
Novel Seleno-Aspirinyl Compound AS-10 Induces Apoptosis, G1 Arrest of Pancreatic Ductal Adenocarcinoma Cells, Inhibits Their NF-κB Signaling, and Synergizes with Gemcitabine Cytotoxicity

 ,
,  ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
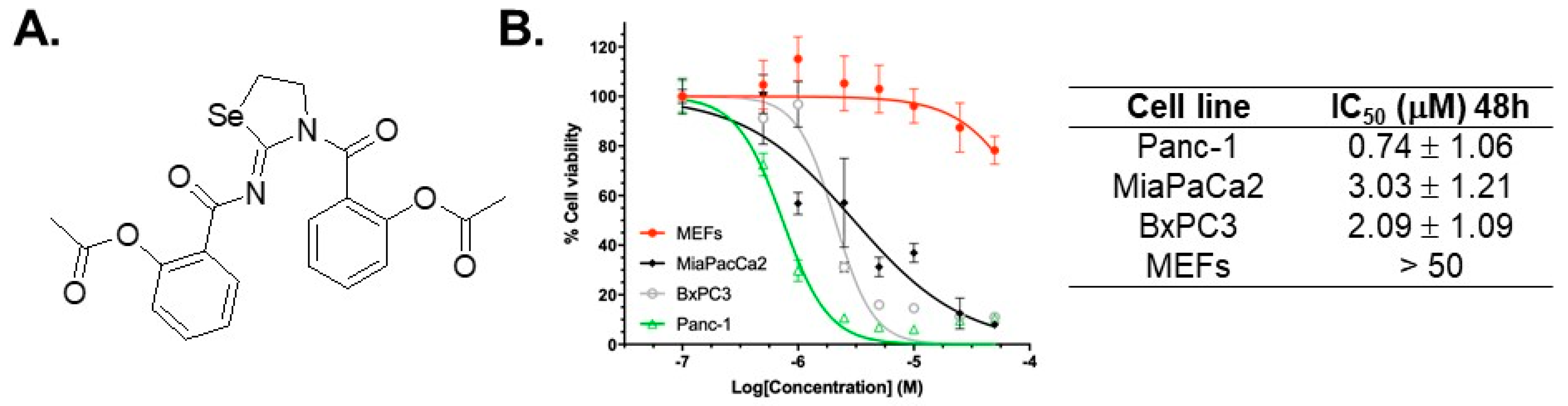
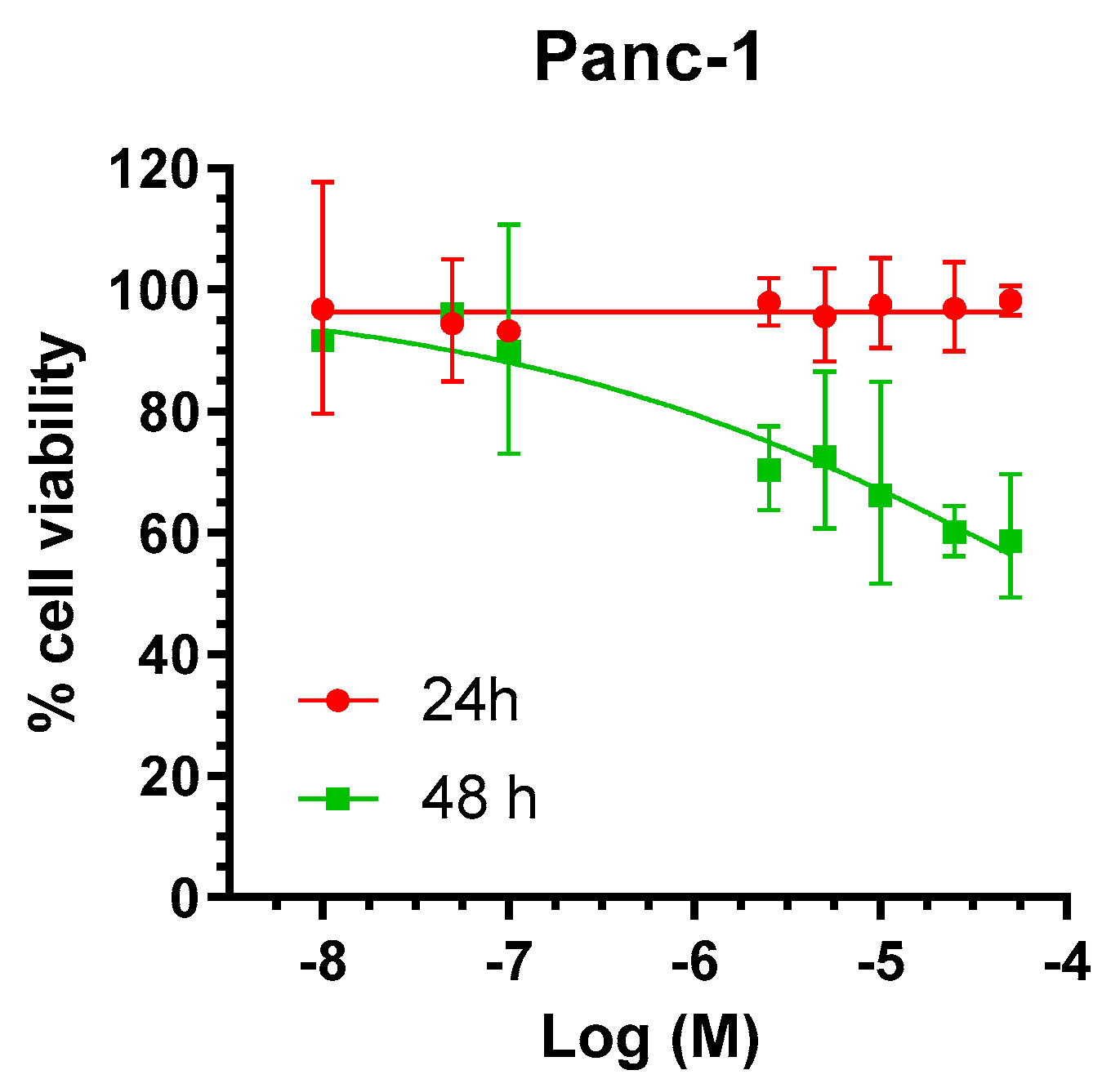
2.1. Selectivity of AS-10 to Inhibit PDAC Cell Growth Over Non-Malignant Cells
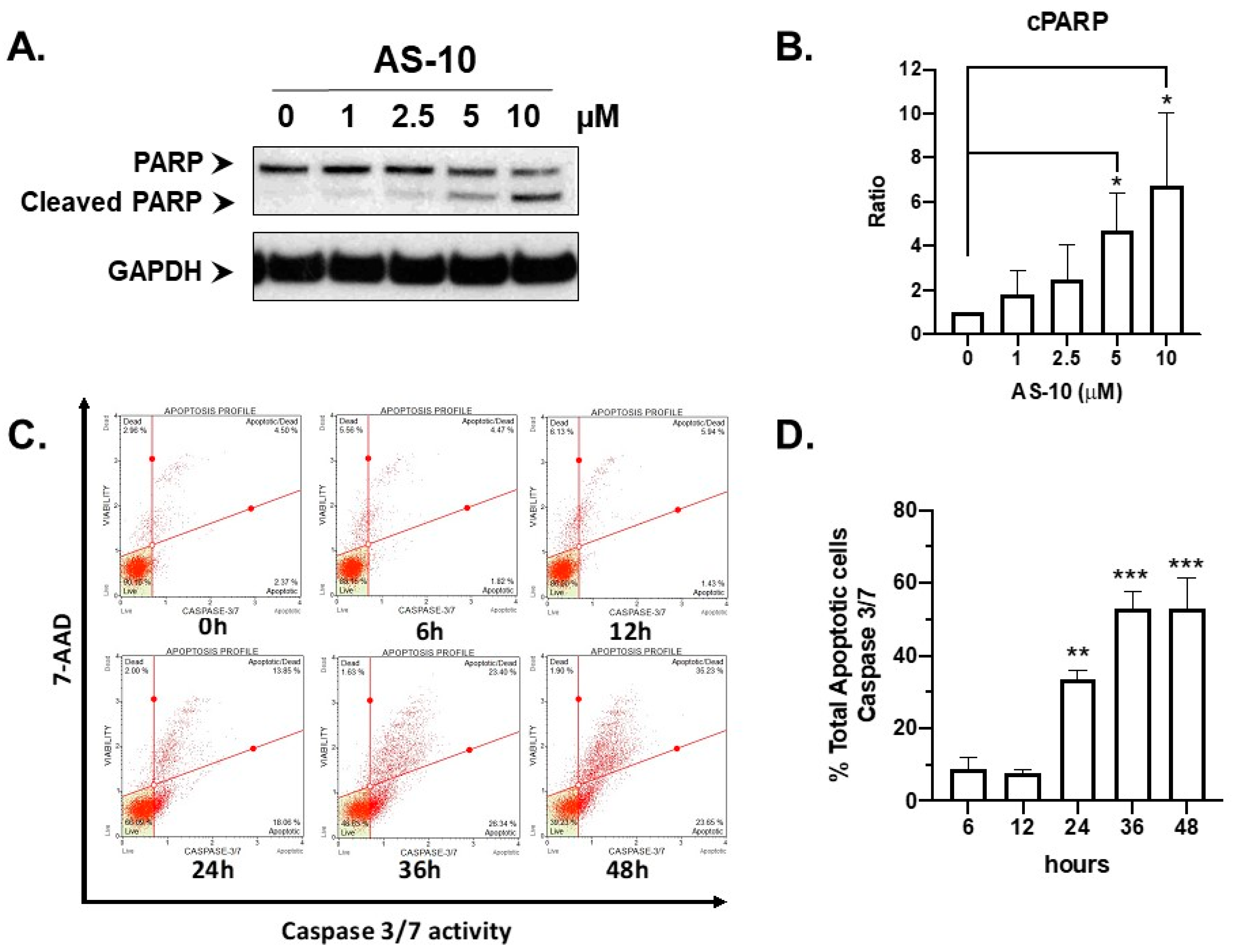
2.2. AS-10 Induced Apoptotic Cell Death in Panc-1 Cells
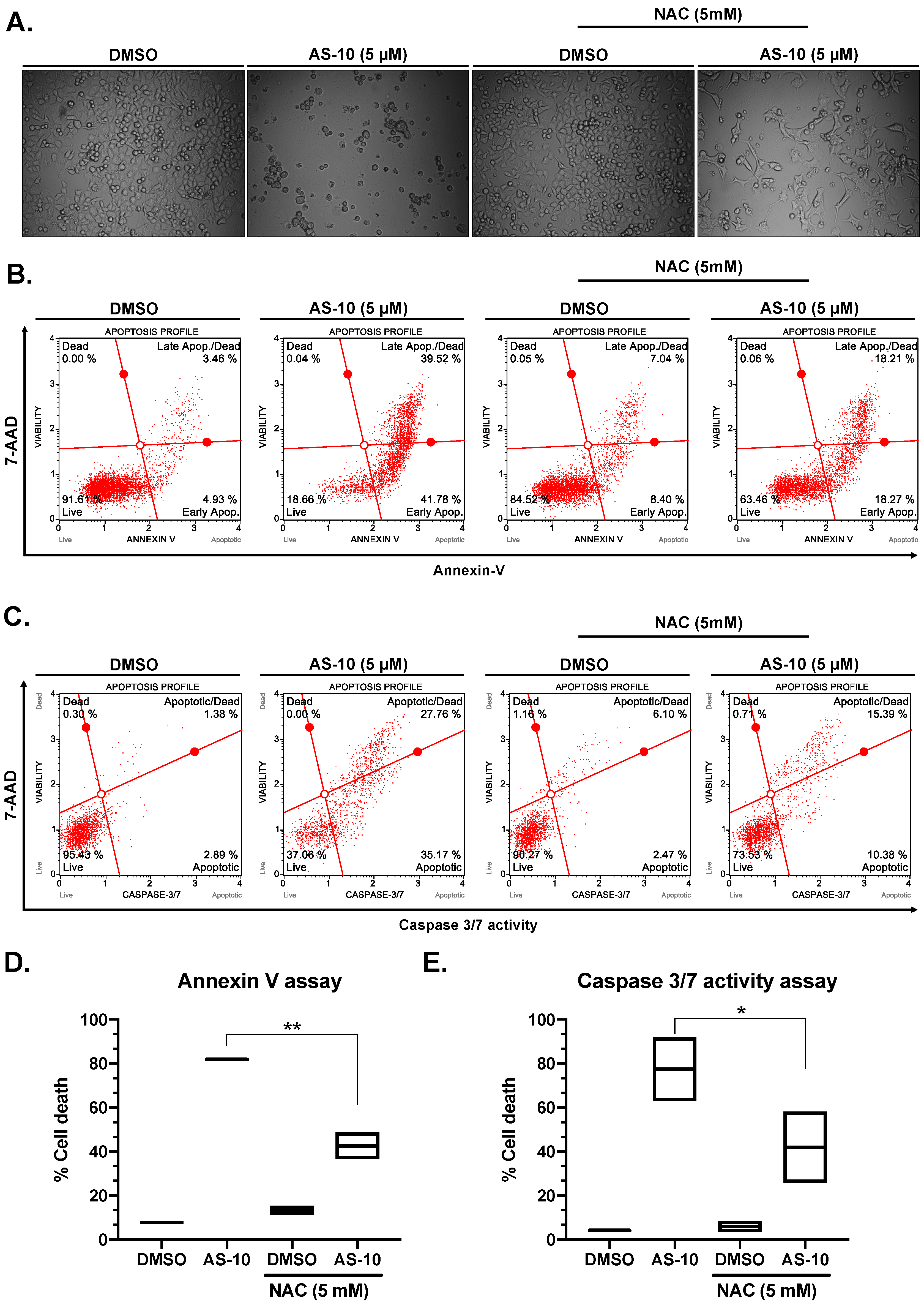
2.3. N-Acetyl Cysteine (NAC) Attenuated AS-10-Induced Apoptosis of Panc-1 Cells
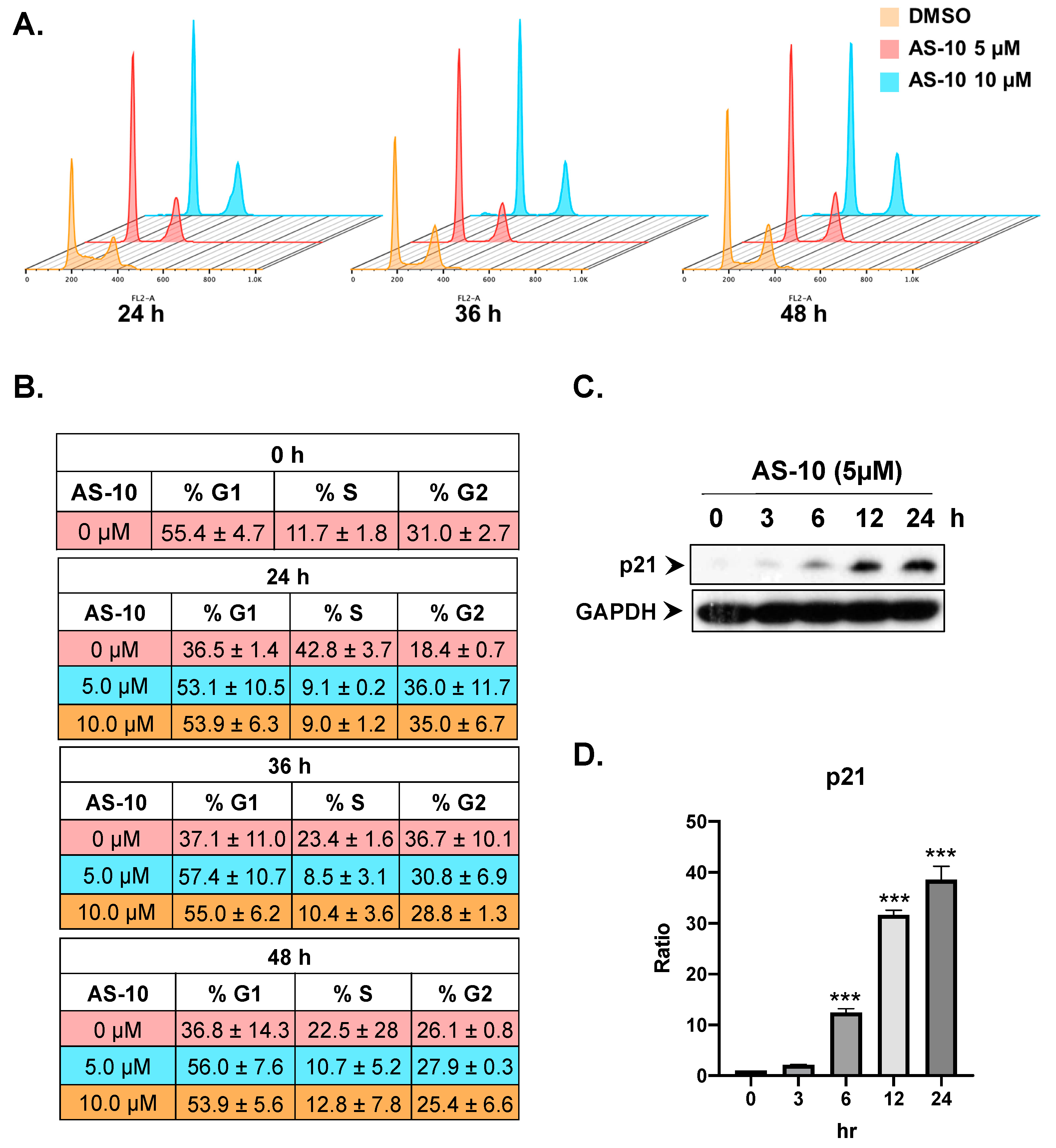
2.4. AS-10 Induced G1/G0 Cell Cycle Arrest in Panc-1 Cells
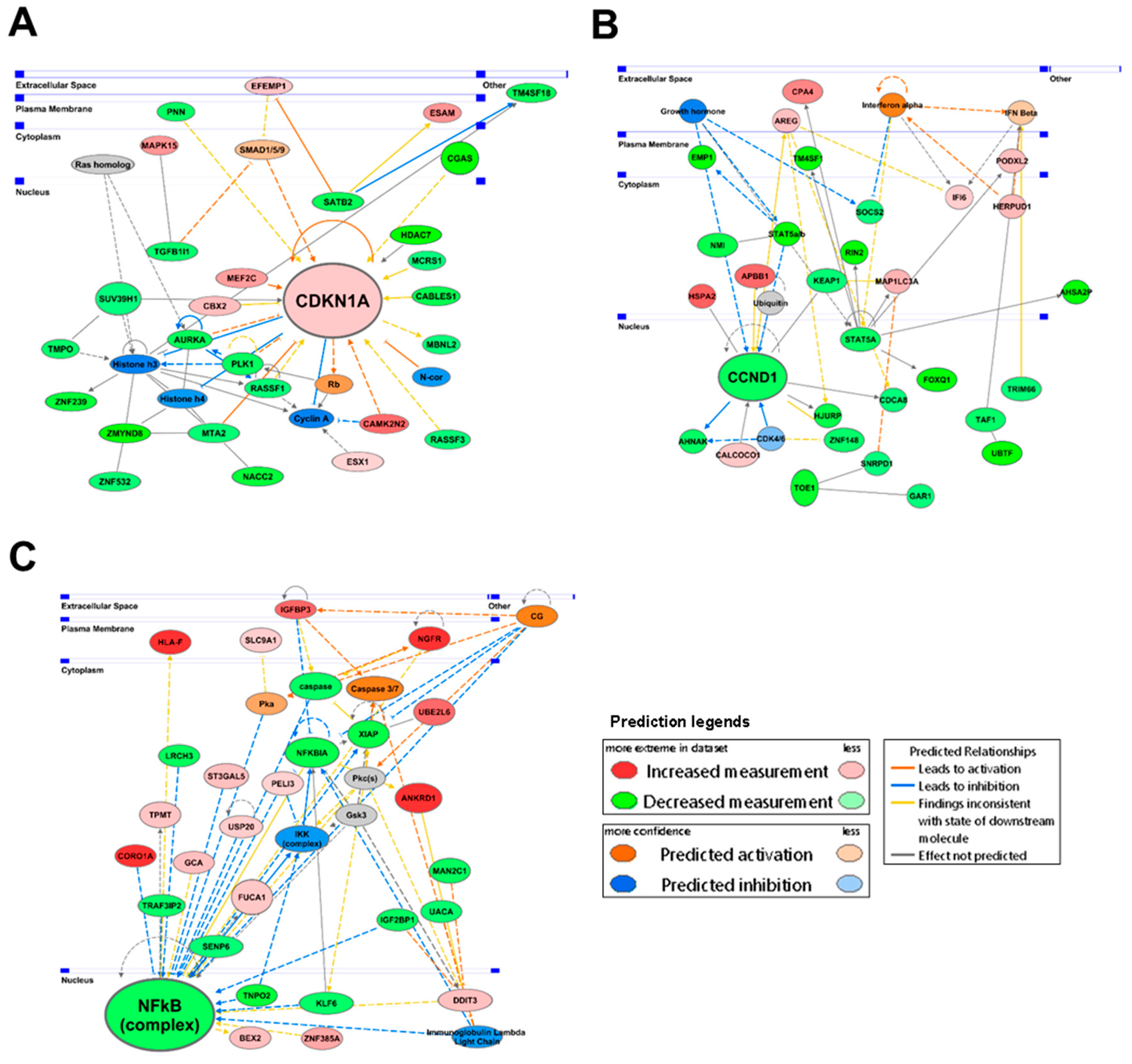
2.5. Transcriptomic Profiling of AS-10 Effects on Panc-1 Cells by RNA-seq
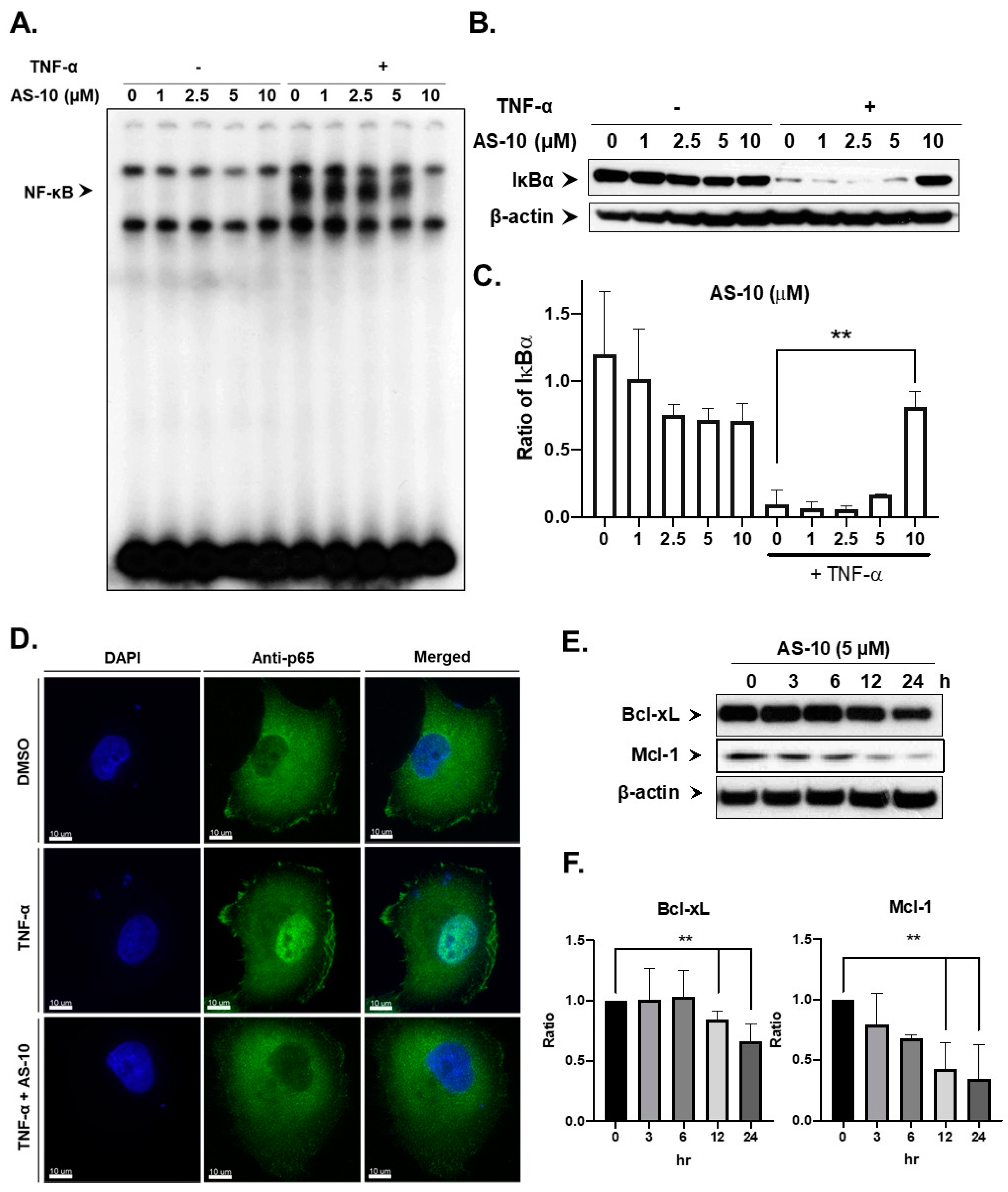
2.6. AS-10 Inhibited Activation of NF-ĸB by the Proinflammatory Cytokine TNF-α in Panc-1 Cells
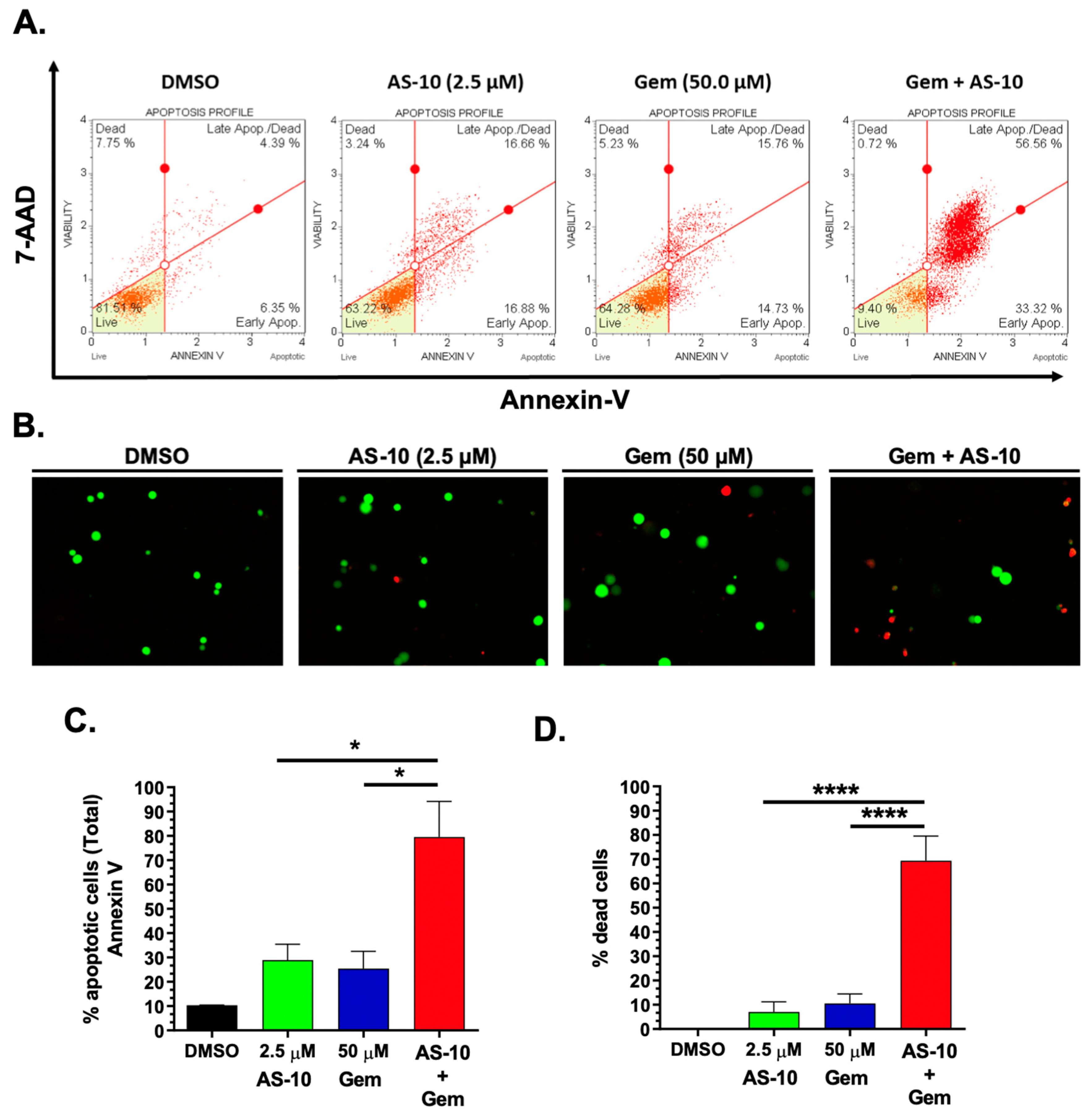
2.7. AS-10 Potentiated the Cytotoxic Effects of Gemcitabine In Vitro
3. Discussion
4. Materials and Methods
4.1. Synthesis and Characterization of 2-((3-(2-acetoxybenzoyl)-1,3-selenazolidin-2-ylidene)carbamoyl)phenyl Acetate (AS-10)
4.2. Reagents
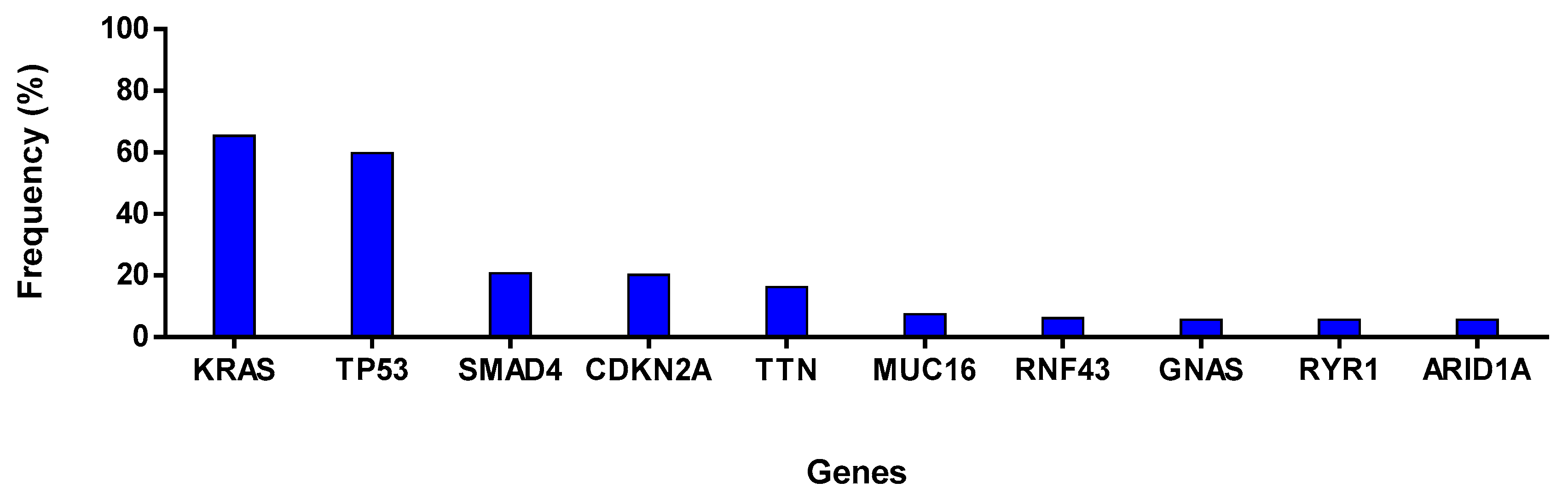
4.3. cBioPortal Database Analysis
4.4. Cell Culture
4.5. Cell Viability Assay
4.6. NAC Studies
4.7. Cell Cycle Analysis
4.8. Western Blot Analysis
4.9. Caspase 3/7 Activity Assay
4.10. Annexin V Assay
4.11. Exploratory RNA Sequencing (RNA-seq), Data Processing, and Data Analysis
4.12. Electrophoretic Mobility Shift Assay (EMSA) for NK-ĸB DNA Binding
4.13. Immunocytochemistry for NF-κB P65 Localization
4.14. Live/Dead Assay with the Viability/Cytotoxicity Kit (L3224, Life Technologies, USA)
4.15. Statistical Analyses
5. Conclusions
6. Patents
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ASA | Acetyl salicylic acid, i.e., Aspirin |
| PDAC | Pancreatic ductal adenocarcinoma |
| NAC | N-acetyl cysteine |
| NSAID | Non-steroidal anti-inflammatory drug |
| PS | Phosphatidylserine |
| EMSA | Electro mobility shift assay |
| MEFs | Mouse embryonic fibroblasts |
| PE | Plating efficiency |
| GI | Gastrointestinal |
| PI | Propidium iodide |
| HDAC | Histone deacetylase |
| 7-AAD | 7-Aminoactinomycin D |
| HER2 | Receptor tyrosine-protein kinase erbB-2 |
| MMPs | Matrix metalloproteinases |
| VEGF | Vascular endothelial growth factor |
| EGFR | Epidermal growth factor receptor |
| MTT | 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide |
Appendix A



References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2019. CA Cancer J. Clin. 2019, 69, 7–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noone, A.M.; Howlader, N.; Krapcho, M.; Miller, D.; Brest, A.; Yu, M.; Ruhl, J.; Tatalovich, Z.; Mariotto, A.; Lewis, D.R.; et al. SEER Cancer Statistics Review, 1975–2015. Available online: https://seer.cancer.gov/csr/1975_2015/ (accessed on 21 August 2018).
- Berlin, J.D.; Catalano, P.; Thomas, J.P.; Kugler, J.W.; Haller, D.G.; Benson, A.B., 3rd. Phase III study of gemcitabine in combination with fluorouracil versus gemcitabine alone in patients with advanced pancreatic carcinoma: Eastern Cooperative Oncology Group Trial E2297. J. Clin. Oncol. 2002, 20, 3270–3275. [Google Scholar] [CrossRef]
- Van Cutsem, E.; van de Velde, H.; Karasek, P.; Oettle, H.; Vervenne, W.L.; Szawlowski, A.; Schoffski, P.; Post, S.; Verslype, C.; Neumann, H.; et al. Phase III trial of gemcitabine plus tipifarnib compared with gemcitabine plus placebo in advanced pancreatic cancer. J. Clin. Oncol. 2004, 22, 1430–1438. [Google Scholar] [CrossRef]
- Safran, H.; Iannitti, D.; Ramanathan, R.; Schwartz, J.D.; Steinhoff, M.; Nauman, C.; Hesketh, P.; Rathore, R.; Wolff, R.; Tantravahi, U.; et al. Herceptin and gemcitabine for metastatic pancreatic cancers that overexpress HER-2/neu. Cancer Investig. 2004, 22, 706–712. [Google Scholar] [CrossRef]
- Bramhall, S.R.; Schulz, J.; Nemunaitis, J.; Brown, P.D.; Baillet, M.; Buckels, J.A. A double-blind placebo-controlled, randomised study comparing gemcitabine and marimastat with gemcitabine and placebo as first line therapy in patients with advanced pancreatic cancer. Br. J. Cancer 2002, 87, 161–167. [Google Scholar] [CrossRef]
- Kindler, H.L.; Friberg, G.; Singh, D.A.; Locker, G.; Nattam, S.; Kozloff, M.; Taber, D.A.; Karrison, T.; Dachman, A.; Stadler, W.M.; et al. Phase II trial of bevacizumab plus gemcitabine in patients with advanced pancreatic cancer. J. Clin. Oncol. 2005, 23, 8033–8040. [Google Scholar] [CrossRef] [PubMed]
- Xiong, H.Q.; Rosenberg, A.; LoBuglio, A.; Schmidt, W.; Wolff, R.A.; Deutsch, J.; Needle, M.; Abbruzzese, J.L. Cetuximab, a monoclonal antibody targeting the epidermal growth factor receptor, in combination with gemcitabine for advanced pancreatic cancer: A multicenter phase II Trial. J. Clin. Oncol. 2004, 22, 2610–2616. [Google Scholar] [CrossRef]
- Philip, P.A.; Benedetti, J.; Corless, C.L.; Wong, R.; O’Reilly, E.M.; Flynn, P.J.; Rowland, K.M.; Atkins, J.N.; Mirtsching, B.C.; Rivkin, S.E.; et al. Phase III study comparing gemcitabine plus cetuximab versus gemcitabine in patients with advanced pancreatic adenocarcinoma: Southwest Oncology Group-directed intergroup trial S0205. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2010, 28, 3605–3610. [Google Scholar] [CrossRef] [Green Version]
- Von Hoff, D.D.; Ervin, T.; Arena, F.P.; Chiorean, E.G.; Infante, J.; Moore, M.; Seay, T.; Tjulandin, S.A.; Ma, W.W.; Saleh, M.N.; et al. Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N. Engl. J. Med. 2013, 369, 1691–1703. [Google Scholar] [CrossRef] [Green Version]
- Conroy, T.; Desseigne, F.; Ychou, M.; Bouche, O.; Guimbaud, R.; Becouarn, Y.; Adenis, A.; Raoul, J.L.; Gourgou-Bourgade, S.; de la Fouchardiere, C.; et al. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N. Engl. J. Med. 2011, 364, 1817–1825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hann, A.; Bohle, W.; Egger, J.; Zoller, W. Feasibility of alternating induction and maintenance chemotherapy in pancreatic cancer. Sci. Rep. 2017, 7, 41549. [Google Scholar] [CrossRef]
- Henriksen, A.; Dyhl-Polk, A.; Chen, I.; Nielsen, D. Checkpoint inhibitors in pancreatic cancer. Cancer Treat. Rev. 2019, 78, 17–30. [Google Scholar] [CrossRef]
- Arlt, A.; Gehrz, A.; Muerkoster, S.; Vorndamm, J.; Kruse, M.L.; Folsch, U.R.; Schafer, H. Role of NF-kappaB and Akt/PI3K in the resistance of pancreatic carcinoma cell lines against gemcitabine-induced cell death. Oncogene 2003, 22, 3243–3251. [Google Scholar] [CrossRef] [Green Version]
- Carbone, C.; Melisi, D. NF-kappaB as a target for pancreatic cancer therapy. Expert Opin. Ther. Targets 2012, 16 (Suppl. 2), S1–S10. [Google Scholar] [CrossRef] [PubMed]
- Gilmore, T.D. Introduction to NF-kappaB: Players, pathways, perspectives. Oncogene 2006, 25, 6680–6684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Streicher, S.A.; Yu, H.; Lu, L.; Kidd, M.S.; Risch, H.A. Case-Control Study of Aspirin Use and Risk of Pancreatic Cancer. Cancer Epidemiol. Biomark. Prev. 2014, 23, 1254–1263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drew, D.A.; Cao, Y.; Chan, A.T. Aspirin and colorectal cancer: The promise of precision chemoprevention. Nat. Rev. Cancer 2016, 16, 173–186. [Google Scholar] [CrossRef] [PubMed]
- Cui, X.J.; He, Q.; Zhang, J.M.; Fan, H.J.; Wen, Z.F.; Qin, Y.R. High-dose aspirin consumption contributes to decreased risk for pancreatic cancer in a systematic review and meta-analysis. Pancreas 2014, 43, 135–140. [Google Scholar] [CrossRef] [PubMed]
- Toruner, M. Aspirin and gastrointestinal toxicity. Anadolu Kardiyol. Derg. AKD Anatol. J. Cardiol. 2007, 7 (Suppl. 2), 27–30. [Google Scholar]
- Huang, Z.; Velazquez, C.A.; Abdellatif, K.R.; Chowdhury, M.A.; Reisz, J.A.; DuMond, J.F.; King, S.B.; Knaus, E.E. Ethanesulfohydroxamic acid ester prodrugs of nonsteroidal anti-inflammatory drugs (NSAIDs): Synthesis, nitric oxide and nitroxyl release, cyclooxygenase inhibition, anti-inflammatory, and ulcerogenicity index studies. J. Med. Chem. 2011, 54, 1356–1364. [Google Scholar] [CrossRef]
- Plano, D.; Karelia, D.N.; Pandey, M.K.; Spallholz, J.E.; Amin, S.; Sharma, A.K. Design, Synthesis, and Biological Evaluation of Novel Selenium (Se-NSAID) Molecules as Anticancer Agents. J. Med. Chem. 2016, 59, 1946–1959. [Google Scholar] [CrossRef]
- Basudhar, D.; Bharadwaj, G.; Cheng, R.Y.; Jain, S.; Shi, S.; Heinecke, J.L.; Holland, R.J.; Ridnour, L.A.; Caceres, V.M.; Spadari-Bratfisch, R.C.; et al. Synthesis and chemical and biological comparison of nitroxyl- and nitric oxide-releasing diazeniumdiolate-based aspirin derivatives. J. Med. Chem. 2013, 56, 7804–7820. [Google Scholar] [CrossRef] [Green Version]
- Lin, T.; Ding, Z.; Li, N.; Xu, J.; Luo, G.; Liu, J.; Shen, J. Seleno-cyclodextrin sensitises human breast cancer cells to TRAIL-induced apoptosis through DR5 induction and NF-kappaB suppression. Eur. J. Cancer 2011, 47, 1890–1907. [Google Scholar] [CrossRef]
- Sharma, A.K.; Amin, S. Post SELECT: Selenium on trial. Future Med. Chem. 2013, 5, 163–174. [Google Scholar] [CrossRef]
- Alcolea, V.; Plano, D.; Karelia, D.N.; Palop, J.A.; Amin, S.; Sanmartin, C.; Sharma, A.K. Novel seleno- and thio-urea derivatives with potent in vitro activities against several cancer cell lines. Eur. J. Med. Chem. 2016, 113, 134–144. [Google Scholar] [CrossRef] [PubMed]
- Sanmartin, C.; Plano, D.; Sharma, A.K.; Palop, J.A. Selenium compounds, apoptosis and other types of cell death: An overview for cancer therapy. Int. J. Mol. Sci. 2012, 13, 9649–9672. [Google Scholar] [CrossRef]
- Abbas, A.M.; Sakr, H.F. Effect of selenium and grape seed extract on indomethacin-induced gastric ulcers in rats. J. Physiol. Biochem. 2013, 69, 527–537. [Google Scholar] [CrossRef]
- Sutaria, D.; Grandhi, B.K.; Thakkar, A.; Wang, J.; Prabhu, S. Chemoprevention of pancreatic cancer using solid-lipid nanoparticulate delivery of a novel aspirin, curcumin and sulforaphane drug combination regimen. Int. J. Oncol. 2012, 41, 2260–2268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deer, E.L.; Gonzalez-Hernandez, J.; Coursen, J.D.; Shea, J.E.; Ngatia, J.; Scaife, C.L.; Firpo, M.A.; Mulvihill, S.J. Phenotype and genotype of pancreatic cancer cell lines. Pancreas 2010, 39, 425–435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor, R.C.; Cullen, S.P.; Martin, S.J. Apoptosis: Controlled demolition at the cellular level. Nat. Rev. Mol. Cell Biol. 2008, 9, 231–241. [Google Scholar] [CrossRef] [PubMed]
- Trachootham, D.; Alexandre, J.; Huang, P. Targeting cancer cells by ROS-mediated mechanisms: A radical therapeutic approach? Nature reviews. Drug Discov. 2009, 8, 579–591. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhang, X.; Song, Z.; Gu, F. An anti-CAPN5 intracellular antibody acts as an inhibitor of CAPN5-mediated neuronal degeneration. Oncotarget 2017, 8, 100312–100325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Voges, D.; Zwickl, P.; Baumeister, W. The 26S Proteasome: A Molecular Machine Designed for Controlled Proteolysis. Annu. Rev. Biochem. 1999, 68, 1015–1068. [Google Scholar] [CrossRef]
- Stiewe, T.; Putzer, B.M. Role of p73 in malignancy: Tumor suppressor or oncogene? Cell Death Differ. 2002, 9, 237–245. [Google Scholar] [CrossRef] [Green Version]
- Guerrero-Martinez, J.A.; Reyes, J.C. High expression of SMARCA4 or SMARCA2 is frequently associated with an opposite prognosis in cancer. Sci. Rep. 2018, 8, 2043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Falvo, J.V.; Tsytsykova, A.V.; Goldfeld, A.E. Transcriptional control of the TNF gene. TNF Pathophysiol. 2010, 11, 27–60. [Google Scholar] [CrossRef] [Green Version]
- Voutsadakis, I.A. Molecular predictors of gemcitabine response in pancreatic cancer. World J. Gastrointest. Oncol. 2011, 3, 153–164. [Google Scholar] [CrossRef]
- Dhillon, N.; Aggarwal, B.B.; Newman, R.A.; Wolff, R.A.; Kunnumakkara, A.B.; Abbruzzese, J.L.; Ng, C.S.; Badmaev, V.; Kurzrock, R. Phase II trial of curcumin in patients with advanced pancreatic cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2008, 14, 4491–4499. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Abbruzzese, J.L.; Evans, D.B.; Larry, L.; Cleary, K.R.; Chiao, P.J. The nuclear factor-kappa B RelA transcription factor is constitutively activated in human pancreatic adenocarcinoma cells. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 1999, 5, 119–127. [Google Scholar]
- Zhang, Y.; Liu, L.; Fan, P.; Bauer, N.; Gladkich, J.; Ryschich, E.; Bazhin, A.V.; Giese, N.A.; Strobel, O.; Hackert, T.; et al. Aspirin counteracts cancer stem cell features, desmoplasia and gemcitabine resistance in pancreatic cancer. Oncotarget 2015, 6, 9999–10015. [Google Scholar] [CrossRef] [Green Version]
- Schumacker, P.T. Reactive oxygen species in cancer cells: Live by the sword, die by the sword. Cancer Cell 2006, 10, 175–176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaltiel, I.A.; Krenning, L.; Bruinsma, W.; Medema, R.H. The same, only different—DNA damage checkpoints and their reversal throughout the cell cycle. J. Cell Sci. 2015, 128, 607–620. [Google Scholar] [CrossRef] [Green Version]
- Marra, D.E.; Simoncini, T.; Liao, J.K. Inhibition of vascular smooth muscle cell proliferation by sodium salicylate mediated by upregulation of p21(Waf1) and p27(Kip1). Circulation 2000, 102, 2124–2130. [Google Scholar] [CrossRef] [Green Version]
- Dikshit, P.; Chatterjee, M.; Goswami, A.; Mishra, A.; Jana, N.R. Aspirin induces apoptosis through the inhibition of proteasome function. J. Biol. Chem. 2006, 281, 29228–29235. [Google Scholar] [CrossRef] [Green Version]
- Passacquale, G.; Phinikaridou, A.; Warboys, C.; Cooper, M.; Lavin, B.; Alfieri, A.; Andia, M.E.; Botnar, R.M.; Ferro, A. Aspirin-induced histone acetylation in endothelial cells enhances synthesis of the secreted isoform of netrin-1 thus inhibiting monocyte vascular infiltration. Br. J. Pharm. 2015, 172, 3548–3564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koutsounas, I.; Giaginis, C.; Patsouris, E.; Theocharis, S. Current evidence for histone deacetylase inhibitors in pancreatic cancer. World J. Gastroenterol. 2013, 19, 813–828. [Google Scholar] [CrossRef] [PubMed]
- Kramer, O.H.; Knauer, S.K.; Zimmermann, D.; Stauber, R.H.; Heinzel, T. Histone deacetylase inhibitors and hydroxyurea modulate the cell cycle and cooperatively induce apoptosis. Oncogene 2008, 27, 732–740. [Google Scholar] [CrossRef] [Green Version]
- Gasparian, A.V.; Yao, Y.J.; Lu, J.; Yemelyanov, A.Y.; Lyakh, L.A.; Slaga, T.J.; Budunova, I.V. Selenium compounds inhibit I kappa B kinase (IKK) and nuclear factor-kappa B (NF-kappa B) in prostate cancer cells. Mol. Cancer 2002, 1, 1079–1087. [Google Scholar]
- Mittal, R.; Peak-Chew, S.Y.; McMahon, H.T. Acetylation of MEK2 and I kappa B kinase (IKK) activation loop residues by YopJ inhibits signaling. Proc. Natl. Acad. Sci. USA 2006, 103, 18574–18579. [Google Scholar] [CrossRef] [Green Version]
- Awasthi, N.; Zhang, C.; Schwarz, A.M.; Hinz, S.; Wang, C.; Williams, N.S.; Schwarz, M.A.; Schwarz, R.E. Comparative benefits of Nab-paclitaxel over gemcitabine or polysorbate-based docetaxel in experimental pancreatic cancer. Carcinogenesis 2013, 34, 2361–2369. [Google Scholar] [CrossRef] [Green Version]
- Fryer, R.A.; Barlett, B.; Galustian, C.; Dalgleish, A.G. Mechanisms underlying gemcitabine resistance in pancreatic cancer and sensitisation by the iMiD lenalidomide. Anticancer Res. 2011, 31, 3747–3756. [Google Scholar] [PubMed]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [Green Version]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq—A Python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef]
- Pandey, M.K.; Sandur, S.K.; Sung, B.; Sethi, G.; Kunnumakkara, A.B.; Aggarwal, B.B. Butein, a tetrahydroxychalcone, inhibits nuclear factor (NF)-kappaB and NF-kappaB-regulated gene expression through direct inhibition of IkappaBalpha kinase beta on cysteine 179 residue. J. Biol. Chem. 2007, 282, 17340–17350. [Google Scholar] [CrossRef] [PubMed] [Green Version]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Karelia, D.N.; Kim, S.; K. Pandey, M.; Plano, D.; Amin, S.; Lu, J.; Sharma, A.K. Novel Seleno-Aspirinyl Compound AS-10 Induces Apoptosis, G1 Arrest of Pancreatic Ductal Adenocarcinoma Cells, Inhibits Their NF-κB Signaling, and Synergizes with Gemcitabine Cytotoxicity. Int. J. Mol. Sci. 2021, 22, 4966. https://doi.org/10.3390/ijms22094966
Karelia DN, Kim S, K. Pandey M, Plano D, Amin S, Lu J, Sharma AK. Novel Seleno-Aspirinyl Compound AS-10 Induces Apoptosis, G1 Arrest of Pancreatic Ductal Adenocarcinoma Cells, Inhibits Their NF-κB Signaling, and Synergizes with Gemcitabine Cytotoxicity. International Journal of Molecular Sciences. 2021; 22(9):4966. https://doi.org/10.3390/ijms22094966
Chicago/Turabian StyleKarelia, Deepkamal N., Sangyub Kim, Manoj K. Pandey, Daniel Plano, Shantu Amin, Junxuan Lu, and Arun K. Sharma. 2021. "Novel Seleno-Aspirinyl Compound AS-10 Induces Apoptosis, G1 Arrest of Pancreatic Ductal Adenocarcinoma Cells, Inhibits Their NF-κB Signaling, and Synergizes with Gemcitabine Cytotoxicity" International Journal of Molecular Sciences 22, no. 9: 4966. https://doi.org/10.3390/ijms22094966
APA StyleKarelia, D. N., Kim, S., K. Pandey, M., Plano, D., Amin, S., Lu, J., & Sharma, A. K. (2021). Novel Seleno-Aspirinyl Compound AS-10 Induces Apoptosis, G1 Arrest of Pancreatic Ductal Adenocarcinoma Cells, Inhibits Their NF-κB Signaling, and Synergizes with Gemcitabine Cytotoxicity. International Journal of Molecular Sciences, 22(9), 4966. https://doi.org/10.3390/ijms22094966