K. Identification of DNA sequences flanking the breakpoint of human t (14q21q) Robertsonian translocations. Am. J. Hum. Genet. 1992, 50, 717. [Google Scholar] [PubMed]
- Meneveri, R.; Agresti, A.; Della Valle, G.; Talarico, D.; Siccardi, A.; Ginelli, E. Identification of a human clustered G+ C-rich DNA family of repeats (Sau3A family). J. Mol. Biol. 1985, 186, 483–489. [Google Scholar] [CrossRef]
- Waye, J.S.; Willard, H.F. Human beta satellite DNA: Genomic organization and sequence definition of a class of highly repetitive tandem DNA. Proc. Natl. Acad. Sci. USA 1989, 86, 6250–6254. [Google Scholar] [CrossRef] [Green Version]
- Meneveri, R.; Agresti, A.; Marozzi, A.; Saccone, S.; Rocchi, M.; Archidiacono, N.; Corneo, G.; Valle, G.D.; Ginelli, E. Molecular organization and chromosomal location of human GC-rich heterochromatic blocks. Gene 1993, 123, 227–234. [Google Scholar] [CrossRef]
- Eichler, E.E.; Hoffman, S.M.; Adamson, A.A.; Gordon, L.A.; McCready, P.; Lamerdin, J.E.; Mohrenweiser, H.W. Complex β-satellite repeat structures and the expansion of the zinc finger gene cluster in 19p12. Genome Res. 1998, 8, 791–808. [Google Scholar] [CrossRef] [Green Version]
- Cardone, M.; Ballarati, L.; Ventura, M.; Rocchi, M.; Marozzi, A.; Ginelli, E.; Meneveri, R. Evolution of beta satellite DNA sequences: Evidence for duplication-mediated repeat amplification and spreading. Mol. Biol. Evol. 2004, 21, 1792–1799. [Google Scholar] [CrossRef]
- Yang, J.; Yuan, B.; Wu, Y.; Li, M.; Li, J.; Xu, D.; Gao, Z.-h.; Ma, G.; Zhou, Y.; Zuo, Y. The wide distribution and horizontal transfers of beta satellite DNA in eukaryotes. Genomics 2020. [Google Scholar] [CrossRef]
- Lin, C.; Sasi, R.; Fan, Y.-S. Isolation and identification of a novel tandemly repeated DNA sequence in the centromeric region of human chromosome 8. Chromosoma 1993, 102, 333–339. [Google Scholar] [CrossRef]
- Lee, C.; Li, X.; Jabs, E.; Lin, C. Human gamma X satellite DNA: An X chromosome specific centromeric DNA sequence. Chromosoma 1995, 104, 103–112. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.-H.; Ebersole, T.; Kouprina, N.; Noskov, V.N.; Ohzeki, J.-I.; Masumoto, H.; Mravinac, B.; Sullivan, B.A.; Pavlicek, A.; Dovat, S. Human gamma-satellite DNA maintains open chromatin structure and protects a transgene from epigenetic silencing. Genome Res. 2009, 19, 533–544. [Google Scholar] [CrossRef] [Green Version]
- Miga, K.H. The Promises and Challenges of Genomic Studies of Human Centromeres. Prog. Mol. Subcell. Biol. 2017, 56, 285–304. [Google Scholar] [CrossRef]
- Jain, M.; Olsen, H.E.; Turner, D.J.; Stoddart, D.; Bulazel, K.V.; Paten, B.; Haussler, D.; Willard, H.F.; Akeson, M.; Miga, K.H. Linear assembly of a human centromere on the Y chromosome. Nat. Biotechnol. 2018, 36, 321–323. [Google Scholar] [CrossRef] [Green Version]
- Miga, K.H. Completing the human genome: The progress and challenge of satellite DNA assembly. Chromosome Res. Int. J. Mol. Supramol. Evol. Asp. Chromosome Biol. 2015, 23, 421–426. [Google Scholar] [CrossRef] [PubMed]
- Gosden, J.; Lawrie, S.; Gosden, C. Satellite DNA sequences in the human acrocentric chromosomes: Information from translocations and heteromorphisms. Am. J. Hum. Genet. 1981, 33, 243. [Google Scholar]
- Levy, S.; Sutton, G.; Ng, P.C.; Feuk, L.; Halpern, A.L.; Walenz, B.P.; Axelrod, N.; Huang, J.; Kirkness, E.F.; Denisov, G. The diploid genome sequence of an individual human. PLoS Biol. 2007, 5, e254. [Google Scholar] [CrossRef] [Green Version]
- Jeffreys, A.J.; Wilson, V.; Thein, S.L. Hypervariable ‘minisatellite’regions in human DNA. Nature 1985, 314, 67–73. [Google Scholar] [CrossRef]
- Litt, M.; Luty, J.A. A hypervariable microsatellite revealed by in vitro amplification of a dinucleotide repeat within the cardiac muscle actin gene. Am. J. Hum. Genet. 1989, 44, 397. [Google Scholar]
- Garrido-Ramos, M.A. Satellite DNA in Plants: More than Just Rubbish. Cytogenet Genome Res 2015, 146, 153–170. [Google Scholar] [CrossRef]
- Black, E.M.; Giunta, S. Repetitive Fragile Sites: Centromere Satellite DNA As a Source of Genome Instability in Human Diseases. Genes 2018, 9, 615. [Google Scholar] [CrossRef] [Green Version]
- Collins, F.S.; Green, E.D.; Guttmacher, A.E.; Guyer, M.S. A vision for the future of genomics research. Nature 2003, 422, 835. [Google Scholar] [CrossRef]
- Eichler, E.E.; Clark, R.A.; She, X. An assessment of the sequence gaps: Unfinished business in a finished human genome. Nat. Rev. Genet. 2004, 5, 345. [Google Scholar] [CrossRef]
- Phillippy, A. The (Near) Complete Sequence of a Human Genome. Available online: https://genomeinformatics.github.io/CHM13v1/ (accessed on 20 April 2021).
- Benson, G. Tandem repeats finder: A program to analyze DNA sequences. Nucleic Acids Res. 1999, 27, 573–580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitsuhashi, S.; Frith, M.C.; Mizuguchi, T.; Miyatake, S.; Toyota, T.; Adachi, H.; Oma, Y.; Kino, Y.; Mitsuhashi, H.; Matsumoto, N. Tandem-genotypes: Robust detection of tandem repeat expansions from long DNA reads. Genome Biol. 2019, 20, 58. [Google Scholar] [CrossRef] [Green Version]
- Mitsuhashi, S.; Matsumoto, N. Long-read sequencing for rare human genetic diseases. J. Hum. Genet. 2020, 65, 11–19. [Google Scholar] [CrossRef]
- Louzada, S.; Lopes, M.; Ferreira, D.; Adega, F.; Escudeiro, A.; Gama-Carvalho, M.; Chaves, R. Decoding the Role of Satellite DNA in Genome Architecture and Plasticity—An Evolutionary and Clinical Affair. Genes 2020, 11, 72. [Google Scholar] [CrossRef] [Green Version]
- Li, H. Toward better understanding of artifacts in variant calling from high-coverage samples. Bioinformatics 2014, 30, 2843–2851. [Google Scholar] [CrossRef] [Green Version]
- Ameur, A.; Kloosterman, W.P.; Hestand, M.S. Single-molecule sequencing: Towards clinical applications. Trends Biotechnol. 2018, 37, 72–85. [Google Scholar] [CrossRef]
- Cao, M.D.; Nguyen, S.H.; Ganesamoorthy, D.; Elliott, A.G.; Cooper, M.A.; Coin, L.J. Scaffolding and completing genome assemblies in real-time with nanopore sequencing. Nat. Commun. 2017, 8, 14515. [Google Scholar] [CrossRef] [Green Version]
- Miga, K.H. Centromere studies in the era of ‘telomere-to-telomere’genomics. Exp. Cell Res. 2020, 112127. [Google Scholar] [CrossRef]
- Li, Z.; Chen, Y.; Mu, D.; Yuan, J.; Shi, Y.; Zhang, H.; Gan, J.; Li, N.; Hu, X.; Liu, B. Comparison of the two major classes of assembly algorithms: Overlap–layout–consensus and de-bruijn-graph. Brief. Funct. Genom. 2012, 11, 25–37. [Google Scholar] [CrossRef] [Green Version]
- Luce, A.C.; Sharma, A.; Mollere, O.S.; Wolfgruber, T.K.; Nagaki, K.; Jiang, J.; Presting, G.G.; Dawe, R.K. Precise centromere mapping using a combination of repeat junction markers and chromatin immunoprecipitation–polymerase chain reaction. Genetics 2006, 174, 1057–1061. [Google Scholar] [CrossRef] [Green Version]
- Miller, J.R.; Koren, S.; Sutton, G. Assembly algorithms for next-generation sequencing data. Genomics 2010, 95, 315–327. [Google Scholar] [CrossRef] [Green Version]
- Schneider, V.A.; Graves-Lindsay, T.; Howe, K.; Bouk, N.; Chen, H.-C.; Kitts, P.A.; Murphy, T.D.; Pruitt, K.D.; Thibaud-Nissen, F.; Albracht, D. Evaluation of GRCh38 and de novo haploid genome assemblies demonstrates the enduring quality of the reference assembly. Genome Res. 2017, 27, 849–864. [Google Scholar] [CrossRef] [Green Version]
- Guo, Y.; Dai, Y.; Yu, H.; Zhao, S.; Samuels, D.C.; Shyr, Y. Improvements and impacts of GRCh38 human reference on high throughput sequencing data analysis. Genomics 2017, 109, 83–90. [Google Scholar] [CrossRef]
- Miga, K.H.; Koren, S.; Rhie, A.; Vollger, M.R.; Gershman, A.; Bzikadze, A.; Brooks, S.; Howe, E.; Porubsky, D.; Logsdon, G.A. Telomere-to-telomere assembly of a complete human X chromosome. Nature 2020, 585, 79–84. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, Y.; Myers, G.; Morishita, S. Long-read Data Revealed Structural Diversity in Human Centromere Sequences. BioRxiv 2019, 784785. [Google Scholar] [CrossRef] [Green Version]
- Mahtani, M.M.; Willard, H.F. Pulsed-field gel analysis of α-satellite DNA at the human X chromosome centromere: High-frequency polymorphisms and array size estimate. Genomics 1990, 7, 607–613. [Google Scholar] [CrossRef]
- Schindelhauer, D.; Schwarz, T. Evidence for a fast, intrachromosomal conversion mechanism from mapping of nucleotide variants within a homogeneous α-satellite DNA array. Genome Res. 2002, 12, 1815–1826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miga, K.H. Breaking through the Unknowns of the Human Reference Genome; Nature Publishing Group: London, UK, 2021. [Google Scholar]
- Heather, J.M.; Chain, B. The sequence of sequencers: The history of sequencing DNA. Genomics 2016, 107, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanger, F.; Air, G.M.; Barrell, B.G.; Brown, N.L.; Coulson, A.R.; Fiddes, J.C.; Hutchison, C.; Slocombe, P.M.; Smith, M. Nucleotide sequence of bacteriophage φX174 DNA. Nature 1977, 265, 687. [Google Scholar] [CrossRef] [PubMed]
- Lander, E.S.; Linton, L.M.; Birren, B.; Nusbaum, C.; Zody, M.C.; Baldwin, J.; Devon, K.; Dewar, K.; Doyle, M.; FitzHugh, W. Initial sequencing and analysis of the human genome. Nature 2001, 409, 860–921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Venter, J.C.; Adams, M.D.; Myers, E.W.; Li, P.W.; Mural, R.J.; Sutton, G.G.; Smith, H.O.; Yandell, M.; Evans, C.A.; Holt, R.A. The sequence of the human genome. Science 2001, 291, 1304–1351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janitz, M. Next-Generation Genome Sequencing: Towards Personalized Medicine; John Wiley & Sons: Hoboken, NJ, USA, 2011. [Google Scholar]
- Ansorge, W.; Sproat, B.; Stegemann, J.; Schwager, C.; Zenke, M. Automated DNA sequencing: Ultrasensitive detection of fluorescent bands during electrophoresis. Nucleic Acids Res. 1987, 15, 4593–4602. [Google Scholar] [CrossRef] [Green Version]
- Luckey, J.A.; Drossman, H.; Kostichka, A.J.; Mead, D.A.; D’Cunha, J.; Norris, T.B.; Smith, L.M. High speed DNA sequencing by capillary electrophoresis. Nucleic Acids Res. 1990, 18, 4417–4421. [Google Scholar] [CrossRef] [Green Version]
- Smith, L.M.; Fung, S.; Hunkapiller, M.W.; Hunkapiller, T.J.; Hood, L.E. The synthesis of oligonucleotides containing an aliphatic amino group at the 5′ terminus: Synthesis of fluorescent DNA primers for use in DNA sequence analysis. Nucleic Acids Res. 1985, 13, 2399–2412. [Google Scholar] [CrossRef]
- Swerdlow, H.; Gesteland, R. Capillary gel electrophoresis for rapid, high resolution DNA sequencing. Nucleic Acids Res. 1990, 18, 1415–1419. [Google Scholar] [CrossRef] [Green Version]
- Anderson, S. Shotgun DNA sequencing using cloned DNase I-generated fragments. Nucleic Acids Res. 1981, 9, 3015–3027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Staden, R. A strategy of DNA sequencing employing computer programs. Nucleic Acids Res. 1979, 6, 2601–2610. [Google Scholar] [CrossRef] [PubMed]
- de Lannoy, C.; de Ridder, D.; Risse, J. The long reads ahead: De novo genome assembly using the MinION. F1000Research 2017, 6, 1023. [Google Scholar] [CrossRef] [PubMed]
- Goodwin, S.; McPherson, J.D.; McCombie, W.R. Coming of age: Ten years of next-generation sequencing technologies. Nat. Rev. Genet. 2016, 17, 333. [Google Scholar] [CrossRef]
- Shendure, J.; Balasubramanian, S.; Church, G.M.; Gilbert, W.; Rogers, J.; Schloss, J.A.; Waterston, R.H. DNA sequencing at 40: Past, present and future. Nature 2017, 550, 345–353. [Google Scholar] [CrossRef] [PubMed]
- Mardis, E.R. DNA sequencing technologies: 2006–2016. Nat. Protoc. 2017, 12, 213. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Yang, Q.; Wang, Z. The evolution of nanopore sequencing. Front. Genet. 2015, 5, 449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harris, R.S.; Cechova, M.; Makova, K.D. Noise-Cancelling Repeat Finder: Uncovering tandem repeats in error-prone long-read sequencing data. Bioinformatics 2019, 35, 4809–4811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jain, M.; Koren, S.; Miga, K.H.; Quick, J.; Rand, A.C.; Sasani, T.A.; Tyson, J.R.; Beggs, A.D.; Dilthey, A.T.; Fiddes, I.T. Nanopore sequencing and assembly of a human genome with ultra-long reads. Nat. Biotechnol. 2018, 36, 338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cacheux, L.; Ponger, L.; Gerbault-Seureau, M.; Richard, F.A.; Escudé, C. Diversity and distribution of alpha satellite DNA in the genome of an Old World monkey: Cercopithecus solatus. BMC Genom. 2016, 17, 916. [Google Scholar] [CrossRef] [Green Version]
- Logsdon, G.A.; Vollger, M.R.; Eichler, E.E. Long-read human genome sequencing and its applications. Nat. Rev. Genet. 2020, 21, 597–614. [Google Scholar] [CrossRef]
- Amarasinghe, S.L.; Su, S.; Dong, X.; Zappia, L.; Ritchie, M.E.; Gouil, Q. Opportunities and challenges in long-read sequencing data analysis. Genome Biol. 2020, 21. [Google Scholar] [CrossRef] [Green Version]
- ONT. At NCM, Announcements Include Single-Read Accuracy of 99.1% on New Chemistry and Sequencing a Record 10 Tb in a Single PromethION Run. Available online: https://nanoporetech.com/about-us/news/ncm-announcements-include-single-read-accuracy-991-new-chemistry-and-sequencing(accessed on 21 March 2021).
- Kraft, F.; Kurth, I. Long-read sequencing to understand genome biology and cell function. Int. J. Biochem. Cell Biol. 2020, 105799. [Google Scholar] [CrossRef] [PubMed]
- Wenger, A.M.; Peluso, P.; Rowell, W.J.; Chang, P.C.; Hall, R.J.; Concepcion, G.T.; Ebler, J.; Fungtammasan, A.; Kolesnikov, A.; Olson, N.D.; et al. Accurate circular consensus long-read sequencing improves variant detection and assembly of a human genome. Nat. Biotechnol. 2019, 37, 1155–1162. [Google Scholar] [CrossRef]
- De Roeck, A.; De Coster, W.; Bossaerts, L.; Cacace, R.; De Pooter, T.; Van Dongen, J.; D’Hert, S.; De Rijk, P.; Strazisar, M.; Van Broeckhoven, C. NanoSatellite: Accurate characterization of expanded tandem repeat length and sequence through whole genome long-read sequencing on PromethION. Genome Biol. 2019, 20, 239. [Google Scholar] [CrossRef] [Green Version]
- Nurk, S.; Walenz, B.P.; Rhie, A.; Vollger, M.R.; Logsdon, G.A.; Grothe, R.; Miga, K.H.; Eichler, E.E.; Phillippy, A.M.; Koren, S. HiCanu: Accurate assembly of segmental duplications, satellites, and allelic variants from high-fidelity long reads. BioRxiv 2020. [Google Scholar] [CrossRef]
- Vollger, M.R.; Logsdon, G.A.; Audano, P.A.; Sulovari, A.; Porubsky, D.; Peluso, P.; Wenger, A.M.; Concepcion, G.T.; Kronenberg, Z.N.; Munson, K.M. Improved assembly and variant detection of a haploid human genome using single-molecule, high-fidelity long reads. Ann. Hum. Genet. 2020, 84, 125–140. [Google Scholar] [CrossRef]
- Cao, H.; Wahlestedt, C.; Kapranov, P. Strategies to annotate and characterize long noncoding RNAs: Advantages and pitfalls. Trends Genet. 2018, 34, 704–721. [Google Scholar] [CrossRef] [PubMed]
- Ulitsky, I. Interactions between short and long noncoding RNAs. FEBS Lett. 2018, 592, 2874–2883. [Google Scholar] [CrossRef] [Green Version]
- Saksouk, N.; Simboeck, E.; Déjardin, J. Constitutive heterochromatin formation and transcription in mammals. Epigenetics Chromatin 2015, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garalde, D.R.; Snell, E.A.; Jachimowicz, D.; Sipos, B.; Lloyd, J.H.; Bruce, M.; Pantic, N.; Admassu, T.; James, P.; Warland, A.; et al. Highly parallel direct RNA sequencing on an array of nanopores. Nat. Methods 2018, 15, 201–206. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.M.; Jain, M.; Mulroney, L.; Garalde, D.R.; Akeson, M. Reading canonical and modified nucleobases in 16S ribosomal RNA using nanopore native RNA sequencing. PLoS ONE 2019, 14, e0216709. [Google Scholar] [CrossRef] [Green Version]
- Stark, R.; Grzelak, M.; Hadfield, J. RNA sequencing: The teenage years. Nat. Rev. Genet. 2019, 20, 631–656. [Google Scholar] [CrossRef]
- Kono, N.; Arakawa, K. Nanopore sequencing: Review of potential applications in functional genomics. Dev. Growth Differ. 2019, 61, 316–326. [Google Scholar] [CrossRef] [Green Version]
- Workman, R.E.; Tang, A.D.; Tang, P.S.; Jain, M.; Tyson, J.R.; Razaghi, R.; Zuzarte, P.C.; Gilpatrick, T.; Payne, A.; Quick, J. Nanopore native RNA sequencing of a human poly (A) transcriptome. Nat. Methods 2019, 16, 1297–1305. [Google Scholar] [CrossRef]
- Weirather, J.L.; de Cesare, M.; Wang, Y.; Piazza, P.; Sebastiano, V.; Wang, X.J.; Buck, D.; Au, K.F. Comprehensive comparison of Pacific Biosciences and Oxford Nanopore Technologies and their applications to transcriptome analysis. F1000Res 2017, 6, 100. [Google Scholar] [CrossRef] [PubMed]
- Chaisson, M.J.; Wilson, R.K.; Eichler, E.E. Genetic variation and the de novo assembly of human genomes. Nat. Rev. Genet. 2015, 16, 627. [Google Scholar] [CrossRef] [PubMed]
- Sevim, V.; Bashir, A.; Chin, C.-S.; Miga, K.H. Alpha-CENTAURI: Assessing novel centromeric repeat sequence variation with long read sequencing. Bioinformatics 2016, 32, 1921–1924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCombie, W.R.; McPherson, J.D.; Mardis, E.R. Next-Generation Sequencing Technologies. Cold Spring Harb. Perspect. Med. 2019, 9. [Google Scholar] [CrossRef]
- Kumar, K.R.; Cowley, M.J.; Davis, R.L. Next-generation sequencing and emerging technologies. Semin. Thromb. Hemost. 2019, 45, 661–673. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.R.; Zhou, P.; Mudge, J.; Gurtowski, J.; Lee, H.; Ramaraj, T.; Walenz, B.P.; Liu, J.; Stupar, R.M.; Denny, R. Hybrid assembly with long and short reads improves discovery of gene family expansions. BMC Genom. 2017, 18, 541. [Google Scholar] [CrossRef] [PubMed]
- Minei, R.; Hoshina, R.; Ogura, A. De novo assembly of middle-sized genome using MinION and Illumina sequencers. BMC Genom. 2018, 19, 700. [Google Scholar] [CrossRef] [PubMed]
- Weissensteiner, M.H.; Pang, A.W.; Bunikis, I.; Höijer, I.; Vinnere-Petterson, O.; Suh, A.; Wolf, J.B. Combination of short-read, long-read, and optical mapping assemblies reveals large-scale tandem repeat arrays with population genetic implications. Genome Res. 2017, 27, 697–708. [Google Scholar] [CrossRef] [Green Version]
- Louzada, S.; Komatsu, J.; Yang, F. Fluorescence in situ hybridization onto DNA fibres generated using molecular combing. In Fluorescence In Situ Hybridization (FISH); Springer: Berlin/Heidelberg, Germany, 2017; pp. 275–293. [Google Scholar]
- Deschamps, S.; Zhang, Y.; Llaca, V.; Ye, L.; Sanyal, A.; King, M.; May, G.; Lin, H. A chromosome-scale assembly of the sorghum genome using nanopore sequencing and optical mapping. Nat. Commun. 2018, 9. [Google Scholar] [CrossRef]
- Etherington, G.J.; Heavens, D.; Baker, D.; Lister, A.; McNelly, R.; Garcia, G.; Clavijo, B.; Macaulay, I.; Haerty, W.; Di Palma, F. Sequencing smart: De novo sequencing and assembly approaches for a non-model mammal. GigaScience 2020, 9, giaa045. [Google Scholar] [CrossRef]
- Watson, M.; Warr, A. Errors in long-read assemblies can critically affect protein prediction. Nat. Biotechnol. 2019, 37, 124–126. [Google Scholar] [CrossRef] [Green Version]
- Yeo, S.; Coombe, L.; Warren, R.L.; Chu, J.; Birol, I. ARCS: Scaffolding genome drafts with linked reads. Bioinformatics 2018, 34, 725–731. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Fan, J.; Sun, Z.; Liu, S. NextPolish: A fast and efficient genome polishing tool for long read assembly. Bioinformatics 2020. [CrossRef]
- Deakin, J.E.; Potter, S.; O’Neill, R.; Ruiz-Herrera, A.; Cioffi, M.B.; Eldridge, M.D.; Fukui, K.; Marshall Graves, J.A.; Griffin, D.; Grutzner, F. Chromosomics: Bridging the gap between genomes and chromosomes. Genes 2019, 10, 627. [Google Scholar] [CrossRef] [Green Version]
- Ma, L.; Li, W.; Song, Q. Chromosome-range whole-genome high-throughput experimental haplotyping by single-chromosome microdissection. In Haplotyping; Springer: Berlin/Heidelberg, Germany, 2017; pp. 161–169. [Google Scholar]
- Seifertova, E.; Zimmerman, L.B.; Gilchrist, M.J.; Macha, J.; Kubickova, S.; Cernohorska, H.; Zarsky, V.; Owens, N.D.; Sesay, A.K.; Tlapakova, T. Efficient high-throughput sequencing of a laser microdissected chromosome arm. BMC Genom. 2013, 14, 357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Traut, W.; Vogel, H.; Glöckner, G.; Hartmann, E.; Heckel, D.G. High-throughput sequencing of a single chromosome: A moth W chromosome. Chromosome Res. 2013, 21, 491–505. [Google Scholar] [CrossRef]
- Makunin, A.I.; Kichigin, I.G.; Larkin, D.M.; O’Brien, P.C.; Ferguson-Smith, M.A.; Yang, F.; Proskuryakova, A.A.; Vorobieva, N.V.; Chernyaeva, E.N.; O’Brien, S.J. Contrasting origin of B chromosomes in two cervids (Siberian roe deer and grey brocket deer) unravelled by chromosome-specific DNA sequencing. BMC Genom. 2016, 17, 618. [Google Scholar] [CrossRef] [Green Version]
- Dudchenko, O.; Batra, S.S.; Omer, A.D.; Nyquist, S.K.; Hoeger, M.; Durand, N.C.; Shamim, M.S.; Machol, I.; Lander, E.S.; Aiden, A.P. De novo assembly of the Aedes aegypti genome using Hi-C yields chromosome-length scaffolds. Science 2017, 356, 92–95. [Google Scholar] [CrossRef] [Green Version]
- Putnam, N.H.; O’Connell, B.L.; Stites, J.C.; Rice, B.J.; Blanchette, M.; Calef, R.; Troll, C.J.; Fields, A.; Hartley, P.D.; Sugnet, C.W. Chromosome-scale shotgun assembly using an in vitro method for long-range linkage. Genome Res. 2016, 26, 342–350. [Google Scholar] [CrossRef] [Green Version]
- Alvarez-Cubero, M.J.; Santiago, O.; Martinez-Labarga, C.; Martinez-Garcia, B.; Marrero-Diaz, R.; Rubio-Roldan, A.; Perez-Gutierrez, A.M.; Carmona-Saez, P.; Lorente, J.A.; Martinez-Gonzalez, L.J. Methodology for Y Chromosome Capture: A complete genome sequence of Y chromosome using flow cytometry, laser microdissection and magnetic streptavidin-beads. Sci. Rep. 2018, 8, 9436. [Google Scholar] [CrossRef]
- Kuderna, L.F.; Lizano, E.; Julià, E.; Gomez-Garrido, J.; Serres-Armero, A.; Kuhlwilm, M.; Alandes, R.A.; Alvarez-Estape, M.; Juan, D.; Simon, H. Selective single molecule sequencing and assembly of a human Y chromosome of African origin. Nat. Commun. 2019, 10. [Google Scholar] [CrossRef] [Green Version]
- Sedlazeck, F.J.; Lee, H.; Darby, C.A.; Schatz, M.C. Piercing the dark matter: Bioinformatics of long-range sequencing and mapping. Nat. Rev. Genet. 2018, 19, 329. [Google Scholar] [CrossRef]
- Singer, M.F. Highly repeated sequences in mammalian genomes. In International Review of Cytology; Elsevier: Amsterdam, The Netherlands, 1982; Volume 76, pp. 67–112. [Google Scholar]
- Gall, J.G. The origin of in situ hybridization–a personal history. Methods 2016, 98, 4–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwarzacher, T.; Heslop-Harrison, P. Practical In Situ Hybridization; BIOS Scientific Publishers Ltd: Milton Park, UK, 2000. [Google Scholar]
- Kim, Y.B.; Oh, J.H.; McIver, L.J.; Rashkovetsky, E.; Michalak, K.; Garner, H.R.; Kang, L.; Nevo, E.; Korol, A.B.; Michalak, P. Divergence of Drosophila melanogaster repeatomes in response to a sharp microclimate contrast in Evolution Canyon, Israel. Proc. Natl. Acad. Sci. USA 2014, 111, 10630–10635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruiz-Ruano, F.J.; López-León, M.D.; Cabrero, J.; Camacho, J.P.M. High-throughput analysis of the satellitome illuminates satellite DNA evolution. Sci. Rep. 2016, 6, 28333. [Google Scholar] [CrossRef] [Green Version]
- Ebert, P.; Audano, P.A.; Zhu, Q.; Rodriguez-Martin, B.; Porubsky, D.; Bonder, M.J.; Sulovari, A.; Ebler, J.; Zhou, W.; Mari, R.S. Haplotype-resolved diverse human genomes and integrated analysis of structural variation. Science 2021, 372. [Google Scholar] [CrossRef]
- Corneo, G.; Ginelli, E.; Polli, E. A satellite DNA isolated from human tissues. J. Mol. Biol. 1967, 23, 619. [Google Scholar] [CrossRef]
- Corneo, G.; Ginelli, E.; Polli, E. Isolation of the complementary strands of a human satellite DNA. J. Mol. Biol. 1968, 33, 331–335. [Google Scholar] [CrossRef]
- Corneo, G.; Ginelli, E.; Polli, E. Repeated sequences in human DNA. J. Mol. Biol. 1970, 48, 319–327. [Google Scholar] [CrossRef]
- Corneo, G.; Ginelli, E.; Polli, E. Renaturation properties and localization in heterochromatin of human satellite DNA’s. Biochim. Et Biophys. Acta (Bba)-Nucleic Acids Protein Synth. 1971, 247, 528–534. [Google Scholar] [CrossRef]
- Mikheenko, A.; Bzikadze, A.V.; Gurevich, A.; Miga, K.H.; Pevzner, P.A. TandemMapper and TandemQUAST: Mapping long reads and assessing/improving assembly quality in extra-long tandem repeats. BioRxiv 2019. [Google Scholar] [CrossRef]
- Mikheenko, A.; Bzikadze, A.V.; Gurevich, A.; Miga, K.H.; Pevzner, P.A. TandemTools: Mapping long reads and assessing/improving assembly quality in extra-long tandem repeats. Bioinformatics 2020, 36, i75–i83. [Google Scholar] [CrossRef] [PubMed]
- Novák, P.; Ávila Robledillo, L.; Koblížková, A.; Vrbová, I.; Neumann, P.; Macas, J. TAREAN: A computational tool for identification and characterization of satellite DNA from unassembled short reads. Nucleic Acids Res. 2017, 45, e111. [Google Scholar] [CrossRef] [PubMed]
- Novák, P.; Neumann, P.; Macas, J. Graph-based clustering and characterization of repetitive sequences in next-generation sequencing data. BMC Bioinform. 2010, 11, 378. [Google Scholar] [CrossRef] [Green Version]
- Novak, P.; Neumann, P.; Pech, J.; Steinhaisl, J.; Macas, J. RepeatExplorer: A Galaxy-based web server for genome-wide characterization of eukaryotic repetitive elements from next-generation sequence reads. Bioinformatics 2013, 29, 792–793. [Google Scholar] [CrossRef] [Green Version]
- Pech, M.; Igo-Kemenes, T.; Zachau, H.G. Nucleotide sequence of a highly repetitive component of rat DNA. Nucleic Acids Res. 1979, 7, 417–432. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
| Repeat Unit Size | Identified Subfamilies | HOR Formation | Chromosomal Presence | Genome Representativity | ||
|---|---|---|---|---|---|---|
| αSAT | 171 bp | SFs; 28 identified (e.g., pTRA-1/2/4/7) | ✓ | All | 3–5% | AT-rich |
| SATI | 42 bp | pTRI-6 | ✓ | 3; 4; All acrocentric | 0.12% | |
| SATII | 5 bp | 3 mentioned, no name identified | ✓ | 11; 2; 5; 7; 10; 13–17; 21; 22 | 1.5% (together w/ SATIII) | GC-rich |
| SATIII | 5 bp | pTRS-47; pTRS-63; | ✓ | Y; 1; 3–5; 7; 92; 10; 13–18 ;20–22 | 1.5% (together w/ SATII) | |
| pTR9-s3; pTRS-2; | ||||||
| pE-1/2; pR-1/2/4; | ||||||
| pK-1; pW-1 | ||||||
| βSAT | 68 bp | pB3/4 | ✓ | Y; 1; 3; 9; 19; All acrocentric | 0.02% | |
| γSAT | 220 bp | GSAT; GSATX; GSATII | - | All | 0.13% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lopes, M.; Louzada, S.; Gama-Carvalho, M.; Chaves, R. Genomic Tackling of Human Satellite DNA: Breaking Barriers through Time. Int. J. Mol. Sci. 2021, 22, 4707. https://doi.org/10.3390/ijms22094707
Lopes M, Louzada S, Gama-Carvalho M, Chaves R. Genomic Tackling of Human Satellite DNA: Breaking Barriers through Time. International Journal of Molecular Sciences. 2021; 22(9):4707. https://doi.org/10.3390/ijms22094707
Chicago/Turabian StyleLopes, Mariana, Sandra Louzada, Margarida Gama-Carvalho, and Raquel Chaves. 2021. "Genomic Tackling of Human Satellite DNA: Breaking Barriers through Time" International Journal of Molecular Sciences 22, no. 9: 4707. https://doi.org/10.3390/ijms22094707
APA StyleLopes, M., Louzada, S., Gama-Carvalho, M., & Chaves, R. (2021). Genomic Tackling of Human Satellite DNA: Breaking Barriers through Time. International Journal of Molecular Sciences, 22(9), 4707. https://doi.org/10.3390/ijms22094707