
Muscarinic Receptors and BK Channels Are Affected by Lipid Raft Disruption of Salivary Gland Cells

 , , and
, , and 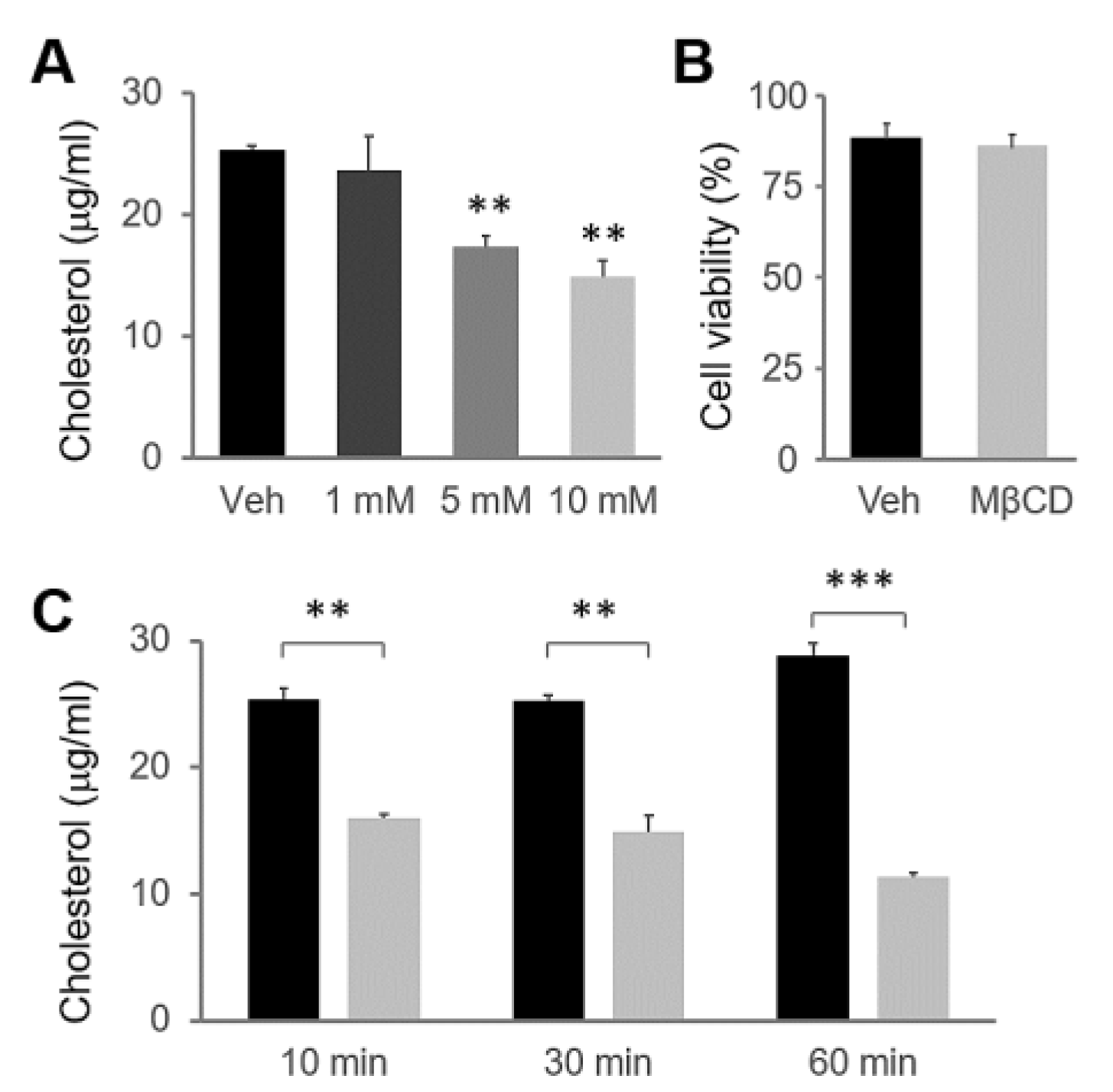{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. MβCD Preincubation Induced Cholesterol Depletion in HSG Cells
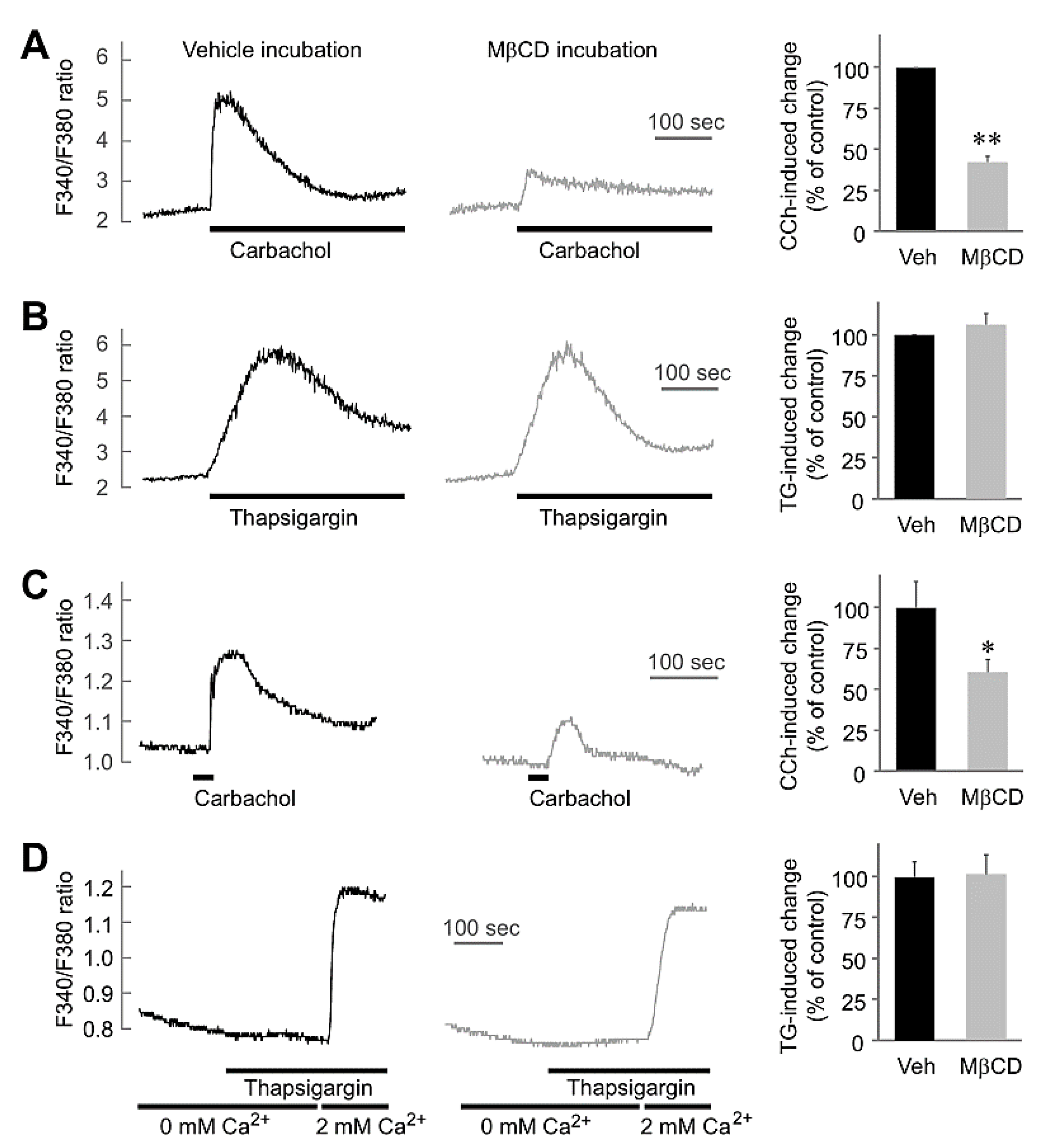
2.2. MβCD Preincubation-Inhibited Muscarinic [Ca2+]i Increases without Any Effect on Thapsigargin-Mediated [Ca2+]i in Salivary Gland Cells
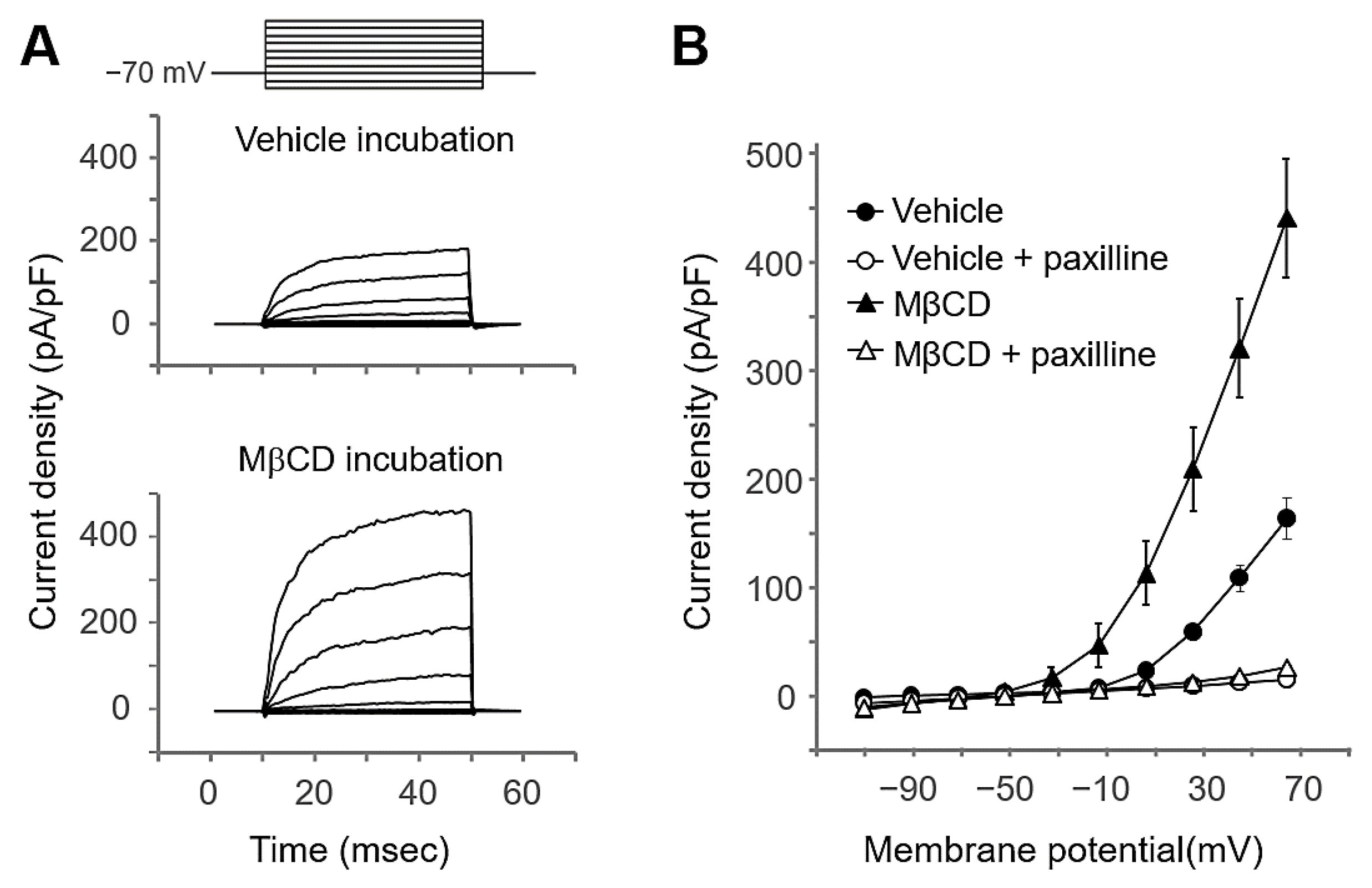
2.3. MβCD Preincubation Increased the BK Channel Activity in Salivary Gland Cells
2.4. MβCD Preincubation Did Not Directly Affect AQP5 Translocation in Salivary Gland Cells
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Cell Preparation
4.3. Cytosolic Free Ca2+ Measurement
4.4. Cholesterol Measurement
4.5. Cell Viability Assay
4.6. Electrophysiological Recording
4.7. Preparation of Membrane Fractions and Western Blotting
4.8. Analysis of AQP5 Protein Expression by Flow Cytometry
4.9. Data Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Dartt, D.A. Neural regulation of lacrimal gland secretory processes: Relevance in dry eye diseases. Prog. Retin. Eye Res. 2009, 28, 155–177. [Google Scholar] [CrossRef] [Green Version]
- Proctor, G.B.; Carpenter, G.H. Regulation of salivary gland function by autonomic nerves. Auton. Neurosci. 2007, 133, 3–18. [Google Scholar] [CrossRef] [PubMed]
- Brown, D.; Hasler, U.; Nunes, P.; Bouley, R.; Lu, H.A. Phosphorylation events and the modulation of aquaporin 2 cell surface expression. Curr. Opin. Nephrol. Hypertens. 2008, 17, 491–498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kitchen, P.; Day, R.E.; Salman, M.M.; Conner, M.T.; Bill, R.M.; Conner, A.C. Beyond water homeostasis: Diverse functional roles of mammalian aquaporins. Biochim. Biophys. Acta 2015, 1850, 2410–2421. [Google Scholar] [CrossRef] [Green Version]
- Olesen, E.T.; Fenton, R.A. Aquaporin-2 membrane targeting: Still a conundrum. Am. J. Physiol. Renal Physiol. 2017, 312, F744–F747. [Google Scholar] [CrossRef] [Green Version]
- King, L.S.; Kozono, D.; Agre, P. From structure to disease: The evolving tale of aquaporin biology. Nat. Rev. Mol. Cell Biol. 2004, 5, 687–698. [Google Scholar] [CrossRef]
- Valenti, G.; Procino, G.; Tamma, G.; Carmosino, M.; Svelto, M. Minireview: Aquaporin 2 trafficking. Endocrinology 2005, 146, 5063–5070. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delporte, C. Aquaporins in salivary glands and pancreas. Biochim. Biophys. Acta 2014, 1840, 1524–1532. [Google Scholar] [CrossRef] [PubMed]
- Tada, J.; Sawa, T.; Yamanaka, N.; Shono, M.; Akamatsu, T.; Tsumura, K.; Parvin, M.N.; Kanamori, N.; Hosoi, K. Involvement of vesicle-cytoskeleton interaction in AQP5 trafficking in AQP5-gene-transfected HSG cells. Biochem. Biophys. Res. Commun. 1999, 266, 443–447. [Google Scholar] [CrossRef] [PubMed]
- Aureli, M.; Grassi, S.; Prioni, S.; Sonnino, S.; Prinetti, A. Lipid membrane domains in the brain. Biochim. Biophys. Acta 2015, 1851, 1006–1016. [Google Scholar] [CrossRef]
- Sebastiao, A.M.; Colino-Oliveira, M.; Assaife-Lopes, N.; Dias, R.B.; Ribeiro, J.A. Lipid rafts, synaptic transmission and plasticity: Impact in age-related neurodegenerative diseases. Neuropharmacology 2013, 64, 97–107. [Google Scholar] [CrossRef]
- Simons, K.; Sampaio, J.L. Membrane organization and lipid rafts. Cold Spring Harb. Perspect. Biol. 2011, 3, a004697. [Google Scholar] [CrossRef]
- Schuck, S.; Simons, K. Polarized sorting in epithelial cells: Raft clustering and the biogenesis of the apical membrane. J. Cell Sci. 2004, 117 Pt 25, 5955–5964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allen, J.A.; Halverson-Tamboli, R.A.; Rasenick, M.M. Lipid raft microdomains and neurotransmitter signalling. Nat. Rev. Neurosci. 2007, 8, 128–140. [Google Scholar] [CrossRef]
- Ambudkar, I.S. Calcium signalling in salivary gland physiology and dysfunction. J. Physiol. 2016, 594, 2813–2824. [Google Scholar] [CrossRef] [Green Version]
- Pani, B.; Liu, X.; Bollimuntha, S.; Cheng, K.T.; Niesman, I.R.; Zheng, C.; Achen, V.R.; Patel, H.H.; Ambudkar, I.S.; Singh, B.B. Impairment of TRPC1-STIM1 channel assembly and AQP5 translocation compromise agonist-stimulated fluid secretion in mice lacking caveolin1. J. Cell Sci. 2013, 126 Pt 2, 667–675. [Google Scholar] [CrossRef] [Green Version]
- Romanenko, V.G.; Nakamoto, T.; Srivastava, A.; Begenisich, T.; Melvin, J.E. Regulation of membrane potential and fluid secretion by Ca2+-activated K+ channels in mouse submandibular glands. J Physiol. 2007, 581 Pt 2, 801–817. [Google Scholar] [CrossRef]
- Ishikawa, Y.; Cho, G.; Yuan, Z.; Inoue, N.; Nakae, Y. Aquaporin-5 water channel in lipid rafts of rat parotid glands. Biochim. Biophys. Acta 2006, 1758, 1053–1060. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishikawa, Y.; Yuan, Z.; Inoue, N.; Skowronski, M.T.; Nakae, Y.; Shono, M.; Cho, G.; Yasui, M.; Agre, P.; Nielsen, S. Identification of AQP5 in lipid rafts and its translocation to apical membranes by activation of M3 mAChRs in interlobular ducts of rat parotid gland. Am. J. Physiol. Cell Physiol. 2005, 289, C1303–C1311. [Google Scholar] [CrossRef]
- Koudinov, A.R.; Koudinova, N.V. Essential role for cholesterol in synaptic plasticity and neuronal degeneration. FASEB J. 2001, 15, 1858–1860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, M.; Hwang, S.M.; Koo, N.Y.; Kim, B.; Kho, H.S.; Choi, S.Y.; Song, Y.W.; Park, K. Autoantibodies in Sjogren’s syndrome patients acutely inhibit muscarinic receptor function. Oral Dis. 2012, 18, 132–139. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.H.; Gauna, A.E.; Perez, G.; Park, Y.J.; Pauley, K.M.; Kawai, T.; Cha, S. Autoantibodies against muscarinic type 3 receptor in Sjogren’s syndrome inhibit aquaporin 5 trafficking. PLoS ONE 2013, 8, e53113. [Google Scholar] [CrossRef] [PubMed]
- Caliceti, C.; Zambonin, L.; Prata, C.; Vieceli Dalla Sega, F.; Hakim, G.; Hrelia, S.; Fiorentini, D. Effect of plasma membrane cholesterol depletion on glucose transport regulation in leukemia cells. PLoS ONE 2012, 7, e41246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bar-On, P.; Rockenstein, E.; Adame, A.; Ho, G.; Hashimoto, M.; Masliah, E. Effects of the cholesterol-lowering compound methyl-beta-cyclodextrin in models of alpha-synucleinopathy. J. Neurochem. 2006, 98, 1032–1045. [Google Scholar] [CrossRef]
- Dopico, A.M.; Bukiya, A.N.; Singh, A.K. Large conductance, calcium- and voltage-gated potassium (BK) channels: Regulation by cholesterol. Pharmacol. Ther. 2012, 135, 133–150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delporte, C.; Steinfeld, S. Distribution and roles of aquaporins in salivary glands. Biochim. Biophys. Acta 2006, 1758, 1061–1070. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.H.; Park, S.H.; Moon, Y.W.; Hwang, S.; Kim, D.; Jo, S.H.; Oh, S.B.; Kim, J.S.; Jahng, J.W.; Lee, J.H.; et al. Histamine H1 receptor induces cytosolic calcium increase and aquaporin translocation in human salivary gland cells. J. Pharmacol. Exp. Ther. 2009, 330, 403–412. [Google Scholar] [CrossRef]
- Lee, K.; Choi, S.; Choi, L.M.; Lee, J.; Kim, J.H.; Chung, G.; Lee, G.; Choi, S.Y.; Park, K. Desipramine inhibits salivary Ca(2+) signaling and aquaporin translocation. Oral Dis. 2015, 21, 530–535. [Google Scholar] [CrossRef]
- Jin, M.; Hwang, S.M.; Davies, A.J.; Shin, Y.; Bae, J.S.; Lee, J.H.; Lee, E.B.; Song, Y.W.; Park, K. Autoantibodies in primary Sjogren’s syndrome patients induce internalization of muscarinic type 3 receptors. Biochim. Biophys. Acta 2012, 1822, 161–167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oldfield, S.; Hancock, J.; Mason, A.; Hobson, S.A.; Wynick, D.; Kelly, E.; Randall, A.D.; Marrion, N.V. Receptor-mediated suppression of potassium currents requires colocalization within lipid rafts. Mol. Pharmacol. 2009, 76, 1279–1289. [Google Scholar] [CrossRef] [Green Version]
- Weaver, A.K.; Olsen, M.L.; McFerrin, M.B.; Sontheimer, H. BK channels are linked to inositol 1,4,5-triphosphate receptors via lipid rafts: A novel mechanism for coupling [Ca(2+)](i) to ion channel activation. J. Biol. Chem. 2007, 282, 31558–31568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tajima, N.; Itokazu, Y.; Korpi, E.R.; Somerharju, P.; Kakela, R. Activity of BK(Ca) channel is modulated by membrane cholesterol content and association with Na+/K+-ATPase in human melanoma IGR39 cells. J. Biol. Chem. 2011, 286, 5624–5638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riddle, M.A.; Hughes, J.M.; Walker, B.R. Role of caveolin-1 in endothelial BKCa channel regulation of vasoreactivity. Am. J. Physiol. Cell Physiol. 2011, 301, C1404–C1414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Besshoh, S.; Bawa, D.; Teves, L.; Wallace, M.C.; Gurd, J.W. Increased phosphorylation and redistribution of NMDA receptors between synaptic lipid rafts and post-synaptic densities following transient global ischemia in the rat brain. J. Neurochem. 2005, 93, 186–194. [Google Scholar] [CrossRef]
- Delint-Ramirez, I.; Salcedo-Tello, P.; Bermudez-Rattoni, F. Spatial memory formation induces recruitment of NMDA receptor and PSD-95 to synaptic lipid rafts. J. Neurochem. 2008, 106, 1658–1668. [Google Scholar] [CrossRef]
- Hering, H.; Lin, C.C.; Sheng, M. Lipid rafts in the maintenance of synapses, dendritic spines, and surface AMPA receptor stability. J. Neurosci. 2003, 23, 3262–3271. [Google Scholar] [CrossRef] [Green Version]
- Choi, T.Y.; Jung, S.; Nah, J.; Ko, H.Y.; Jo, S.H.; Chung, G.; Park, K.; Jung, Y.K.; Choi, S.Y. Low levels of methyl beta-cyclodextrin disrupt GluA1-dependent synaptic potentiation but not synaptic depression. J. Neurochem. 2015, 132, 276–285. [Google Scholar] [CrossRef] [Green Version]
- Dionisio, N.; Galan, C.; Jardin, I.; Salido, G.M.; Rosado, J.A. Lipid rafts are essential for the regulation of SOCE by plasma membrane resident STIM1 in human platelets. Biochim. Biophys. Acta 2011, 1813, 431–437. [Google Scholar] [CrossRef] [Green Version]
- Galan, C.; Woodard, G.E.; Dionisio, N.; Salido, G.M.; Rosado, J.A. Lipid rafts modulate the activation but not the maintenance of store-operated Ca(2+) entry. Biochim. Biophys. Acta 2010, 1803, 1083–1093. [Google Scholar] [CrossRef] [Green Version]
- Jardin, I.; Salido, G.M.; Rosado, J.A. Role of lipid rafts in the interaction between hTRPC1, Orai1 and STIM1. Channels 2008, 2, 401–403. [Google Scholar] [CrossRef] [Green Version]
- Cheng, K.T.; Liu, X.; Ong, H.L.; Ambudkar, I.S. Functional requirement for Orai1 in store-operated TRPC1-STIM1 channels. J. Biol. Chem. 2008, 283, 12935–12940. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, K.; Kim, Y.J.; Choi, L.M.; Choi, S.; Nam, H.; Ko, H.Y.; Chung, G.; Lee, J.H.; Jo, S.H.; Lee, G.; et al. Human salivary gland cells express bradykinin receptors that modulate the expression of proinflammatory cytokines. Eur. J. Oral Sci. 2017, 125, 18–27. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Zhang, G.; Cui, J. BK channels: Multiple sensors, one activation gate. Front. Physiol. 2015, 6, 29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bukiya, A.N.; Vaithianathan, T.; Kuntamallappanavar, G.; Asuncion-Chin, M.; Dopico, A.M. Smooth muscle cholesterol enables BK beta1 subunit-mediated channel inhibition and subsequent vasoconstriction evoked by alcohol. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 2410–2423. [Google Scholar] [CrossRef] [Green Version]
- Bukiya, A.N.; Belani, J.D.; Rychnovsky, S.; Dopico, A.M. Specificity of cholesterol and analogs to modulate BK channels points to direct sterol-channel protein interactions. J. Gen. Physiol. 2011, 137, 93–110. [Google Scholar] [CrossRef] [Green Version]
- Asakura, K.; Ueda, A.; Shima, S.; Ishikawa, T.; Hikichi, C.; Hirota, S.; Fukui, T.; Ito, S.; Mutoh, T. Targeting of aquaporin 4 into lipid rafts and its biological significance. Brain Res. 2014, 1583, 237–244. [Google Scholar] [CrossRef]
- Sirtori, C.R. The pharmacology of statins. Pharmacol. Res. 2014, 88, 3–11. [Google Scholar] [CrossRef]
- Habbab, K.M.; Moles, D.R.; Porter, S.R. Potential oral manifestations of cardiovascular drugs. Oral Dis. 2010, 16, 769–773. [Google Scholar] [CrossRef]
- Pascual Cruz, M.; Chimenos Kustner, E.; Garcia Vicente, J.A.; Mezquiriz Ferrero, X.; Borrell Thio, E.; Lopez Lopez, J. Adverse side effects of statins in the oral cavity. Med. Oral Patol. Oral Cir. Bucal. 2008, 13, E98–E101. [Google Scholar]
- Shirasuna, K.; Sato, M.; Miyazaki, T. A neoplastic epithelial duct cell line established from an irradiated human salivary gland. Cancer 1981, 48, 745–752. [Google Scholar] [CrossRef]
- Wang, C.S.; Wee, Y.; Yang, C.H.; Melvin, J.E.; Baker, O.J. ALX/FPR2 Modulates Anti-Inflammatory Responses in Mouse Submandibular Gland. Sci. Rep. 2016, 6, 24244. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, J.; Kim, Y.-J.; Choi, L.-M.; Lee, K.; Park, H.-K.; Choi, S.-Y. Muscarinic Receptors and BK Channels Are Affected by Lipid Raft Disruption of Salivary Gland Cells. Int. J. Mol. Sci. 2021, 22, 4780. https://doi.org/10.3390/ijms22094780
Lee J, Kim Y-J, Choi L-M, Lee K, Park H-K, Choi S-Y. Muscarinic Receptors and BK Channels Are Affected by Lipid Raft Disruption of Salivary Gland Cells. International Journal of Molecular Sciences. 2021; 22(9):4780. https://doi.org/10.3390/ijms22094780
Chicago/Turabian StyleLee, Jisoo, Yoon-Jung Kim, La-Mee Choi, Keimin Lee, Hee-Kyung Park, and Se-Young Choi. 2021. "Muscarinic Receptors and BK Channels Are Affected by Lipid Raft Disruption of Salivary Gland Cells" International Journal of Molecular Sciences 22, no. 9: 4780. https://doi.org/10.3390/ijms22094780
APA StyleLee, J., Kim, Y. -J., Choi, L. -M., Lee, K., Park, H. -K., & Choi, S. -Y. (2021). Muscarinic Receptors and BK Channels Are Affected by Lipid Raft Disruption of Salivary Gland Cells. International Journal of Molecular Sciences, 22(9), 4780. https://doi.org/10.3390/ijms22094780