
Dystrophin Deficiency Causes Progressive Depletion of Cardiovascular Progenitor Cells in the Heart

 ,
,  , , , , and
, , , , and 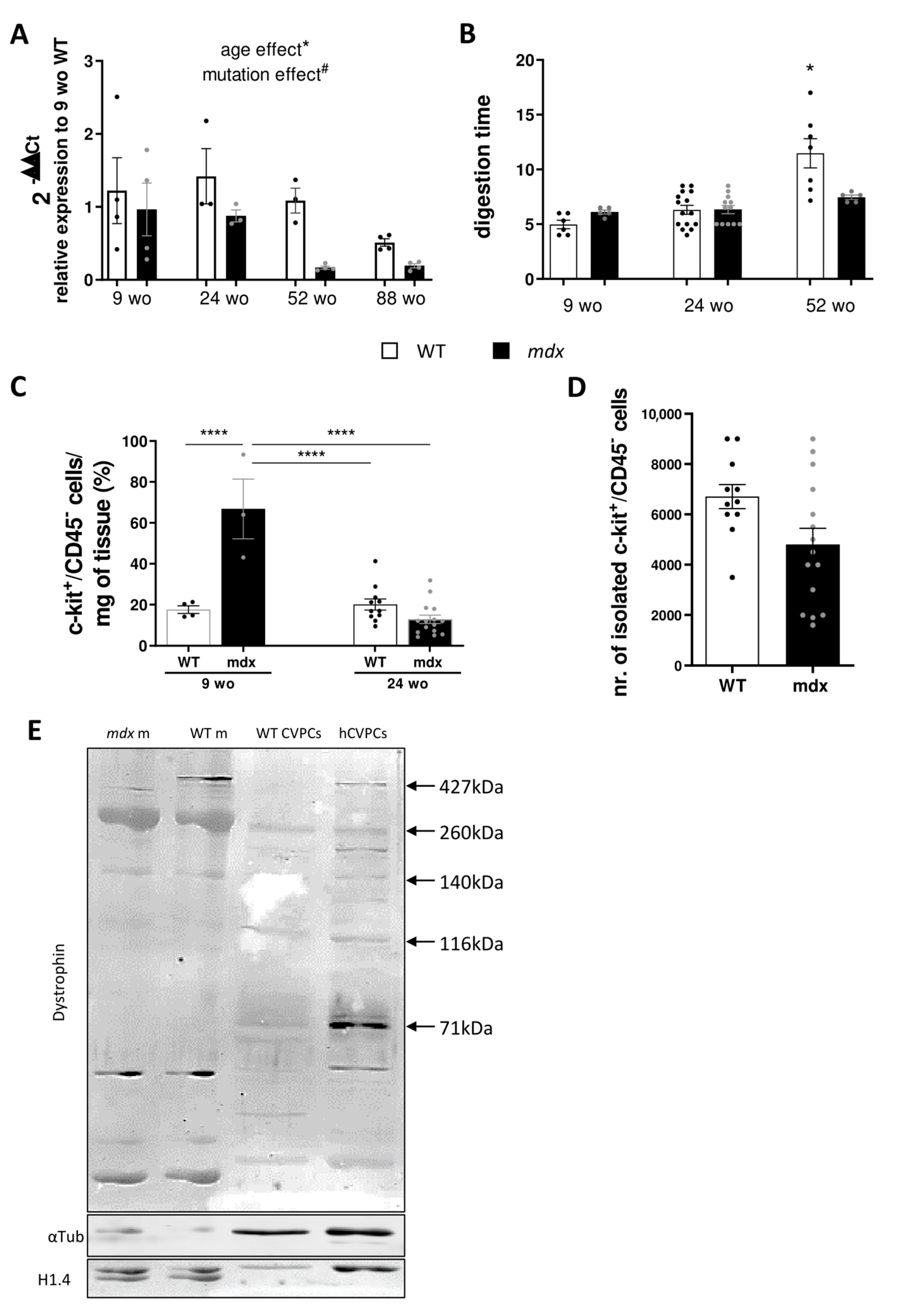{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Mdx Cardiac Tissue Displays Decreasing Levels in C-Kit Receptor mRNA Expression with Ageing
2.2. Mdx CVPCs Show Early In Vivo Amplification Followed by Depletion and Low Resilience in In Vitro Culture
2.3. Different Expressed Dystrophin Isoforms in Mouse and Human CVPCs
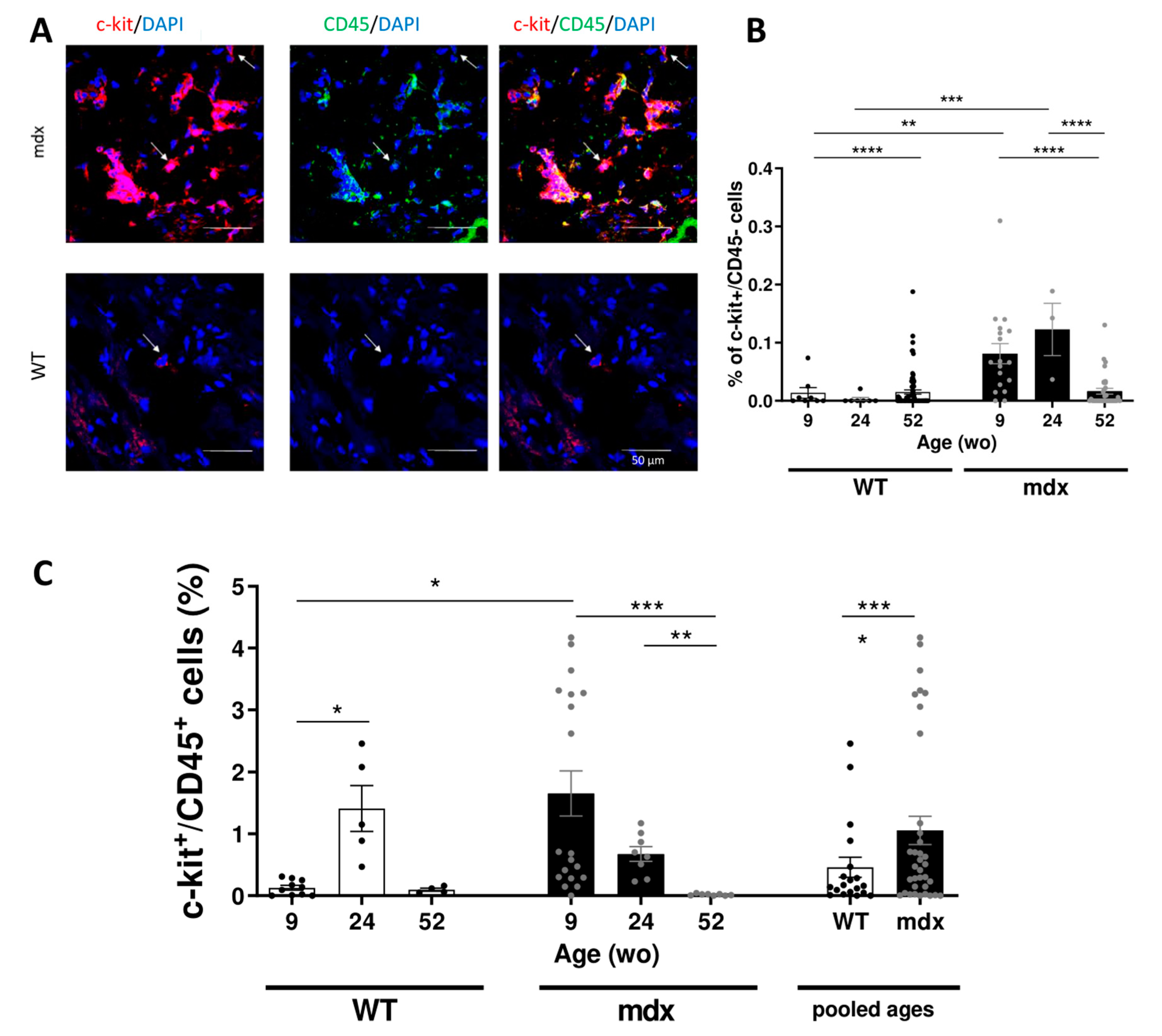
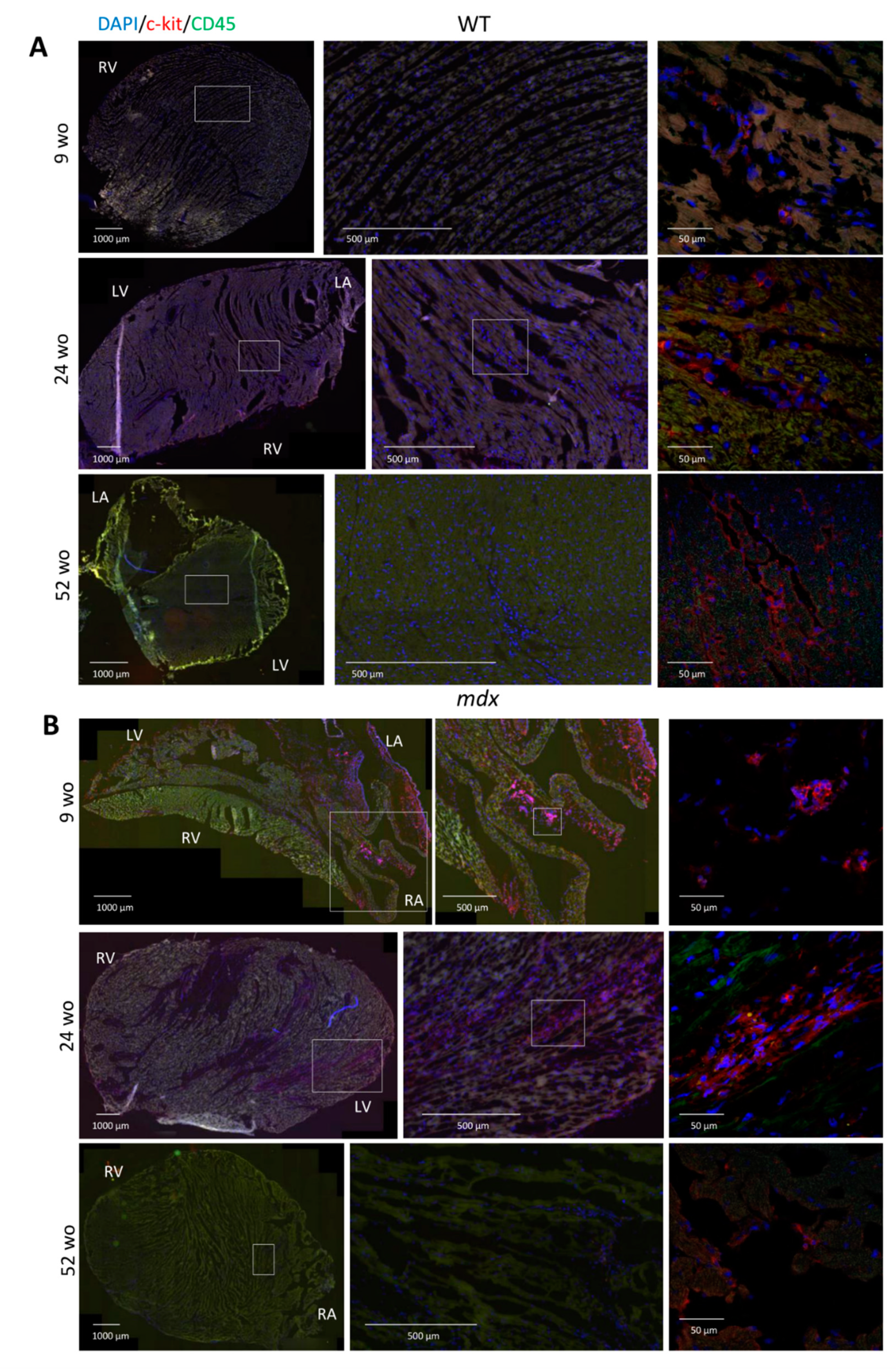
2.4. Increased Resident CVPCs at Early Stage Followed by Premature Depletion in Dystrophin-Deficient Heart
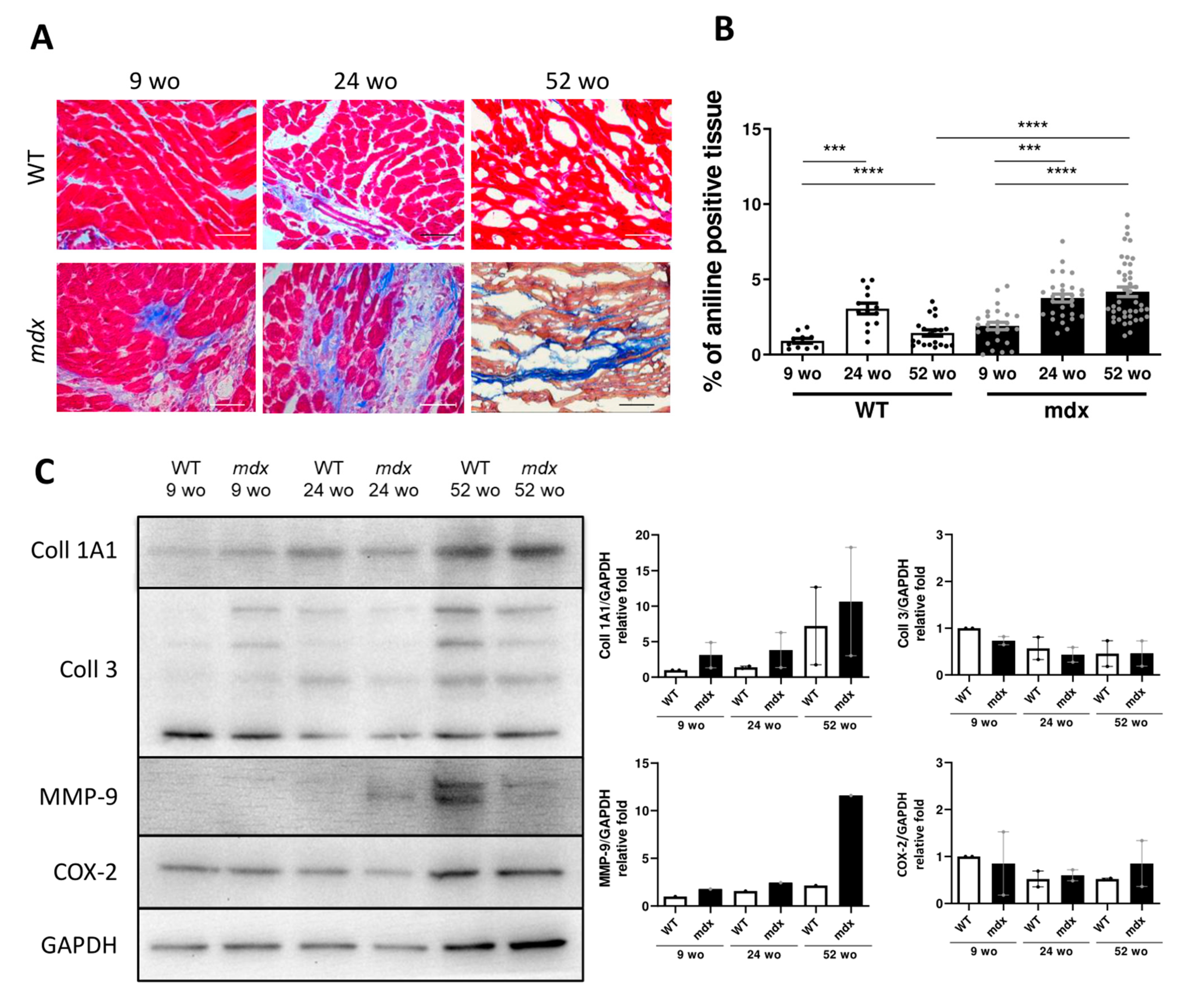
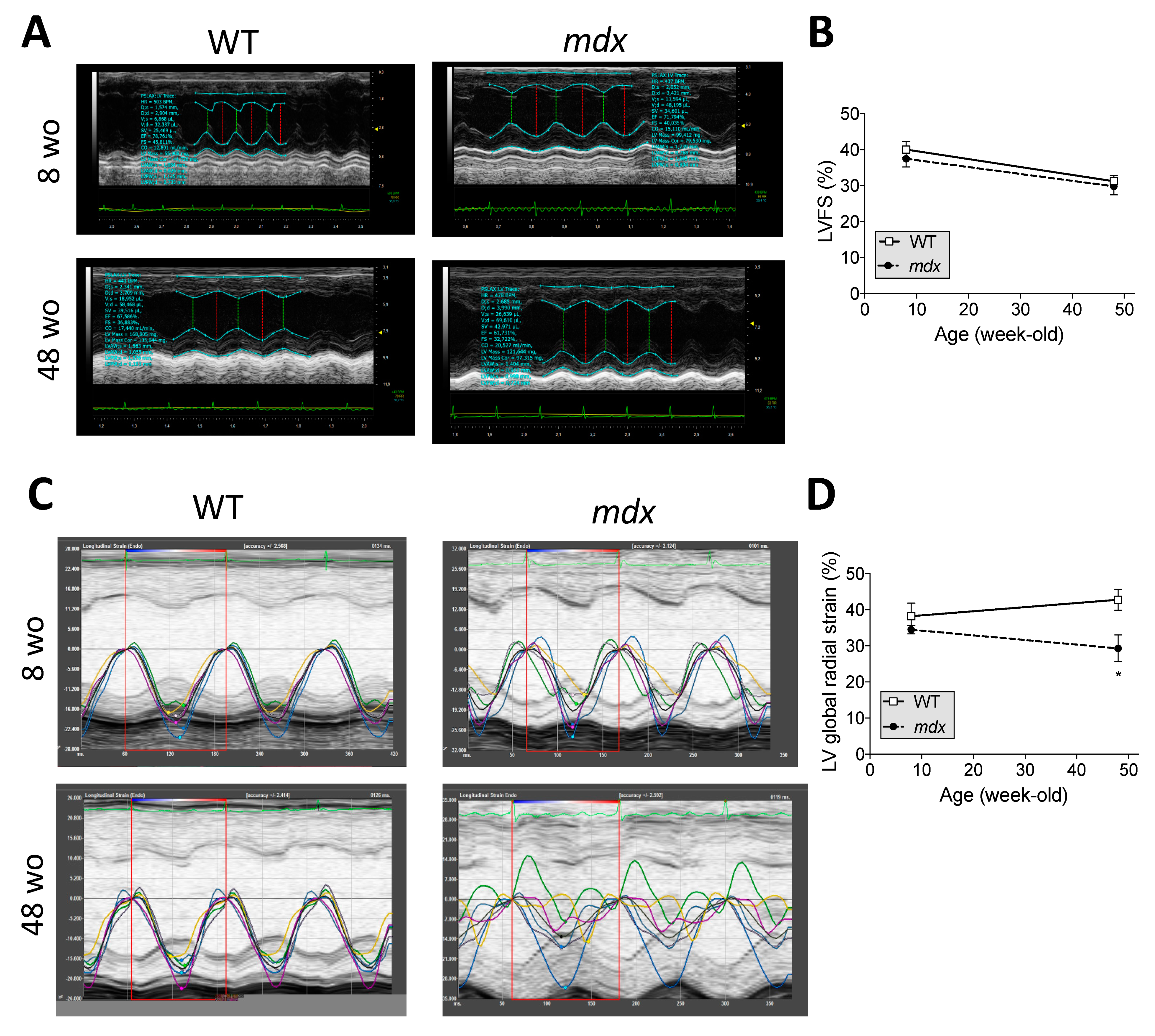
2.5. Early-Stage CVPC Recruitment Followed by Depletion Is Correlated with Fibrotic Development and Cardiac Dysfunction in Mdx Hearts
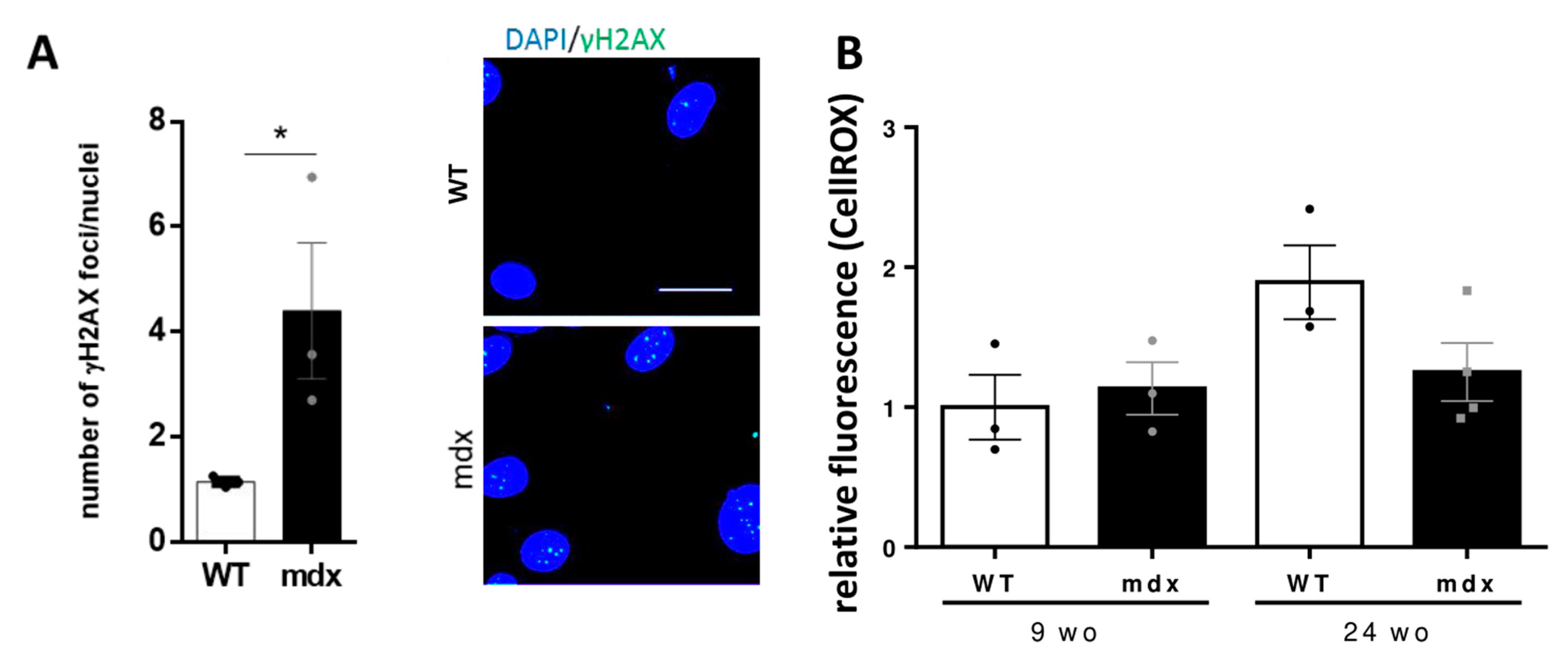
2.6. DNA Damage and Oxidative Stress in CVPCs
3. Discussion
4. Materials and Methods
4.1. Ethical Statements
4.2. Immunohistology
4.3. Mouse Heart Dissociation
4.4. CVPC Analysis
4.5. ROS and DNA Damage Analysis
4.6. Quantitative RT-PCR
4.7. Western Blot Analysis
4.8. Echocardiography
4.9. Statistical Analysis
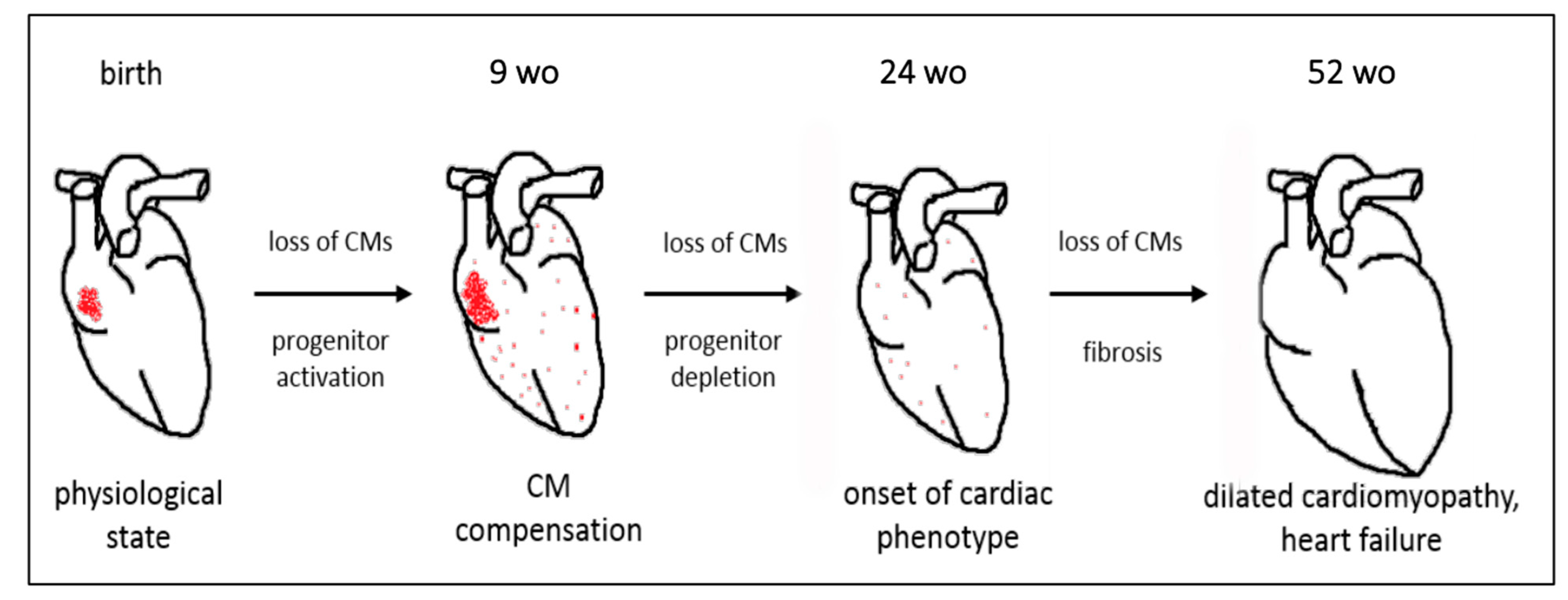
5. Conclusions
6. Contribution to the Field
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Mah, J.K.; Korngut, L.; Dykeman, J.; Day, L.; Pringsheim, T.; Jette, N. A Systematic Review and Meta-Analysis on the Epidemiology of Duchenne and Becker Muscular Dystrophy. Neuromuscul. Disord. 2014, 24, 482–491. [Google Scholar] [CrossRef]
- Nigro, G.; Comi, L.I.; Politano, L.; Bain, R.J. The Incidence and Evolution of Cardiomyopathy in Duchenne Muscular Dystrophy. Int. J. Cardiol. 1990, 26, 271–277. [Google Scholar] [CrossRef]
- Clarac, F.; Massion, J.; Smith, A.M. Duchenne, Charcot and Babinski, Three Neurologists of La Salpetrière Hospital, and Their Contribution to Concepts of the Central Organization of Motor Synergy. J. Physiol. 2009, 103, 361–376. [Google Scholar] [CrossRef]
- Amedro, P.; Vincenti, M.; De La Villeon, G.; Lavastre, K.; Barrea, C.; Guillaumont, S.; Bredy, C.; Gamon, L.; Meli, A.C.; Cazorla, O.; et al. Speckle-Tracking Echocardiography in Children with Duchenne Muscular Dystrophy: A Prospective Multicenter Controlled Cross-Sectional Study. J. Am. Soc. Echocardiogr. 2019, 32, 412–422. [Google Scholar] [CrossRef] [PubMed]
- Manzur, A.Y.; Kinali, M.; Muntoni, F. Update on the Management of Duchenne Muscular Dystrophy. Arch. Dis. Child. 2008, 93, 986–990. [Google Scholar] [CrossRef]
- Fayssoil, A.; Nardi, O.; Orlikowski, D.; Annane, D. Cardiomyopathy in Duchenne Muscular Dystrophy: Pathogenesis and Therapeutics. Heart Fail. Rev. 2010, 15, 103–107. [Google Scholar] [CrossRef] [PubMed]
- Straub, V.; Rafael, J.A.; Chamberlain, J.S.; Campbell, K.P. Animal Models for Muscular Dystrophy Show Different Patterns of Sarcolemmal Disruption. J. Cell Biol. 1997, 139, 375–385. [Google Scholar] [CrossRef]
- Niggli, E.; Shirokova, N. A Guide to Sparkology: The Taxonomy of Elementary Cellular Ca2+ Signaling Events. Cell Calcium 2007, 42, 379–387. [Google Scholar] [CrossRef] [PubMed]
- Williams, I.A.; Allen, D.G. Intracellular Calcium Handling in Ventricular Myocytes from Mdx Mice. Am. J. Physiol. Heart Circ. Physiol. 2007, 292, H846–H855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, C.; Martins, A.S.; Niggli, E.; Shirokova, N. Dystrophic Cardiomyopathy: Amplification of Cellular Damage by Ca2+ Signalling and Reactive Oxygen Species-Generating Pathways. Cardiovasc. Res. 2008, 77, 766–773. [Google Scholar] [CrossRef]
- Poláková, E.; Shirokova, N. Abnormal Sodium Handling and Mitochondrial Metabolism in Cardiac Dystrophy. Biophys. J. 2011, 100, 81a. [Google Scholar] [CrossRef] [Green Version]
- Bellinger, A.M.; Reiken, S.; Carlson, C.; Mongillo, M.; Liu, X.; Rothman, L.; Matecki, S.; Lacampagne, A.; Marks, A.R. Hypernitrosylated Ryanodine Receptor Calcium Release Channels Are Leaky in Dystrophic Muscle. Nat. Med. 2009, 15, 325–330. [Google Scholar] [CrossRef] [PubMed]
- Fauconnier, J.; Thireau, J.; Reiken, S.; Cassan, C.; Richard, S.; Matecki, S.; Marks, A.R.; Lacampagne, A. Leaky RyR2 Trigger Ventricular Arrhythmias in Duchenne Muscular Dystrophy. Proc. Natl. Acad. Sci. USA 2010, 107, 1559–1564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koenig, X.; Dysek, S.; Kimbacher, S.; Mike, A.K.; Cervenka, R.; Lukacs, P.; Nagl, K.; Dang, X.B.; Todt, H.; Bittner, R.E.; et al. Voltage-Gated Ion Channel Dysfunction Precedes Cardiomyopathy Development in the Dystrophic Heart. PLoS ONE 2011, 6, e20300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cassano, M.; Berardi, E.; Crippa, S.; Toelen, J.; Barthelemy, I.; Micheletti, R.; Chuah, M.; Vandendriessche, T.; Debyser, Z.; Blot, S.; et al. Alteration of Cardiac Progenitor Cell Potency in GRMD Dogs. Cell Transplant. 2012, 21, 1945–1967. [Google Scholar] [CrossRef] [Green Version]
- Dumont, N.A.; Wang, Y.X.; von Maltzahn, J.; Pasut, A.; Bentzinger, C.F.; Brun, C.E.; Rudnicki, M.A. Dystrophin Expression in Muscle Stem Cells Regulates Their Polarity and Asymmetric Division. Nat. Med. 2015, 21, 1455–1463. [Google Scholar] [CrossRef] [Green Version]
- Sohn, J.; Lu, A.; Tang, Y.; Wang, B.; Huard, J. Activation of Non-Myogenic Mesenchymal Stem Cells during the Disease Progression in Dystrophic Dystrophin/Utrophin Knockout Mice. Hum. Mol. Genet. 2015, 24, 3814–3829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colussi, C.; Gurtner, A.; Rosati, J.; Illi, B.; Ragone, G.; Piaggio, G.; Moggio, M.; Lamperti, C.; D’Angelo, G.; Clementi, E.; et al. Nitric Oxide Deficiency Determines Global Chromatin Changes in Duchenne Muscular Dystrophy. FASEB J. 2009, 23, 2131–2141. [Google Scholar] [CrossRef] [Green Version]
- Saccone, V.; Consalvi, S.; Giordani, L.; Mozzetta, C.; Barozzi, I.; Sandoná, M.; Ryan, T.; Rojas-Muñoz, A.; Madaro, L.; Fasanaro, P.; et al. HDAC-Regulated MyomiRs Control BAF60 Variant Exchange and Direct the Functional Phenotype of Fibro-Adipogenic Progenitors in Dystrophic Muscles. Genes Dev. 2014, 28, 841–857. [Google Scholar] [CrossRef] [Green Version]
- Lu, A.; Poddar, M.; Tang, Y.; Proto, J.D.; Sohn, J.; Mu, X.; Oyster, N.; Wang, B.; Huard, J. Rapid Depletion of Muscle Progenitor Cells in Dystrophic Mdx/Utrophin-/- Mice. Hum. Mol. Genet. 2014, 23, 4786–4800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Erp, C.; Loch, D.; Laws, N.; Trebbin, A.; Hoey, A.J. Timeline of Cardiac Dystrophy in 3-18-Month-Old MDX Mice. Muscle Nerve 2010, 42, 504–513. [Google Scholar] [CrossRef]
- Nitahara-Kasahara, Y.; Hayashita-Kinoh, H.; Chiyo, T.; Nishiyama, A.; Okada, H.; Takeda, S.; Okada, T. Dystrophic Mdx Mice Develop Severe Cardiac and Respiratory Dysfunction Following Genetic Ablation of the Anti-Inflammatory Cytokine IL-10. Hum. Mol. Genet. 2014, 23, 3990–4000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Furtado, M.B.; Nim, H.T.; Boyd, S.E.; Rosenthal, N.A. View from the Heart: Cardiac Fibroblasts in Development, Scarring and Regeneration. Development 2016, 143, 387–397. [Google Scholar] [CrossRef] [Green Version]
- Furtado Milena, B.; Costa Mauro, W.; Pranoto Edward, A.; Salimova, E.; Pinto, A.R.; Lam, N.T.; Park, A.; Snider, P.; Chandran, A.; Harvey, R.P.; et al. Cardiogenic Genes Expressed in Cardiac Fibroblasts Contribute to Heart Development and Repair. Circ. Res. 2014, 114, 1422–1434. [Google Scholar] [CrossRef] [PubMed]
- Lajiness, J.D.; Conway, S.J. The Dynamic Role of Cardiac Fibroblasts in Development and Disease. J. Cardiovasc. Transl. Res. 2012, 5, 739–748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Talman, V.; Kivelä, R. Cardiomyocyte—Endothelial Cell Interactions in Cardiac Remodeling and Regeneration. Front. Cardiovasc. Med. 2018, 5, 101. [Google Scholar] [CrossRef] [Green Version]
- Mathison, M.; Rosengart, T.K. Heart Regeneration: The Endothelial Cell Comes First. J. Thorac. Cardiovasc. Surg. 2018, 155, 1128–1129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Lui, K.O.; Zhou, B. Endocardial Cell Plasticity in Cardiac Development, Diseases and Regeneration. Circ. Res. 2018, 122, 774–789. [Google Scholar] [CrossRef]
- Gray, G.; Toor, I.; Castellan, R.; Crisan, M.; Meloni, M. Resident Cells of the Myocardium: More than Spectators in Cardiac Injury, Repair and Regeneration. Curr. Opin. Physiol. 2018, 1, 46–51. [Google Scholar] [CrossRef]
- Leong, Y.Y.; Ng, W.H.; Ellison-Hughes, G.M.; Tan, J.J. Cardiac Stem Cells for Myocardial Regeneration: They Are Not Alone. Front. Cardiovasc. Med. 2017, 4, 47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hodgkinson, C.P.; Bareja, A.; Gomez, J.A.; Dzau, V.J. Emerging Concepts in Paracrine Mechanisms in Regenerative Cardiovascular Medicine and Biology. Circ. Res. 2016, 118, 95–107. [Google Scholar] [CrossRef] [Green Version]
- Maxwell, J.T.; Trac, D.; Shen, M.; Brown, M.E.; Davis, M.E.; Chao, M.S.; Supapannachart, K.J.; Zaladonis, C.A.; Baker, E.; Li, M.L.; et al. Electrical Stimulation of Pediatric Cardiac-Derived c-Kit+ Progenitor Cells Improves Retention and Cardiac Function in Right Ventricular Heart Failure. Stem Cells 2019, 37, 1528–1541. [Google Scholar] [CrossRef] [Green Version]
- Zhou, B.; Wu, S.M. Reassessment of C-Kit in Cardiac Cells: A Complex Interplay Between Expression, Fate, and Function. Circ. Res. 2018, 123, 9–11. [Google Scholar] [CrossRef] [PubMed]
- Finan, A.; Demion, M.; Sicard, P.; Guisiano, M.; Bideaux, P.; Monceaux, K.; Thireau, J.; Richard, S. Prolonged Elevated Levels of C-Kit+ Progenitor Cells after a Myocardial Infarction by Beta 2 Adrenergic Receptor Priming. J. Cell. Physiol. 2019, 234, 18283–18296. [Google Scholar] [CrossRef]
- Hong, K.U.; Guo, Y.; Li, Q.-H.; Cao, P.; Al-Maqtari, T.; Vajravelu, B.N.; Du, J.; Book, M.J.; Zhu, X.; Nong, Y.; et al. C-Kit+ Cardiac Stem Cells Alleviate Post-Myocardial Infarction Left Ventricular Dysfunction Despite Poor Engraftment and Negligible Retention in the Recipient Heart. PLoS ONE 2014, 9, e96725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davis, D.R. Cardiac Stem Cells in the Post-Anversa Era. Eur. Heart J. 2019, 40, 1039–1041. [Google Scholar] [CrossRef]
- Pesl, M.; Jelinkova, S.; Caluori, G.; Holicka, M.; Krejci, J.; Nemec, P.; Kohutova, A.; Zampachova, V.; Dvorak, P.; Rotrekl, V. Cardiovascular Progenitor Cells and Tissue Plasticity Are Reduced in a Myocardium Affected by Becker Muscular Dystrophy. Orphanet J. Rare Dis. 2020, 15, 65. [Google Scholar] [CrossRef] [Green Version]
- Meyers, T.A.; Townsend, D. Early Right Ventricular Fibrosis and Reduction in Biventricular Cardiac Reserve in the Dystrophin-Deficient Mdx Heart. Am. J. Physiol. Heart Circ. Physiol. 2015, 308, H303–H315. [Google Scholar] [CrossRef] [Green Version]
- Chisholm, K.M.; Merker, J.D.; Gotlib, J.R.; Gitana, G.; Lefterova, M.; Zehnder, J.L.; George, T.I.; Arber, D.A.; Ohgami, R.S. Mast Cells in Systemic Mastocytosis Have Distinctly Brighter CD45 Expression by Flow Cytometry. Am. J. Clin. Pathol. 2015, 143, 527–534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shiba, N.; Miyazaki, D.; Yoshizawa, T.; Fukushima, K.; Shiba, Y.; Inaba, Y.; Imamura, M.; Takeda, S.; Koike, K.; Nakamura, A. Differential Roles of MMP-9 in Early and Late Stages of Dystrophic Muscles in a Mouse Model of Duchenne Muscular Dystrophy. Biochim. Biophys. Acta 2015, 1852, 2170–2182. [Google Scholar] [CrossRef] [Green Version]
- De Oliveira, F.; Flavia, D.O.; Quintana, H.T.; Bortolin, J.A.; Gomes, O.A.; Liberti, E.A.; Ribeiro, D.A. Cyclooxygenase-2 Expression in Skeletal Muscle of Knockout Mice Suffering Duchenne Muscular Dystrophy. Histochem. Cell Biol. 2013, 139, 685–689. [Google Scholar] [CrossRef] [Green Version]
- Jelinkova, S.; Fojtik, P.; Kohutova, A.; Vilotic, A.; Marková, L.; Pesl, M.; Jurakova, T.; Kruta, M.; Vrbsky, J.; Gaillyova, R.; et al. Dystrophin Deficiency Leads to Genomic Instability in Human Pluripotent Stem Cells via NO Synthase-Induced Oxidative Stress. Cells 2019, 8, 53. [Google Scholar] [CrossRef] [Green Version]
- Van Berlo, J.H.; Kanisicak, O.; Maillet, M.; Vagnozzi, R.J.; Karch, J.; Lin, S.-C.J.; Middleton, R.C.; Marbán, E.; Molkentin, J.D. C-Kit+ Cells Minimally Contribute Cardiomyocytes to the Heart. Nature 2014, 509, 337–341. [Google Scholar] [CrossRef]
- Sultana, N.; Zhang, L.; Yan, J.; Chen, J.; Cai, W.; Razzaque, S.; Jeong, D.; Sheng, W.; Bu, L.; Xu, M.; et al. Resident C-Kit(+) Cells in the Heart Are Not Cardiac Stem Cells. Nat. Commun. 2015, 6, 8701. [Google Scholar] [CrossRef] [Green Version]
- Chimenti, I.; Smith, R.R.; Li, T.-S.; Gerstenblith, G.; Messina, E.; Giacomello, A.; Marbán, E. Relative Roles of Direct Regeneration versus Paracrine Effects of Human Cardiosphere-Derived Cells Transplanted into Infarcted Mice. Circ. Res. 2010, 106, 971–980. [Google Scholar] [CrossRef] [PubMed]
- Li, T.-S.; Cheng, K.; Lee, S.-T.; Matsushita, S.; Davis, D.; Malliaras, K.; Zhang, Y.; Matsushita, N.; Smith, R.R.; Marbán, E. Cardiospheres Recapitulate a Niche-Like Microenvironment Rich in Stemness and Cell-Matrix Interactions, Rationalizing Their Enhanced Functional Potency for Myocardial Repair. Stem Cells 2010, 28, 2088–2098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tseliou, E.; Pollan, S.; Malliaras, K.; Terrovitis, J.; Sun, B.; Galang, G.; Marbán, L.; Luthringer, D.; Marbán, E. Allogeneic Cardiospheres Safely Boost Cardiac Function and Attenuate Adverse Remodeling After Myocardial Infarction in Immunologically Mismatched Rat Strains. J. Am. Coll. Cardiol. 2013, 61, 1108–1119. [Google Scholar] [CrossRef] [Green Version]
- Rogers, R.G.; Fournier, M.; Sanchez, L.; Ibrahim, A.G.; Aminzadeh, M.A.; Lewis, M.I.; Marbán, E. Disease-Modifying Bioactivity of Intravenous Cardiosphere-Derived Cells and Exosomes in Mdx Mice. JCI Insight 2019, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aminzadeh, M.A.; Rogers, R.G.; Fournier, M.; Tobin, R.E.; Guan, X.; Childers, M.K.; Andres, A.M.; Taylor, D.J.; Ibrahim, A.; Ding, X.; et al. Exosome-Mediated Benefits of Cell Therapy in Mouse and Human Models of Duchenne Muscular Dystrophy. Stem Cell Rep. 2018, 10, 942–955. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kubo, H.; Jaleel, N.; Kumarapeli, A.; Berretta, R.M.; Bratinov, G.; Shan, X.; Wang, H.; Houser, S.R.; Margulies, K.B. Increased Cardiac Myocyte Progenitors in Failing Human Hearts. Circulation 2008, 118, 649–657. [Google Scholar] [CrossRef] [Green Version]
- Matuszczak, S.; Czapla, J.; Jarosz-Biej, M.; Wiśniewska, E.; Cichoń, T.; Smolarczyk, R.; Kobusińska, M.; Gajda, K.; Wilczek, P.; Sliwka, J.; et al. Characteristic of C-Kit+ Progenitor Cells in Explanted Human Hearts. Clin. Res. Cardiol. 2014, 103, 711–718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leite, C.F.; Lopes, C.S.; Alves, A.C.; Fuzaro, C.S.C.; Silva, M.V.; de Oliveira, L.F.; Garcia, L.P.; Farnesi, T.S.; de Cuba, M.B.; Rocha, L.B.; et al. Endogenous Resident C-Kit Cardiac Stem Cells Increase in Mice with an Exercise-Induced, Physiologically Hypertrophied Heart. Stem Cell Res. 2015, 15, 151–164. [Google Scholar] [CrossRef] [Green Version]
- Tidball, J.G.; Albrecht, D.E.; Lokensgard, B.E.; Spencer, M.J. Apoptosis Precedes Necrosis of Dystrophin-Deficient Muscle. J. Cell. Sci. 1995, 108, 2197–2204. [Google Scholar] [CrossRef] [PubMed]
- Mikhaĭlov, V.M.; Komarov, S.A.; Nilova, V.K.; Shteĭn, G.I.; Baranov, V.S. Ultrastructural and morphometrical analysis of apoptosis stages in cardiomyocytes of MDX mice. Tsitologiia 2001, 43, 729–737. [Google Scholar] [PubMed]
- Bridges, L.R. The Association of Cardiac Muscle Necrosis and Inflammation with the Degenerative and Persistent Myopathy of MDX Mice. J. Neurol. Sci. 1986, 72, 147–157. [Google Scholar] [CrossRef]
- Megeney, L.A.; Kablar, B.; Perry, R.L.S.; Ying, C.; May, L.; Rudnicki, M.A. Severe Cardiomyopathy in Mice Lacking Dystrophin and MyoD. Proc. Natl. Acad. Sci. USA 1999, 96, 220–225. [Google Scholar] [CrossRef] [Green Version]
- Rubi, L.; Todt, H.; Kubista, H.; Koenig, X.; Hilber, K. Calcium Current Properties in Dystrophin-deficient Ventricular Cardiomyocytes from Aged Mdx Mice. Physiol. Rep. 2018, 6, e13567. [Google Scholar] [CrossRef] [Green Version]
- Jelinkova, S.; Vilotic, A.; Pribyl, J.; Aimond, F.; Salykin, A.; Acimovic, I.; Pesl, M.; Caluori, G.; Klimovic, S.; Urban, T.; et al. DMD Pluripotent Stem Cell Derived Cardiac Cells Recapitulate in Vitro Human Cardiac Pathophysiology. Front. Bioeng. Biotechnol. 2020, 8, 535. [Google Scholar] [CrossRef]
- Messina, S.; Altavilla, D.; Aguennouz, M.; Seminara, P.; Minutoli, L.; Monici, M.C.; Bitto, A.; Mazzeo, A.; Marini, H.; Squadrito, F.; et al. Lipid Peroxidation Inhibition Blunts Nuclear Factor-KappaB Activation, Reduces Skeletal Muscle Degeneration, and Enhances Muscle Function in Mdx Mice. Am. J. Pathol. 2006, 168, 918–926. [Google Scholar] [CrossRef] [Green Version]
- Radley, H.G.; Davies, M.J.; Grounds, M.D. Reduced Muscle Necrosis and Long-Term Benefits in Dystrophic Mdx Mice after CV1q (Blockade of TNF) Treatment. Neuromuscul. Disord. 2008, 18, 227–238. [Google Scholar] [CrossRef]
- Delfín, D.A.; Zang, K.E.; Schill, K.E.; Patel, N.T.; Janssen, P.M.L.; Raman, S.V.; Rafael-Fortney, J.A. Cardiomyopathy in the Dystrophin/Utrophin-Deficient Mouse Model of Severe Muscular Dystrophy Is Characterized by Dysregulation of Matrix Metalloproteinases. Neuromuscul. Disord. 2012, 22, 1006–1014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dahiya, S.; Givvimani, S.; Bhatnagar, S.; Qipshidze, N.; Tyagi, S.C.; Kumar, A. Osteopontin-Stimulated Expression of Matrix Metalloproteinase-9 Causes Cardiomyopathy in the Mdx Model of Duchenne Muscular Dystrophy. J. Immunol. 2011, 187, 2723–2731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pioner, J.M.; Guan, X.; Klaiman, J.M.; Racca, A.W.; Pabon, L.; Muskheli, V.; Macadangdang, J.; Ferrantini, C.; Hoopmann, M.R.; Moritz, R.L.; et al. Absence of Full-Length Dystrophin Impairs Normal Maturation and Contraction of Cardiomyocytes Derived from Human-Induced Pluripotent Stem Cells. Cardiovasc. Res. 2020, 116, 368–382. [Google Scholar] [CrossRef]
- Khouzami, L.; Bourin, M.-C.; Christov, C.; Damy, T.; Escoubet, B.; Caramelle, P.; Perier, M.; Wahbi, K.; Meune, C.; Pavoine, C.; et al. Delayed Cardiomyopathy in Dystrophin Deficient Mdx Mice Relies on Intrinsic Glutathione Resource. Am. J. Pathol. 2010, 177, 1356–1364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siemionow, M.; Malik, M.; Langa, P.; Cwykiel, J.; Brodowska, S.; Heydemann, A. Cardiac Protection after Systemic Transplant of Dystrophin Expressing Chimeric (DEC) Cells to the Mdx Mouse Model of Duchenne Muscular Dystrophy. Stem Cell Rev. Rep. 2019, 15, 827–841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Au, C.G.; Butler, T.L.; Sherwood, M.C.; Egan, J.R.; North, K.N.; Winlaw, D.S. Increased Connective Tissue Growth Factor Associated with Cardiac Fibrosis in the Mdx Mouse Model of Dystrophic Cardiomyopathy. Int. J. Exp. Pathol. 2011, 92, 57–65. [Google Scholar] [CrossRef] [PubMed]
- Fayssoil, A.; Renault, G.; Guerchet, N.; Marchiol-Fournigault, C.; Fougerousse, F.; Richard, I. Cardiac Characterization of Mdx Mice Using High-Resolution Doppler Echocardiography. J. Ultrasound Med. 2013, 32, 757–761. [Google Scholar] [CrossRef]
- Stuckey, D.J.; Carr, C.A.; Camelliti, P.; Tyler, D.J.; Davies, K.E.; Clarke, K. In Vivo MRI Characterization of Progressive Cardiac Dysfunction in the Mdx Mouse Model of Muscular Dystrophy. PLoS ONE 2012, 7, e28569. [Google Scholar] [CrossRef]
- Spurney, C.; Yu, Q.; Nagaraju, K. Speckle Tracking Analysis of the Left Ventricular Anterior Wall Shows Significantly Decreased Relative Radial Strain Patterns in Dystrophin Deficient Mice after 9 Months of Age. PLoS Curr. 2011, 3, RRN1273. [Google Scholar] [CrossRef]
- Taqatqa, A.; Bokowski, J.; Al-Kubaisi, M.; Khalil, A.; Miranda, C.; Alaksham, H.; Fughhi, I.; Kenny, D.; Diab, K.A. The Use of Speckle Tracking Echocardiography for Early Detection of Myocardial Dysfunction in Patients with Duchenne Muscular Dystrophy. Pediatr. Cardiol. 2016, 37, 1422–1428. [Google Scholar] [CrossRef]
- Soslow, J.H.; Xu, M.; Slaughter, J.C.; Stanley, M.; Crum, K.; Markham, L.W.; Parra, D.A. Evaluation of Echocardiographic Measures of Left Ventricular Function in Patients with Duchenne Muscular Dystrophy: Assessment of Reproducibility and Comparison to Cardiac Magnetic Resonance Imaging. J. Am. Soc. Echocardiogr. 2016, 29, 983–991. [Google Scholar] [CrossRef] [Green Version]
- Mertens, L.; Ganame, J.; Claus, P.; Goemans, N.; Thijs, D.; Eyskens, B.; Van Laere, D.; Bijnens, B.; D’hooge, J.; Sutherland, G.R.; et al. Early Regional Myocardial Dysfunction in Young Patients with Duchenne Muscular Dystrophy. J. Am. Soc. Echocardiogr. 2008, 21, 1049–1054. [Google Scholar] [CrossRef] [PubMed]
- Sahoo, S.; Losordo, D.W. Exosomes and Cardiac Repair after Myocardial Infarction. Circ. Res. 2014, 114, 333–344. [Google Scholar] [CrossRef] [PubMed]
- Kishore, R.; Khan, M. More Than Tiny Sacks: Stem Cell Exosomes as Cell-Free Modality for Cardiac Repair. Circ. Res. 2016, 118, 330–343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singla, D.K. Stem Cells and Exosomes in Cardiac Repair. Curr. Opin. Pharmacol. 2016, 27, 19–23. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, E.; Fujita, D.; Takahashi, M.; Oba, S.; Nishimatsu, H. Stem Cell-Derived Exosomes as a Therapeutic Tool for Cardiovascular Disease. World J. Stem Cells 2016, 8, 297–305. [Google Scholar] [CrossRef] [Green Version]
- Saha, P.; Sharma, S.; Korutla, L.; Datla, S.R.; Shoja-Taheri, F.; Mishra, R.; Bigham, G.E.; Sarkar, M.; Morales, D.; Bittle, G.; et al. Circulating Exosomes Derived from Transplanted Progenitor Cells Aid the Functional Recovery of Ischemic Myocardium. Sci. Transl. Med. 2019, 11. [Google Scholar] [CrossRef]
- Fang, S.; Wei, J.; Pentinmikko, N.; Leinonen, H.; Salven, P. Generation of Functional Blood Vessels from a Single C-Kit+ Adult Vascular Endothelial Stem Cell. PLoS Biol. 2012, 10, e1001407. [Google Scholar] [CrossRef]
- Hosoda, T. C-Kit-Positive Cardiac Stem Cells and Myocardial Regeneration. Am. J. Cardiovasc. Dis. 2012, 2, 58–67. [Google Scholar]
- Wang, Y.X.; Feige, P.; Brun, C.E.; Hekmatnejad, B.; Dumont, N.A.; Renaud, J.-M.; Faulkes, S.; Guindon, D.E.; Rudnicki, M.A. EGFR-Aurka Signaling Rescues Polarity and Regeneration Defects in Dystrophin-Deficient Muscle Stem Cells by Increasing Asymmetric Divisions. Cell Stem Cell 2019, 24, 419–432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsie, A.W.; Recio, L.; Katz, D.S.; Lee, C.Q.; Wagner, M.; Schenley, R.L. Evidence for Reactive Oxygen Species Inducing Mutations in Mammalian Cells. Proc. Natl. Acad. Sci. USA 1986, 83, 9616–9620. [Google Scholar] [CrossRef] [Green Version]
- Douki, T.; Rivière, J.; Cadet, J. DNA Tandem Lesions Containing 8-Oxo-7,8-Dihydroguanine and Formamido Residues Arise from Intramolecular Addition of Thymine Peroxyl Radical to Guanine. Chem. Res. Toxicol. 2002, 15, 445–454. [Google Scholar] [CrossRef] [PubMed]
- Khairallah, M.; Khairallah, R.; Young, M.E.; Dyck, J.R.B.; Petrof, B.J.; Des Rosiers, C. Metabolic and Signaling Alterations in Dystrophin-Deficient Hearts Precede Overt Cardiomyopathy. J. Mol. Cell. Cardiol. 2007, 43, 119–129. [Google Scholar] [CrossRef]
- El Haddad, M.; Jean, E.; Turki, A.; Hugon, G.; Vernus, B.; Bonnieu, A.; Passerieux, E.; Hamade, A.; Mercier, J.; Laoudj-Chenivesse, D.; et al. Glutathione Peroxidase 3, a New Retinoid Target Gene, Is Crucial for Human Skeletal Muscle Precursor Cell Survival. J. Cell. Sci. 2012, 125, 6147–6156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsiao, L.-C.; Perbellini, F.; Gomes, R.S.M.; Tan, J.J.; Vieira, S.; Faggian, G.; Clarke, K.; Carr, C.A. Murine Cardiosphere-Derived Cells Are Impaired by Age but Not by Cardiac Dystrophic Dysfunction. Stem Cells Dev. 2014, 23, 1027–1036. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saito, Y.; Takahashi, K. Selenoprotein P: Its Structure and Functions. J. Health Sci. 2000, 46, 409–413. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, K.; Avissar, N.; Whitin, J.; Cohen, H. Purification and Characterization of Human Plasma Glutathione Peroxidase: A Selenoglycoprotein Distinct from the Known Cellular Enzyme. Arch. Biochem. Biophys. 1987, 256, 677–686. [Google Scholar] [CrossRef]
- Yarimizu, J.; Nakamura, H.; Yodoi, J.; Takahashi, K. Efficiency of Selenocysteine Incorporation in Human Thioredoxin Reductase. Antioxid. Redox Signal. 2000, 2, 643–651. [Google Scholar] [CrossRef]
- Williams, I.A.; Allen, D.G. The Role of Reactive Oxygen Species in the Hearts of Dystrophin-Deficient Mdx Mice. Am. J. Physiol. Heart Circ. Physiol. 2007, 293, H1969–H1977. [Google Scholar] [CrossRef] [Green Version]
- Dick, E.; Kalra, S.; Anderson, D.; George, V.; Ritso, M.; Laval, S.H.; Barresi, R.; Aartsma-Rus, A.; Lochmüller, H.; Denning, C. Exon Skipping and Gene Transfer Restore Dystrophin Expression in Human Induced Pluripotent Stem Cells-Cardiomyocytes Harboring DMD Mutations. Stem Cells Dev. 2013, 22, 2714–2724. [Google Scholar] [CrossRef] [Green Version]
- Inomata, K.; Aoto, T.; Binh, N.T.; Okamoto, N.; Tanimura, S.; Wakayama, T.; Iseki, S.; Hara, E.; Masunaga, T.; Shimizu, H.; et al. Genotoxic Stress Abrogates Renewal of Melanocyte Stem Cells by Triggering Their Differentiation. Cell 2009, 137, 1088–1099. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rossi, D.J.; Bryder, D.; Seita, J.; Nussenzweig, A.; Hoeijmakers, J.; Weissman, I.L. Deficiencies in DNA Damage Repair Limit the Function of Haematopoietic Stem Cells with Age. Nature 2007, 447, 725–729. [Google Scholar] [CrossRef]
- Pilzecker, B.; Buoninfante, O.A.; van den Berk, P.; Lancini, C.; Song, J.-Y.; Citterio, E.; Jacobs, H. DNA Damage Tolerance in Hematopoietic Stem and Progenitor Cells in Mice. Proc. Natl. Acad. Sci. USA 2017, 114, E6875–E6883. [Google Scholar] [CrossRef] [Green Version]
- Mandal, P.K.; Rossi, D.J. DNA-Damage-Induced Differentiation in Hematopoietic Stem Cells. Cell 2012, 148, 847–848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burkhalter, M.D.; Rudolph, K.L.; Sperka, T. Genome Instability of Ageing Stem Cells--Induction and Defence Mechanisms. Ageing Res. Rev. 2015, 23, 29–36. [Google Scholar] [CrossRef]
- Amancio, G.D.C.S.; Grabe-Guimarães, A.; Haikel, D.; Moreau, J.; Barcellos, N.M.S.; Lacampagne, A.; Matecki, S.; Cazorla, O. Effect of Pyridostigmine on in Vivo and in Vitro Respiratory Muscle of Mdx Mice. Respir. Physiol. Neurobiol. 2017, 243, 107–114. [Google Scholar] [CrossRef]
- Bankhead, P.; Loughrey, M.B.; Fernández, J.A.; Dombrowski, Y.; McArt, D.G.; Dunne, P.D.; McQuaid, S.; Gray, R.T.; Murray, L.J.; Coleman, H.G.; et al. QuPath: Open Source Software for Digital Pathology Image Analysis. Sci. Rep. 2017, 7, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Broadley, K.J. The Langendorff Heart Preparation—Reappraisal of Its Role as a Research and Teaching Model for Coronary Vasoactive Drugs. J. Pharmacol. Methods 1979, 2, 143–156. [Google Scholar] [CrossRef]
- He, J.-Q.; Vu, D.M.; Hunt, G.; Chugh, A.; Bhatnagar, A.; Bolli, R. Human Cardiac Stem Cells Isolated from Atrial Appendages Stably Express C-Kit. PLoS ONE 2011, 6, e27719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chazalette, D.; Hnia, K.; Rivier, F.; Hugon, G.; Mornet, D. Alpha7B Integrin Changes in Mdx Mouse Muscles after L-Arginine Administration. FEBS Lett. 2005, 579, 1079–1084. [Google Scholar] [CrossRef] [PubMed] [Green Version]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jelinkova, S.; Sleiman, Y.; Fojtík, P.; Aimond, F.; Finan, A.; Hugon, G.; Scheuermann, V.; Beckerová, D.; Cazorla, O.; Vincenti, M.; et al. Dystrophin Deficiency Causes Progressive Depletion of Cardiovascular Progenitor Cells in the Heart. Int. J. Mol. Sci. 2021, 22, 5025. https://doi.org/10.3390/ijms22095025
Jelinkova S, Sleiman Y, Fojtík P, Aimond F, Finan A, Hugon G, Scheuermann V, Beckerová D, Cazorla O, Vincenti M, et al. Dystrophin Deficiency Causes Progressive Depletion of Cardiovascular Progenitor Cells in the Heart. International Journal of Molecular Sciences. 2021; 22(9):5025. https://doi.org/10.3390/ijms22095025
Chicago/Turabian StyleJelinkova, Sarka, Yvonne Sleiman, Petr Fojtík, Franck Aimond, Amanda Finan, Gerald Hugon, Valerie Scheuermann, Deborah Beckerová, Olivier Cazorla, Marie Vincenti, and et al. 2021. "Dystrophin Deficiency Causes Progressive Depletion of Cardiovascular Progenitor Cells in the Heart" International Journal of Molecular Sciences 22, no. 9: 5025. https://doi.org/10.3390/ijms22095025
APA StyleJelinkova, S., Sleiman, Y., Fojtík, P., Aimond, F., Finan, A., Hugon, G., Scheuermann, V., Beckerová, D., Cazorla, O., Vincenti, M., Amedro, P., Richard, S., Jaros, J., Dvorak, P., Lacampagne, A., Carnac, G., Rotrekl, V., & Meli, A. C. (2021). Dystrophin Deficiency Causes Progressive Depletion of Cardiovascular Progenitor Cells in the Heart. International Journal of Molecular Sciences, 22(9), 5025. https://doi.org/10.3390/ijms22095025