
Neutral Dissociation of Pyridine Evoked by Irradiation of Ionized Atomic and Molecular Hydrogen Beams


Abstract
:1. Introduction
2. Results and Discussion
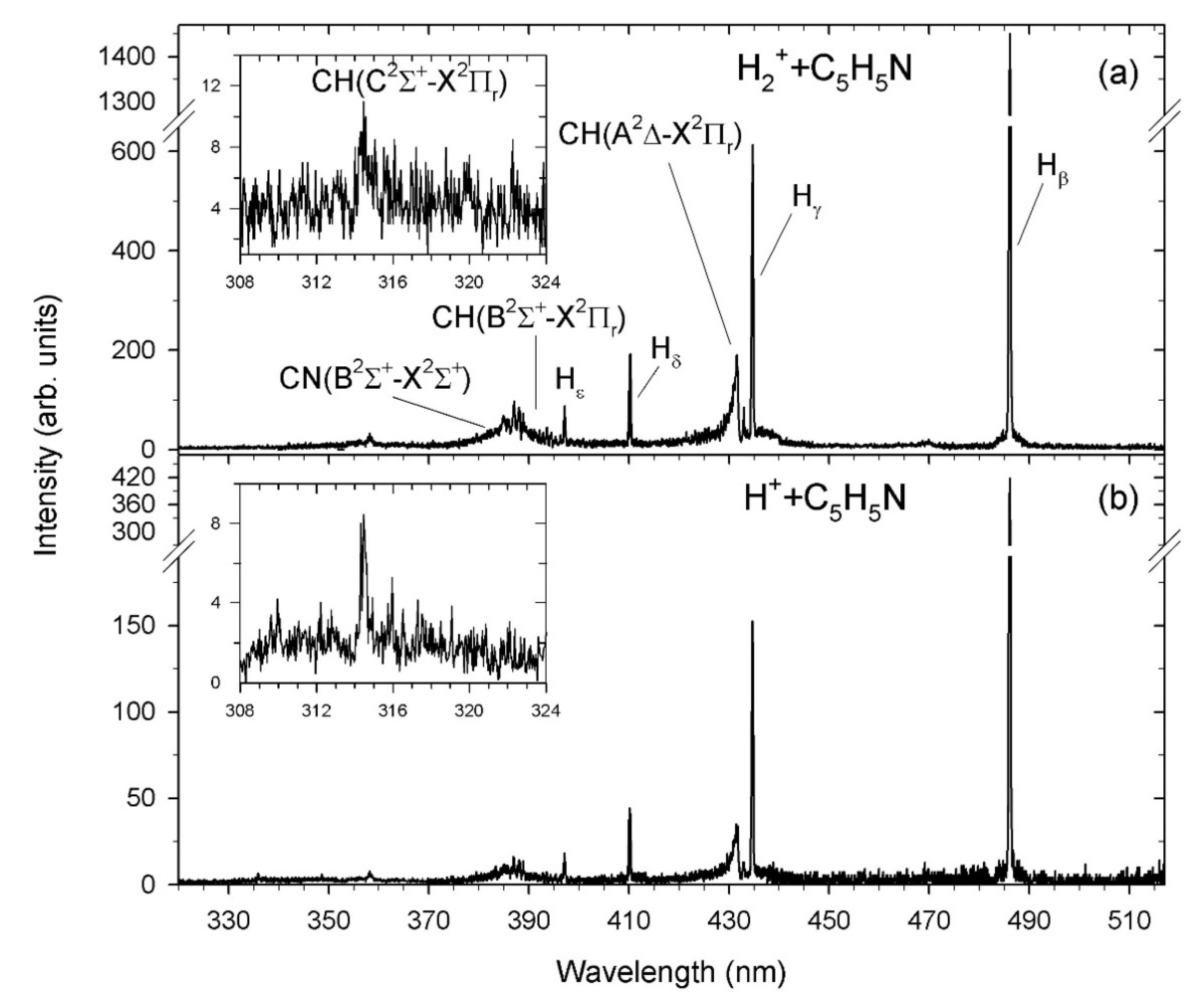
2.1. Fragmentation Spectra
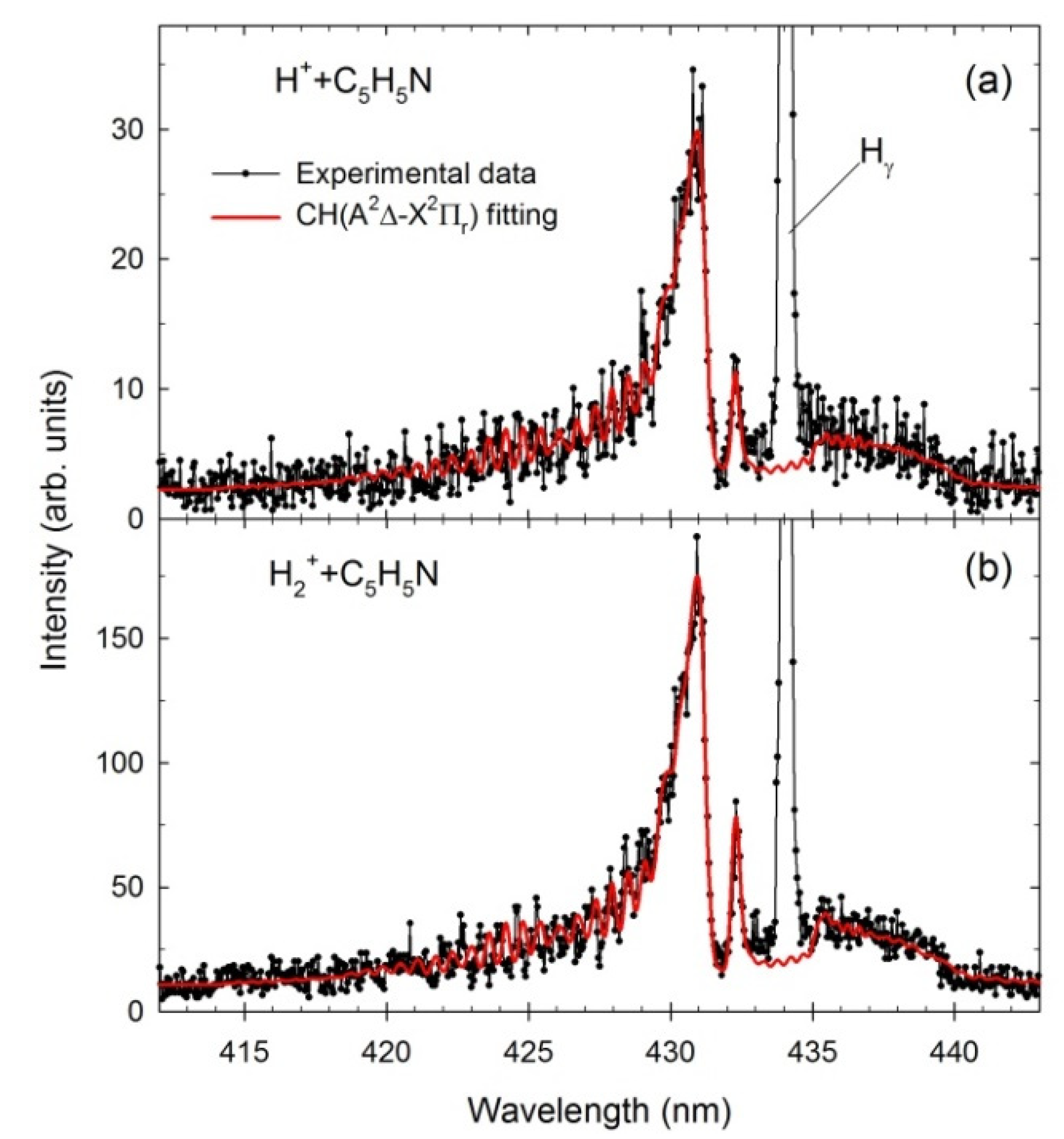
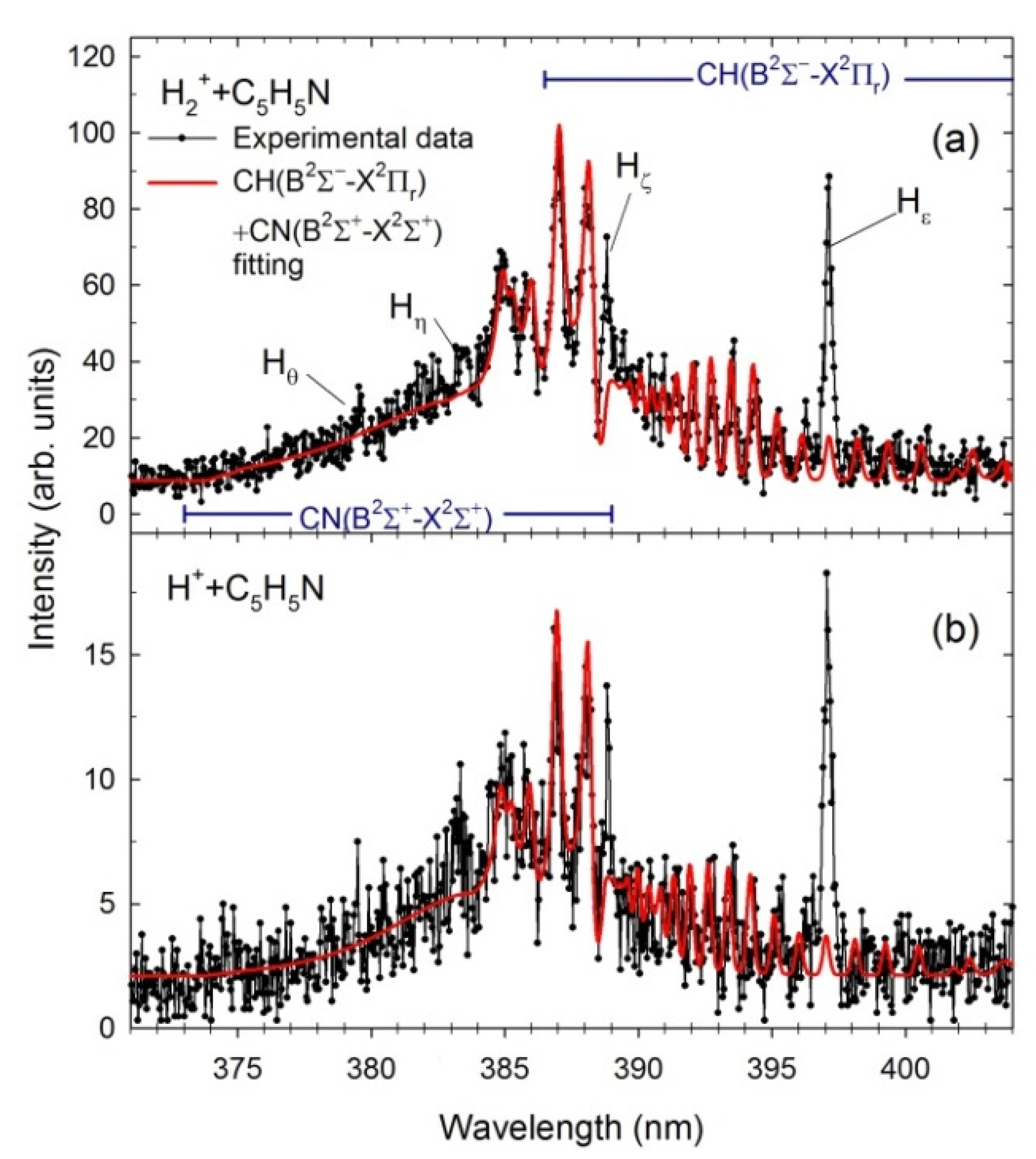
2.2. Theoretical Spectra of CN and CH Radicals
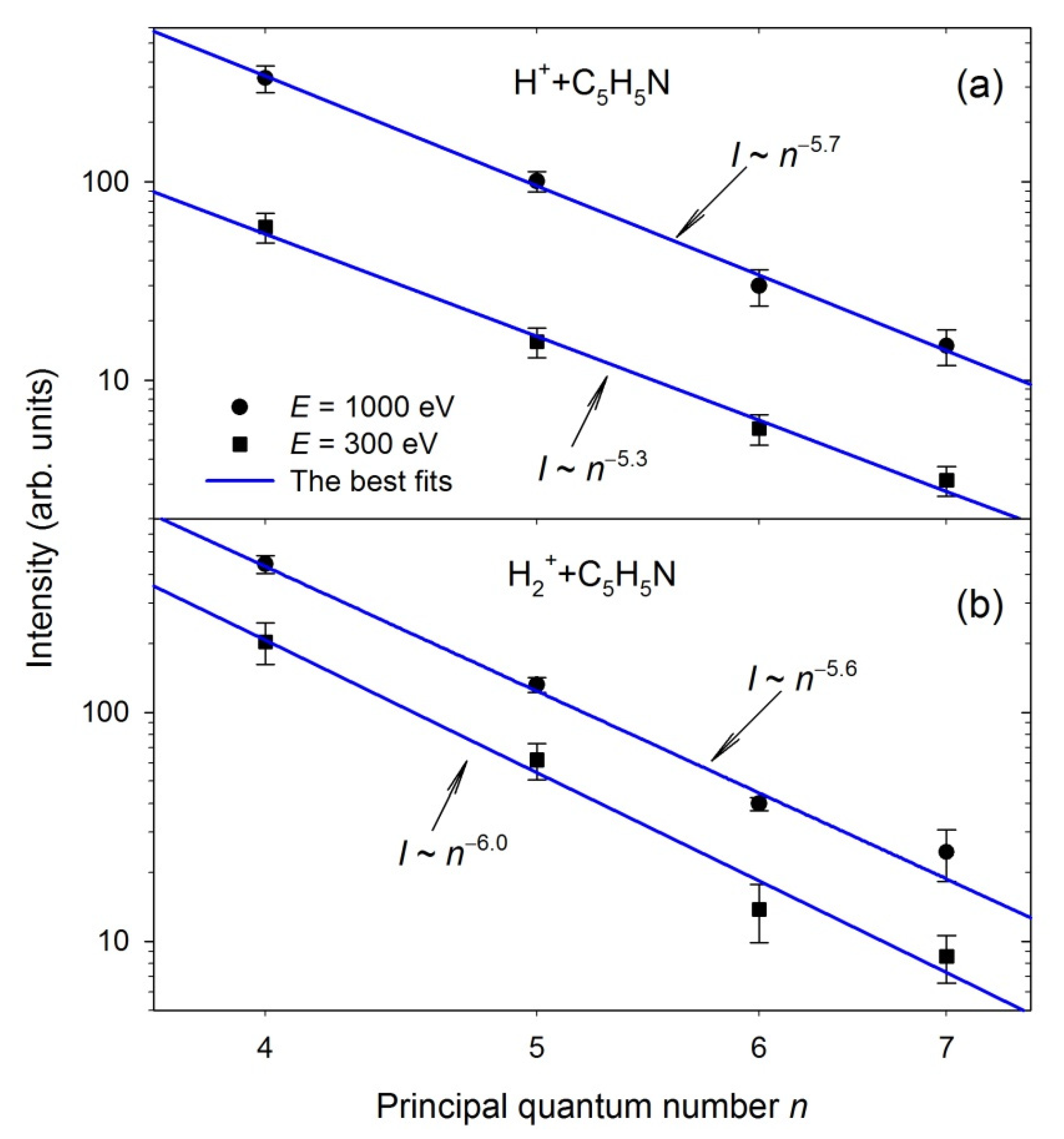
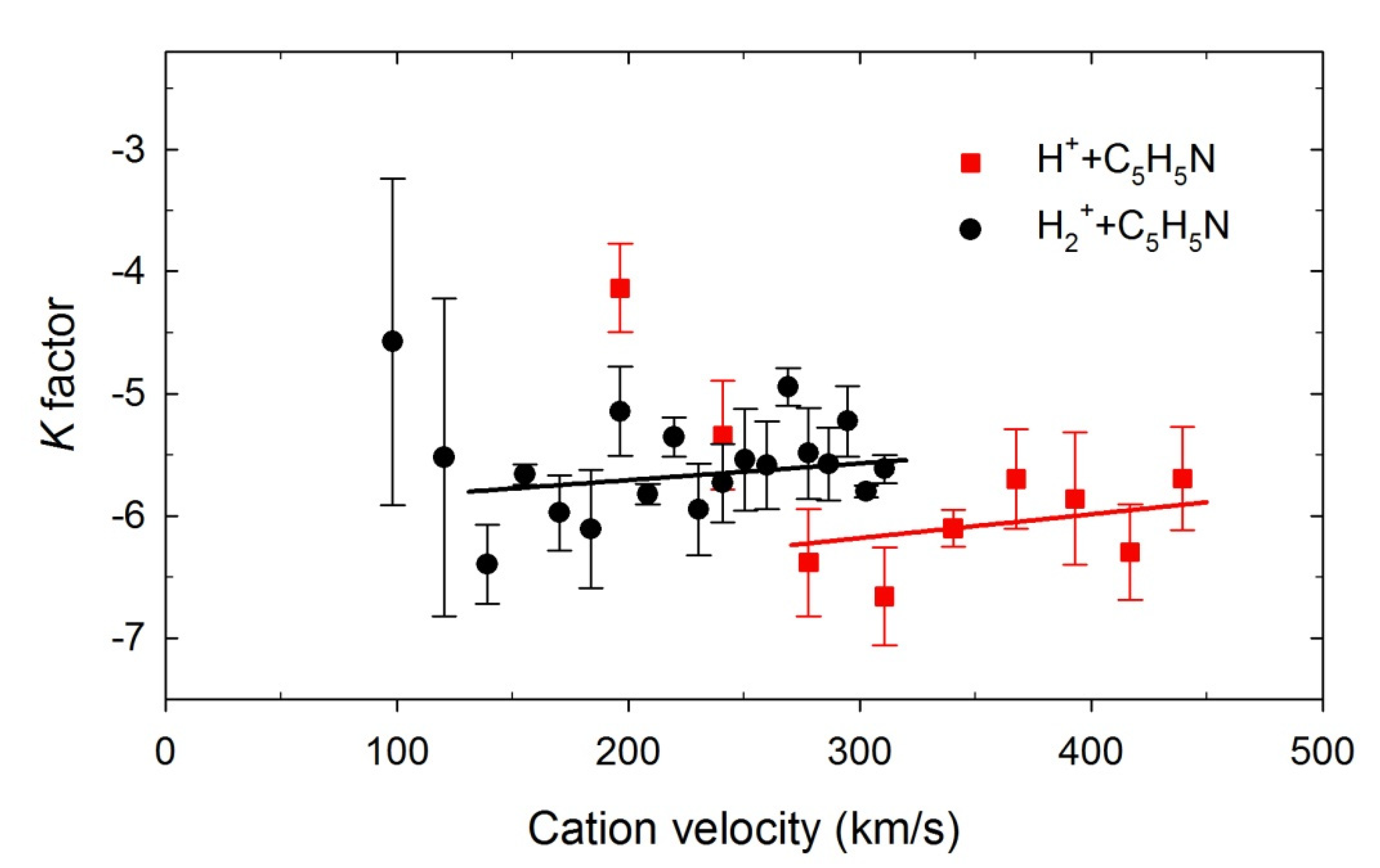
2.3. The H(n) Intensity Ratios
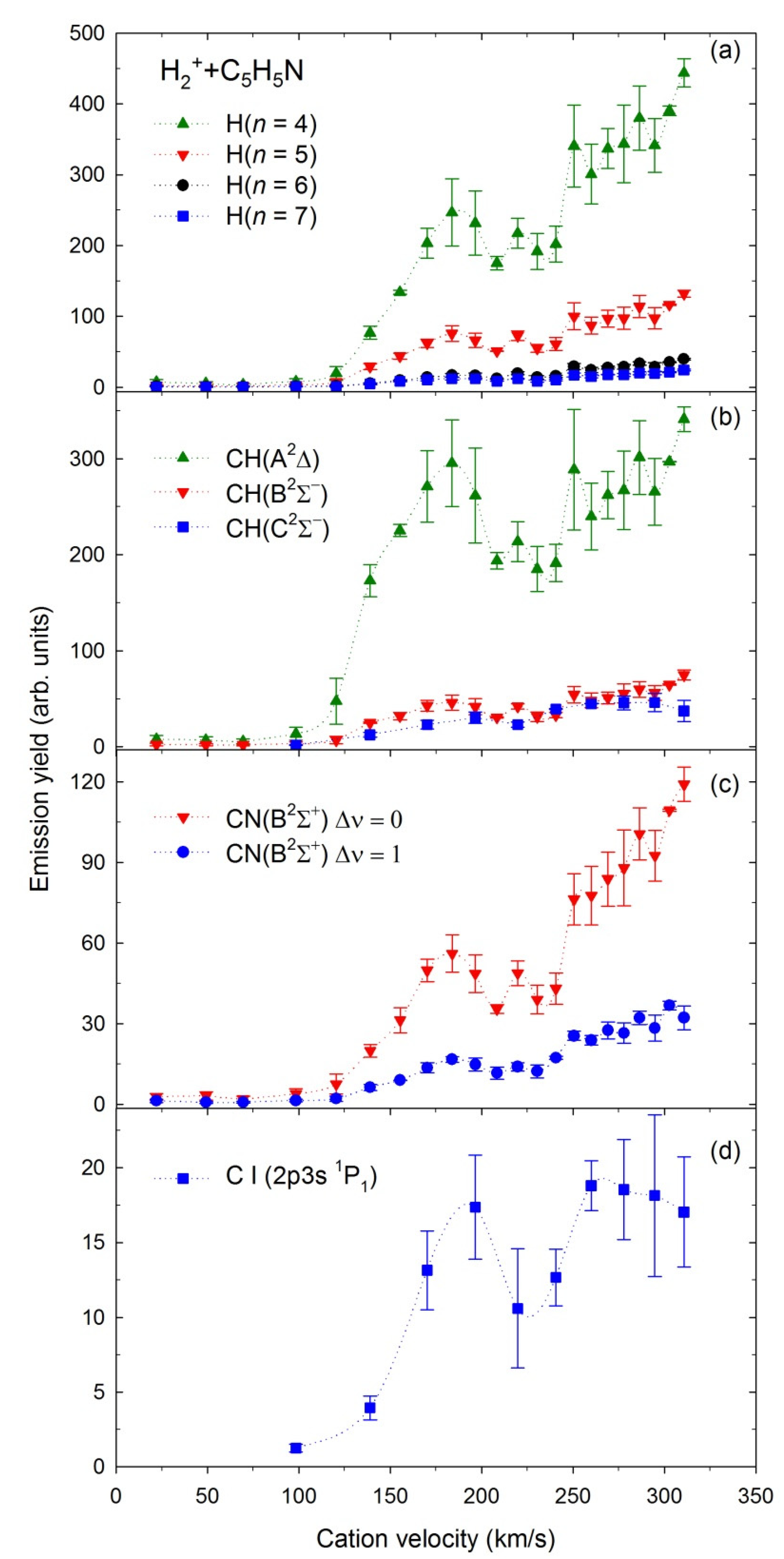
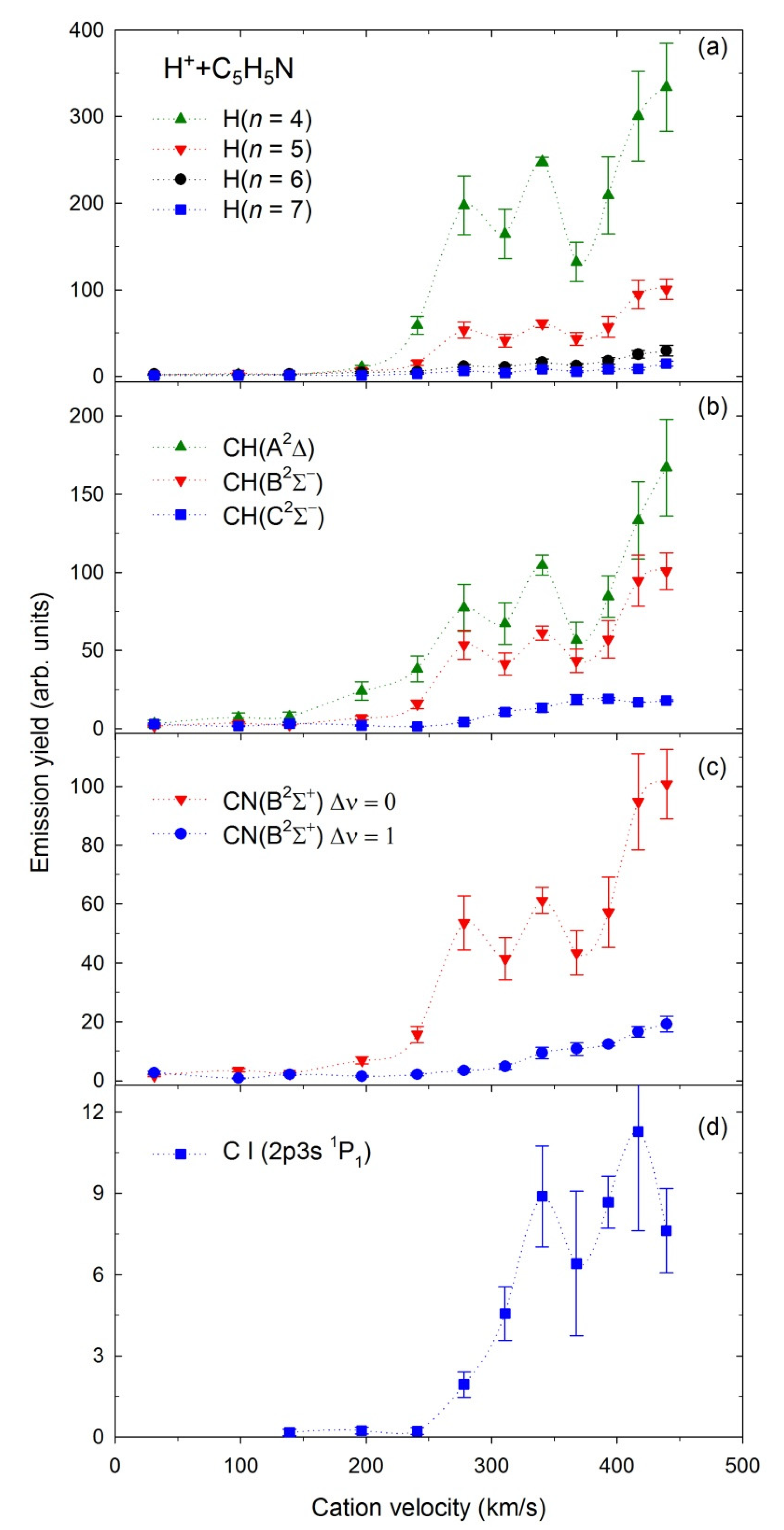
2.4. Emission Yields
2.5. Elucidation of Collisional and Fragmentation Processes
- (i)
- Charge-transfer (CT) that ensues via an electron relocation from C5H5N to the H+/H2+ cations, followed by fragmentation of C5H5N+ parent cation of pyridine. This reaction is usually exothermic [104], which enables the transfer of a significant amount of energy into internal degrees of freedom of molecular products. CT reaction occurs readily at relatively long projectile−target distances [18,19,31,103].
- (ii)
- Dissociative excitation (DE) involves excitation and further fragmentation of pyridine molecules.
- (iii)
- Dissociative ionization (DI) represents direct ionization of the pyridine molecule accompanied by excitation and fragmentation of the pyridine cation. Alvarado et al. [105], in their investigations on interactions of keV H+ and Heq+ with isolated deoxyribose molecules, assumed that the creation of small fragments is associated with violent close collisions involving mainly direct ionization accompanied by electronic and vibrational excitation.
- (iv)
- The fourth reaction is a transient cation–molecule complex formation (TC) owing to an ion−dipole interaction [106]. The constituent units interact electrostatically due to the attractive force between the charge of the H2+ ions and the permanent dipole moment (2.21 D [107]) of pyridine. Akin to the charge transfer reaction, the complex formation occurs at a relatively long projectile−target distance. The ab initio quantum chemical calculations of the collisions of He+/He2+ cations with furan [31] have recently shown significant changes of the wave functions leading to avoided crossings at the potential energy curves around R = 1.5–2.0 Å. Physically this means that electronic clouds of the target and the projectile start overlapping at this length, thus merging both reactants into [He−C4H4O]+/2+ temporary cluster. Note that the CT mechanism also occurs via avoided crossings and, in principle, can also be regarded as the formation of a quasimolecular complex [31].
- (v)
- The fifth mechanism that we can ascertain is a direct dissociative excitation of an H2+ projectile (DP) since the H2+ is a molecule that can be decomposed during collisions.
3. Materials and Methods
4. Summary
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Fulvio, D.; Potapov, A.; He, J.; Henning, T. Astrochemical Pathways to Complex Organic and Prebiotic Molecules: Experimental Perspectives for In Situ Solid-State Studies. Life 2021, 11, 568. [Google Scholar] [CrossRef] [PubMed]
- Vasconcelos, F.A.; Pilling, S.; Agnihotri, A.; Rothard, H.; Boduch, P. Methylenimine and cyanomethanimine synthesis from ion irradiation of N2-CH4 ice: Implication on the formation of prebiotic molecules in outer solar system bodies. Icarus 2020, 351, 113944. [Google Scholar] [CrossRef]
- Kaiser, R.I.; Hansen, N. An Aromatic Universe—A Physical Chemistry Perspective. J. Phys. Chem. A 2021, 125, 3826–3840. [Google Scholar] [CrossRef]
- Cooke, I.R.; Sims, I.R. Experimental Studies of Gas Phase Reactivity in Relation to Complex Organic Molecules in Star-Forming Regions. ACS Earth Space Chem. 2019, 3, 1109–1134. [Google Scholar] [CrossRef]
- Rothard, H.; Domaracka, A.; Boduch, P.; Palumbo, M.E.; Strazzulla, G.; Da Silveira, E.F.; Dartois, E. Modification of ices by cosmic rays and solar wind. J. Phys. B Atom. Mol. Opt. Phys. 2017, 50, 062001. [Google Scholar] [CrossRef]
- Larsson, M.; Geppert, W.D.; Nyman, G. Ion chemistry in space. Rep. Prog. Phys. 2012, 75, 066901. [Google Scholar] [CrossRef]
- Ali, A.; Sittler, E.C.J.; Chornay, D.; Rowe, B.R.; Puzzarini, C. Organic chemistry in Titan’s upper atmosphere and its astrobiological consequences: I. Views towards Cassini plasma spectrometer (CAPS) and ion neutral mass spectrometer (INMS) experiments in space. Planet. Space Sci. 2015, 109–110, 46–63. [Google Scholar] [CrossRef]
- Semo, N.M.; Koski, W.S. Some ion–molecule reactions pertinent to combustion. J. Phys. Chem. 1984, 88, 5320–5324. [Google Scholar] [CrossRef]
- Fialkov, A.B. Investigations on ions in flames. Prog. Energy Combust. Sci. 1997, 23, 399–528. [Google Scholar] [CrossRef]
- Skuratov, V.A.; Gun, K.J.; Stano, J.; Zagorski, D.L. In situ luminescence as monitor of radiation damage under swift heavy ion irradiation. Nucl. Instrum. Methods Phys. Res. Sect. B 2006, 245, 194–200. [Google Scholar] [CrossRef]
- Townsend, P.D. Variations on the use of ion beam luminescence. Nucl. Instrum. Methods Phys. Res. Sect. B 2012, 286, 35–39. [Google Scholar] [CrossRef]
- Townsend, P.D.; Crespillo, M.L. An ideal system for analysis and interpretation of ion beam induced luminescence. Phys. Procedia 2016, 66, 345–351. [Google Scholar] [CrossRef] [Green Version]
- Utke, I.; Hoffmann, P.; Melngailis, J. Gas-Assisted Focused Electron Beam and Ion Beam Processing and Fabrication. J. Vac. Sci. Technol. B 2008, 26, 1197–1276. [Google Scholar] [CrossRef] [Green Version]
- He, S.; Tian, R.; Wu, W.; Li, W.-D.; Wang, D. Helium-Ion-Beam Nanofabrication: Extreme Processes and Applications. Int. J. Extrem. Manuf. 2021, 3, 012001. [Google Scholar] [CrossRef]
- Hayles, M.F.; De Winter, D.A.M. An introduction to cryo-FIB-SEM cross-sectioning of frozen, hydrated Life Science samples. J. Microsc. 2021, 281, 138–156. [Google Scholar] [CrossRef] [PubMed]
- Veligura, V.; Hlawacek, G.; van Gastel, R.; Zandvliet, H.J.W.; Poelsema, B. A high resolution ionoluminescence study of defect creation and interaction. J. Phys. Condens. Matter 2014, 26, 165401. [Google Scholar] [CrossRef]
- Hlawacek, G.; Veligura, V.; van Gastel, R.; Poelsema, B. Helium Ion Microscopy. J. Vac. Sci. Technol. B 2014, 32, 020801. [Google Scholar] [CrossRef] [Green Version]
- de Vries, J.; Hoekstra, R.; Morgenstern, R.; Schlatholter, T. Charge Driven Fragmentation of Nucleobases. Phys. Rev. Lett. 2003, 91, 053401. [Google Scholar] [CrossRef]
- Schlathölter, T.; Hoekstra, R.; Morgenstern, R. Charge Driven Fragmentation of Biologically Relevant Molecules. Int. J. Mass Spectrom. 2004, 233, 173–179. [Google Scholar] [CrossRef]
- Deng, Z.; Bald, I.; Illenberger, E.; Huels, M.A. Beyond the Bragg Peak: Hyperthermal Heavy Ion Damage to DNA Components. Phys. Rev. Lett. 2005, 95, 153201. [Google Scholar] [CrossRef]
- Schlathölter, T.; Alvarado, F.; Bari, S.; Hoekstra, R. Ion-Induced Ionization and Fragmentation of DNA Building Blocks. Phys. Scr. 2006, 73, C113. [Google Scholar] [CrossRef]
- Zettergren, H.; Domaracka, A.; Schlathölter, T.; Bolognesi, P.; Diaz-Tendero, S.; Łabuda, M.; Tosic, S.; Maclot, S.; Johnsson, P.; Steber, A.; et al. Roadmap on dynamics of molecules and clusters in the gas phase. Eur. Phys. J. D 2021, 75, 152. [Google Scholar] [CrossRef]
- Bibang, P.C.J.A.; Agnihotri, A.N.; Boduch, P.; Domaracka, A.; Kanuchova, Z.; Rothard, H. Radiolysis of pyridine in solid water. Eur. Phys. J. D 2021, 75, 57. [Google Scholar] [CrossRef]
- Wasowicz, T.J. Photon luminescence studies of tetrahydrofuran following trihydrogen cations impact in the 20–1000 eV energy range. Rom. Rep. Phys. 2021, 73, 203. [Google Scholar]
- Wasowicz, T.J.; Pranszke, B. Optical spectroscopic studies of tetrahydrofuran fragmentation induced by collisions with dihydrogen cations. Acta Phys. Pol. A 2021, 140, 228. [Google Scholar] [CrossRef]
- Wasowicz, T.J.; Pranszke, B. Interactions of protons with furan molecules studied by collision-induced emission spectroscopy at the incident energy range of 50–1000 eV. Eur. Phys. J. D 2016, 70, 175. [Google Scholar] [CrossRef] [Green Version]
- Wasowicz, T.J.; Pranszke, B. Observation of the hydrogen migration in the cation-induced fragmentation of the pyridine molecules. J. Phys. Chem. A 2016, 120, 964–971. [Google Scholar] [CrossRef]
- Wasowicz, T.J.; Pranszke, B. Fragmentation of Tetrahydrofuran Molecules by H+, C+, and O+ Collisions at the Incident energy Range of 25−1000 eV. J. Phys. Chem. A 2015, 119, 581–589. [Google Scholar] [CrossRef]
- Wasowicz, T.J.; Pranszke, B. Charge transfer and formation of complexes in the He+ collisions with the furan molecules. J. Phys. Conf. Ser. 2015, 635, 032055. [Google Scholar] [CrossRef]
- Wasowicz, T.J. Hydrogen migration observed in fragmentation of the pyridine molecules in collisions with the H+, H2+, He+ and He++ cations. J. Phys. Conf. Ser. 2015, 635, 032114. [Google Scholar] [CrossRef]
- Wasowicz, T.J.; Łabuda, M.; Pranszke, B. Charge Transfer, Complexes Formation and Furan Fragmentation Induced by Collisions with Low-Energy Helium Cations. Int. J. Mol. Sci. 2019, 20, 6022. [Google Scholar] [CrossRef] [Green Version]
- Wasowicz, T.J. Collision-induced luminescence spectra of pyridine bombarded by 1000 eV He+ cations. Res. Phys. 2020, 18, 103244. [Google Scholar] [CrossRef]
- Amaldi, U.; Kraft, G. Radiotherapy with Beams of Carbon Ions. Rep. Prog. Phys. 2005, 68, 1861–1882. [Google Scholar] [CrossRef]
- Tinganelli, W.; Durante, M. Carbon Ion Radiobiology. Cancers 2020, 12, 3022. [Google Scholar] [CrossRef] [PubMed]
- Thariat, J.; Valable, S.; Laurent, C.; Haghdoost, S.; Peres, E.A.; Bernaudin, M.; Sichel, F.; Lesueur, P.; Cesaire, M.; Petit, E.; et al. Hadrontherapy Interactions in Molecular and Cellular Biology. Int. J. Mol. Sci. 2019, 21, 133. [Google Scholar] [CrossRef] [Green Version]
- Cucinotta, F.; Durante, M. Cancer risk from exposure to galactic cosmic rays: Implications for space exploration by human beings. Lancet Oncol. 2006, 7, 431–435. [Google Scholar] [CrossRef] [Green Version]
- Durante, M.; Cucinotta, F.A. Physical basis of radiation protection in space travel. Rev. Mod. Phys. 2011, 83, 1245–1281. [Google Scholar] [CrossRef]
- Durante, M.; Cucinotta, F.A. Heavy Ion Carcinogenesis and Human Space Exploration. Nat. Rev. Cancer 2008, 8, 465–472. [Google Scholar] [CrossRef]
- Wasowicz, T.J.; Kivimäki, A.; Catone, D.; Richter, R. Vacuum ultraviolet photoionization and ionic fragmentation of the isoxazole molecules. Int. J. Mass Spectrom. 2020, 449, 116276. [Google Scholar] [CrossRef]
- Kivimäki, A.; Stråhlman, C.; Wasowicz, T.J.; Kettunen, J.A.; Richter, R. Yields and Time-of-Flight Spectra of Neutral High-Rydberg Fragments at the K Edges of the CO2 Molecule. J. Phys. Chem. A. 2016, 120, 4360–4367. [Google Scholar] [CrossRef]
- Kivimäki, A.; Wasowicz, T.J.; Richter, R. Soft X-ray Induced Production of Neutral Fragments in High-Rydberg States at the O 1s Ionization Threshold of the Water Molecule. J. Phys. Chem. A. 2021, 125, 713–720. [Google Scholar] [CrossRef]
- McConkey, J.W.; Malone, C.P.; Johnson, P.V.; Winstead, C.; McKoy, V.; Kanik, I. Electron Impact Dissociation of Oxygen-Containing Molecules. A Critical Review. Phys. Rep. 2008, 466, 1–103. [Google Scholar] [CrossRef]
- Hatano, H. Interaction of Vacuum Ultraviolet Photons with Molecules. Formation and Dissociation Dynamics of Molecular Superexcited States. Phys. Rep. 1999, 313, 109–169. [Google Scholar] [CrossRef]
- Linert, I.; Lachowicz, I.; Wasowicz, T.J.; Zubek, M. Fragmentation of Isoxazole Molecules by Electron Impact in the Energy Range 10−85 eV. Chem. Phys. Lett. 2010, 498, 27–31. [Google Scholar] [CrossRef]
- Wasowicz, T.J.; Linert, I.; Lachowicz, I.; Zubek, M. Electron impact fragmentation of pyrrole molecules studied by fluorescence emission spectroscopy. Photonics Lett. Pol. 2011, 3, 110–112. [Google Scholar]
- Hein, J.D.; Al-Khazraji, H.; Tiessen, C.J.; Lukic, D.; Trocchi, J.A.; McConkey, J.W. Excited atomic fragments following electron dissociation of pyrimidine. J. Phys. B At. Mol. Opt. Phys. 2013, 46, 045202. [Google Scholar] [CrossRef]
- Erdevdi, N.M.; Zvenigorodskii, V.V.; Shpenik, O.B.; Romanova, L.G. Excitation of Adenine Molecules by Slow Electrons. Opt. Spectrosc. 2013, 114, 47–51. [Google Scholar] [CrossRef]
- Shpenik, O.B.; Erdevdy, N.M.; Zvenighorodsky, V.V.; Romanova, L.G. Luminescence of cytosine vapor excited by slow electrons. J. Appl. Spectrosc. 2013, 80, 43–46. [Google Scholar] [CrossRef]
- Hein, J.D.; Al-Khazraji, H.; Tiessen, C.J.; Lukic, D.; Trocchi, J.A.; McConkey, J.W. VUV study of electron impact dissociative excitation of thymine. J. Phys. B At. Mol. Opt. Phys. 2016, 49, 125204. [Google Scholar]
- Trocchi, J.A.; Dech, J.; Kedzierski, W.; McConkey, J.W. Production of excited H–atoms in electron collisions with adenine. J. Phys. B At. Mol. Opt. Phys. 2019, 52, 055204. [Google Scholar] [CrossRef]
- Erdevdi, N.M.; Bulhakova, A.I.; Shpenik, O.B.; Zavilopulo, A.N. Electron-Impact-Induced Excitation of Glutamine Molecules. Tech. Phys. Lett. 2020, 46, 815–818. [Google Scholar] [CrossRef]
- Shpenik, O.B.; Maslyuk, V.T.; Zavilopulo, A.N.; Erdevdi, N.M.; Bulhakova, A.I.; Megela, I.G. Electron impact excitation of glutamine molecules irradiated with an M-30 microtron with an energy of 11.5 MeV. J. Phys. B At. Mol. Opt. Phys. 2021, 54, 145201. [Google Scholar] [CrossRef]
- Wasowicz, T.J.; Kivimaki, A.; Dampc, M.; Coreno, M.; De Simone, M.; Zubek, M. Photofragmentation of Tetrahydrofuran Molecules in the Vacuum-Ultraviolet Region via Superexcited States Studied by Fluorescence Spectroscopy. Phys. Rev. A 2011, 83, 033411. [Google Scholar] [CrossRef]
- Wasowicz, T.J.; Kivimaki, A.; Coreno, M.; Zubek, M. Superexcited States in the Vacuum-Ultraviolet Photofragmentation of Isoxazole Molecules. J. Phys. B At. Mol. Opt. Phys. 2012, 45, 205103. [Google Scholar] [CrossRef]
- Zubek, M.; Wasowicz, T.J.; Dąbkowska, I.; Kivimaki, A.; Coreno, M. Hydrogen Migration in Formation of NH(A3Π) Radicals Via Superexcited States in Photodissociation of Isoxazole Molecules. J. Chem. Phys. 2014, 141, 064301. [Google Scholar] [CrossRef]
- Wasowicz, T.J.; Kivimaki, A.; Coreno, M.; Zubek, M. Formation of CN(B2Σ+) Radicals in the Vacuum-Ultraviolet Photodissociation of Pyridine and Pyrimidine Molecules. J. Phys. B At. Mol. Opt. Phys. 2014, 47, 055103. [Google Scholar] [CrossRef]
- Wasowicz, T.J.; Kivimäki, A.; Coreno, M.; Zubek, M. Hydrogen migration in photodissociation of the pyridine molecules. J. Phys. Conf. Ser. 2015, 635, 112049. [Google Scholar] [CrossRef]
- Wasowicz, T.J.; Dabkowska, I.; Kivimäki, A.; Coreno, M.; Zubek, M. Elimination and migration of hydrogen in the vacuum-ultraviolet photodissociation of pyridine molecules. J. Phys. B Atom. Mol. Opt. Phys. 2017, 50, 015101. [Google Scholar] [CrossRef]
- Ozga, C.; Reiß, P.; Kielich, W.; Klumpp, S.; Knie, A.; Ehresmann, A. Fluorescence cascades after excitation of Xe II 5p46p satellite states by synchrotron radiation. J. Phys. B Atom. Mol. Opt. Phys. 2015, 48, 015004. [Google Scholar] [CrossRef]
- Wasowicz, T.J.; Kivimäki, A.; Stupar, M.; Coreno, M. Study of ultraviolet-visible fluorescence emission following resonant Auger decay of the 2p-1nl core-excited states of argon atoms. J. Elec. Spectrosc. Rel. Phenom. 2018, 226, 35–40. [Google Scholar] [CrossRef]
- Reiß, P.; Schmidt, P.; Ozga, C.; Kniel, A.; Ehresmann, A. Dispersed fluorescence spectrometry from the VIS to VUV spectral range for experiments at heavy-ion storage facilities. Phys. Scr. 2015, T166, 014031. [Google Scholar] [CrossRef]
- Kivimäki, A.; Alvarez-Ruiz, J.; Wasowicz, T.J.; Callegari, C.; de Simone, M.; Alagia, M.; Richter, R.; Coreno, M. O 1s excitation and ionization processes in the CO2 molecule studied via detection of low-energy fluorescence emission. J. Phys. B Atom. Mol. Opt. Phys. 2011, 44, 165103. [Google Scholar] [CrossRef] [Green Version]
- Kojima, T.; Aihara, H.; Kodashima, Y.; Makishima, H.; Nakiri, S.; Takada, S.; Shimada, H.; Ukai, M.; Ozga, C.; Holzapfel, X.; et al. Novel analytical study for reaction intermediates in the primary radiation interaction of DNA using a synchrotron radiation-induced luminescence spectroscopy. Radiat. Protec. Dosim. 2019, 183, 32–35. [Google Scholar] [CrossRef]
- Hans, A.; Schmidt, P.; Ozga, C.; Hartmann, G.; Holzapfel, X.; Ehresmann, A.; Knie, A. Extreme Ultraviolet to Visible Dispersed Single Photon Detection for Highly Sensitive Sensing of Fundamental Processes in Diverse Samples. Materials 2018, 11, 869. [Google Scholar] [CrossRef] [Green Version]
- Rashid, S.; Sit, A.; West, B.; Mayer, P.M. Colliding the hydrocarbon building blocks of astrochemical polycyclic aromatic hydrocarbons with 8 keV and ions: Luminescence from methane, acetylene, benzene and naphthalene. Chem. Phys. Lett. 2017, 667, 129–136. [Google Scholar] [CrossRef]
- Baumann, M.; Baxendale, I. An overview of the synthetic routes to the best selling drugs containing 6-membered heterocycles. Beilstein J. Org. Chem. 2013, 9, 2265–2319. [Google Scholar] [CrossRef] [Green Version]
- Kiuru, P.; Yli-Kauhaluoma, J. Pyridine and Its Derivatives. In Heterocycles in Natural Product Synthesis; Majumdar, K., Chattopadhyay, S.K., Eds.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2011; pp. 267–297. [Google Scholar]
- Callaghan, M.P.; Smith, K.E.; Cleaves, H.J.; Ruzicka, J.; Stern, J.C.; Glavin, D.P.; House, C.H.; Dworkin, J.P. Carbonaceous meteorites contain a wide range of extraterrestrial nucleobases. Proc. Natl. Acad. Sci. USA 2011, 108, 13995–13999. [Google Scholar] [CrossRef] [Green Version]
- Smith, K.E.; Callahan, M.P.; Gerakines, P.A.; Dworkin, J.P.; House, C.H. Investigation of pyridine carboxylic acids in CM2 carbonaceous chondrites: Potential precursor molecules for ancient coezymes. Geochim. Cosmichim. Acta 2014, 136, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Charnley, S.B.; Kuan, Y.-J.; Huang, H.-C.; Botta, O.; Butner, H.M.; Cox, N.; Despois, D.; Ehrenfreund, P.; Kisiel, Z.; Lee, Y.-Y.; et al. Astronomical Searches for Nitrogen Heterocycles. Adv. Space Res. 2005, 36, 137–145. [Google Scholar] [CrossRef]
- Zdanovskaia, M.A.; Dorman, P.M.; Orr, V.L.; Owen, A.N.; Kougias, S.M.; Esselman, B.J.; Woods, R.C.; McMahon, R.J. Rotational Spectra of Three Cyanobutadiene Isomers (C5H5N) of Relevance to Astrochemistry and Other Harsh Reaction Environments. J. Am. Chem. Soc. 2021, 143, 9551–9564. [Google Scholar] [CrossRef]
- Zhai, Y.-E.; Shi, D.-Q. Synthesis and Herbicidal Activity of 2-Alkyl(aryl)-4-amino-3-[alkyl(alkoxy)carbonyl]- 5-cyano-6-[(3-trifluoromethyl)phenoxy]-pyridines. J. Heterocycl. Chem. 2013, 50, 1039–1042. [Google Scholar] [CrossRef]
- Fondren, L.D.; McLain, J.; Jackson, D.M.; Adams, N.G.; Babcock, L.M. Studies of Reactions of a Series of Ions With Nitrogen Containing Heterocyclic Molecules Using a Selected Ion Flow Tube. Int. J. Mass Spectrom. 2007, 265, 60–67. [Google Scholar] [CrossRef]
- Lucas, M.; Thomas, A.M.; Kaiser, R.I.; Bashkirov, E.K.; Azyazov, V.N.; Mebel, A.M. Combined Experimental and Computational Investigation of the Elementary Reaction of Ground State Atomic Carbon (C; 3Pj) with Pyridine (C5H5N; X1A1) via Ring Expansion and Ring Degradation Pathways. J. Phys. Chem. A 2018, 122, 3128–3139. [Google Scholar] [CrossRef]
- Recio, P.; Marchione, D.; Caracciolo, A.; Murray, V.J.; Mancini, L.; Rosi, M.; Casavecchia, P.; Balucani, N. A crossed molecular beam investigation of the N(2D) + pyridine reaction and implications for prebiotic chemistry. Chem. Phys. Lett. 2021, 779, 138852. [Google Scholar] [CrossRef]
- Feldman, U.; Landi, E.; Schwadron, N.A. On the sources of fast and slow solar wind. J. Geophys. Res. 2005, 110, A07109. [Google Scholar] [CrossRef] [Green Version]
- Zeitlin, C.; Hassler, D.M.; Cucinotta, F.A.; Ehresmann, B.; Wimmer-Schweingruber, R.F.; Brinza, D.E.; Kang, S.; Weigle, G.; Böttcher, S.; Böhm, E.; et al. Measurements of Energetic Particle Radiation in Transit to Mars on the Mars Science Laboratory. Science 2013, 340, 1080–1084. [Google Scholar] [CrossRef] [Green Version]
- Yogo, A.; Sato, K.; Nishikino, M.; Mori, M.; Teshima, T.; Numasaki, H.; Murakami, M.; Demizu, Y.; Akagi, S.; Nagayama, S.; et al. Application of laser-accelerated protons to the demonstration of DNA double-strand breaks in human cancer cells. Appl. Phys. Lett. 2009, 94, 181502. [Google Scholar] [CrossRef]
- Allison, R.R.; Sibata, C.; Patel, R. Future radiation therapy: Photons, protons and particles. Future Oncol. 2013, 9, 493. [Google Scholar] [CrossRef]
- Mein, S.; Tessonnier, T.; Kopp, B.; Harrabi, S.; Abdollahi, A.; Debus, J.; Haberer, T.; Mairani, A. Spot-Scanning Hadron Arc (SHArc) Therapy: A Study With Light and Heavy Ions. Adv. Rad. Oncol. 2021, 6, 100661. [Google Scholar] [CrossRef]
- Loeffler, J.S.; Durante, M. Charged particle therapy—optimization, challenges and future directions. Nat. Rev. Clin. Oncol. 2013, 10, 411. [Google Scholar] [CrossRef]
- Schulz-Ertner, D.; Jäkel, O.; Schlegel, W. Radiation Therapy with Charged Particles. Semin. Radiat. Oncol. 2006, 16, 249. [Google Scholar] [CrossRef]
- Tsujii, H.; Kamada, T.; Baba, M.; Tsuji, H.; Kato, H.; Kato, S.; Yamada, S.; Yasuda, S.; Yanagi, T.; Kato, H.; et al. Clinical advantages of carbon-ion radiotherapy. New J. Phys. 2008, 10, 075009. [Google Scholar] [CrossRef]
- Luque, J.; Crosley, D.R. Lifbase: Database and Spectral Simulation (Version 1.5), SRI International Report MP 99-009. 1999. Available online: https://www.sri.com/case-studies/lifbase-spectroscopy-tool/ (accessed on 29 October 2021).
- Brzozowski, J.; Bunker, P.; Elander, N.; Erman, P. Predissociation efects in the A, B, and C states of CH and the interstellar formation rate of CH via inverse predissociation. Astrophys. J. 1976, 207, 414–424. [Google Scholar] [CrossRef]
- Zachwieja, M. New Investigations of the A2Δ-X2Π Band System in the CH Radical and a New Reduction of the Vibration-Rotation Spectrum of CH from the ATMOS Spectra. J. Mol. Spectroc. 1995, 170, 285–309. [Google Scholar] [CrossRef]
- Luque, J.; Crosley, D.R. Electronic transition moment and rotational transition probabilities in CH. I. A2Δ-X2Π system. J. Chem. Phys. 1996, 104, 2146. [Google Scholar] [CrossRef]
- Cerny, D.; Bacis, R.; Guelavchvili, G.; Roux, F. Extensive analysis of the red system of the CN molecule with a high resolution Fourier Spectrometer. J. Mol. Spectrosc. 1978, 73, 154. [Google Scholar] [CrossRef]
- Ito, H.; Ozaki, Y.; Suzuki, K.; Kondow, T.; Kuchitsu, K. Analysis of the B2Σ+ ~ A2Πi perturbations in the CN(B2Σ+-X2Σ+) main band system: I. Molecular constants for B2Σ+ and A2Πi. J. Mol. Spectrosc. 1988, 127, 283. [Google Scholar] [CrossRef]
- Mogyorosi, K.; Sarosi, K.; Chikan, V. Direct Production of CH(A2Δ) Radical from Intense Femtosecond Near-IR Laser Pulses. J. Phys. Chem. A 2020, 124, 8112–8119. [Google Scholar] [CrossRef] [PubMed]
- Mogyorosi, K.; Sarosi, K.; Seres, I.; Jojart, P.; Fule, M.; Chikan, V. Formation of CN Radical from Nitrogen and Carbon Condensation and from Photodissociation in Femtosecond Laser-Induced Plasmas: Time-Resolved FT-UV−Vis Spectroscopic Study of the Violet Emission of CN Radical. J. Phys. Chem. A 2020, 124, 2755–2767. [Google Scholar] [CrossRef] [PubMed]
- Trentelman, K.A.; Kable, S.H.; Moss, D.B.; Houston, P.L. Photodissociation dynamics of acetone at 193 nm: Photofragment internal and translational energy distributions. J. Chem. Phys. 1989, 91, 7498. [Google Scholar] [CrossRef]
- Lin, M.-F.; Dyakov, Y.A.; Tseng, C.-M.; Mebel, A.M.; Lin, S.H.; Lee, Y.T.; Ni, C.-K. Photodissociation dynamics of pyridine. J. Chem. Phys. 2005, 123, 054309. [Google Scholar] [CrossRef] [Green Version]
- Chikan, V.; Fournier, F.; Leone, S.R.; Nizamov, B. State-Resolved Dynamics of the CH(A2Δ) Channels from Single and Multiple Photon Dissociation of Bromoform in the 10–20 eV Energy Range. J. Phys. Chem. A 2006, 110, 2850–2857. [Google Scholar] [CrossRef]
- Pei, L.; Farrar, J.M. Imaging ion-molecule reactions: Charge transfer and C–N bond formation in the C+ + NH3 system. J. Chem. Phys. 2012, 136, 204305. [Google Scholar] [CrossRef] [Green Version]
- Bethe, H.E.; Salpeter, E.E. Quantum Mechanics of One-and Two-Electron Atoms; Plenum: New York, NY, USA, 1977. [Google Scholar]
- Windholz, L.; Winklhofer, E.; Drozdowski, R.; Kwela, J.; Wasowicz, T.J.; Heldt, J. Stark effect of atomic Helium second triplet series in electric fields up to 1600 kV/cm. Phys. Scr. 2008, 78, 065303. [Google Scholar] [CrossRef]
- Windholz, L.; Drozdowski, R.; Wasowicz, T.J.; Kwela, J. Anticrossing effects in Stark spectra of helium. Proc. SPIE 2005, 5849, 24–28. [Google Scholar]
- Windholz, L.; Drozdowski, R.; Wasowicz, T.; Kwela, J. Stark effect in He I in extremely high electric field. Opt. Appl. 2006, 36, 569–574. [Google Scholar]
- Windholz, L.; Wasowicz, T.J.; Drozdowski, R.; Kwela, J. Stark effect of atomic Helium singlet lines. J. Opt. Soc. Am. B 2012, 29, 934–943. [Google Scholar] [CrossRef]
- Hoekstra, R.; de Heer, F.J.; Morgenstern, R. State-selective electron capture in collisions of He2+ with H. J. Phys. B Mol. Opt. Phys. 1991, 24, 4025. [Google Scholar] [CrossRef]
- Ramsey, N.F. Thermal Beam Sources. In Atomic, Molecular, and Optical Physics: Atoms and Molecules; Dunning, F.B., Hulet, R.D., Eds.; Academic Press, Inc.: San Diego, CA, USA, 1996; Volume 29B. [Google Scholar]
- Bacchus-Montabonel, M.-C. Looking at Radiation Damage on Prebiotic Building Blocks. J. Phys. Chem. A 2013, 117, 14169–14175. [Google Scholar] [CrossRef]
- Akbulut, M.; Sack, N.J.; Madey, T.E. Elastic and Inelastic Processes in the Interaction of l-10 eV Ions with Solids: Ion Transport through Surface Layers. Surf. Sci. Rep. 1997, 28, 177–245. [Google Scholar] [CrossRef]
- Alvarado, F.; Bari, S.; Hoekstra, R.; Schlathölter, T. Quantification of Ion-Induced Molecular Fragmentation of Isolated 2-Deoxy-D-ribose Molecules. Phys. Chem. Chem. Phys. 2006, 8, 1922–1928. [Google Scholar] [CrossRef] [PubMed]
- Bowen, R.D. Ion-Neutral Complexes. Acc. Chem. Res. 1991, 24, 364–371. [Google Scholar] [CrossRef]
- Sladek, V.; Skorna, P.; Poliak, P.; Lukes, V. The ab initio Study of Halogen and Hydrogen σN-Bonded Para-Substituted Pyridine (X2/XY/HX) Complexes. Chem. Phys. Lett. 2015, 619, 7–13. [Google Scholar] [CrossRef]
- Wren, S.W.; Vogelhuber, K.M.; Garver, J.M.; Kato, S.; Sheps, L.; Bierbaum, V.M.; Lineberger, W.C. C–H Bond Strengths and Acidities in Aromatic Systems: Effects of Nitrogen Incorporation in Mono-, Di-, and Triazines. J. Am. Chem. Soc. 2012, 134, 6584. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Salumbides, E.J.; Hollenstein, U.; Koelemeij, J.C.J.; Eikema, K.S.E.; Ubachs, W.; Merkt, F. Determination of the Ionization and Dissociation Energies of the Hydrogen Molecule. J. Chem. Phys. 2009, 130, 174306. [Google Scholar] [CrossRef] [Green Version]
- Jiao, C.Q.; DeJoseph, J.C.A.; Lee, R.; Garscadden, A. Kinetics of electron impact ionization and ion-molecule reactions of pyridine. Int. J. Mass Spectrom. 2006, 257, 34–40. [Google Scholar] [CrossRef]
- NIST Chemistry WebBook. NIST Standard Reference Database Number 69; Linstrom, P.J., Mallard, W.G., Eds.; National Institute of Standards and Technology: Gaithersburg, MD, USA, 2021.
- Smialek, M.A.; MacDonald, M.A.; Ptasinska, S.; Zuin, L.; Mason, N.J. Photoelectron and Threshold Photoelectron Valence Spectra of Pyridine. Eur. Phys. J. D 2016, 70, 42. [Google Scholar] [CrossRef] [Green Version]
- Blanksby, S.J.; Ellison, G.B. Bond Dissociation Energies of Organic Molecules. Acc. Chem. Res. 2003, 36, 255. [Google Scholar] [CrossRef]
- Vall-llosera, G.; Coreno, M.; Erman, P.; Huels, M.A.; Jakubowska, K.; Kivimäki, A.; Rachlew, E.; Stankiewicz, M. VUV photoionization of free azabenzenes: Pyridine, pyrazine, pyrimidine, pyridazine, and s-triazine. Int. J. Mass Spectrom. 2008, 275, 55–63. [Google Scholar] [CrossRef]
- Gappa, A.; Herpers, E.; Herrmann, R.; Huelsewede, V.; Kappert, W.; Klar, M.; Kirmse, W. Ion−Molecule Complexes in 1,2-Alkyl Shifts. J. Am. Chem. Soc. 1995, 117, 12096–12106. [Google Scholar] [CrossRef]
- Ijaz, W.; Gregg, Z.; Barnes, G.L. Complex Formation during SID and Its Effect on Proton Mobility. J. Phys. Chem. Lett. 2013, 4, 3935–3939. [Google Scholar] [CrossRef]
- Amunugama, R.; Rodgers, M.T. Absolute Alkali Metal Ion Binding Affinities of Several Azines Determined by Threshold Collision-Induced Dissociation and ab initio Theory. Int. J. Mass Spectrom. 2000, 195–196, 439–457. [Google Scholar] [CrossRef]
- Wang, Z.; Zheng, B.; Yu, X.; Li, X.; Yi, P. Structure, Properties, and Nature of the Pyridine-XY (X, Y = F, Cl, Br) Complexes: An ab initio Study. J. Chem. Phys. 2010, 132, 164104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, J.; Yan, H.; Jin, Y.; Dai, G.; Zhong, A. Characteristics and Nature of the Intermolecular Interactions between Pyridine and Various Hydrides: A Theoretical Study. J. Mol. Struct. THEOCHEM 2010, 944, 70–75. [Google Scholar] [CrossRef]
- El-Shall, M.S.; Ibrahim, Y.; Alsharaeh, E.H.; Meot-Ner, M.; Watson, S.P. Reactions between Aromatic Hydrocarbons and Heterocycles: Covalent and Proton-bond Dimer Cations of Benzene/Pyridine. J. Am. Chem. Soc. 2009, 131, 10066–10076. [Google Scholar] [CrossRef]
- Feng, J.-Y.; Lee, Y.-P.; Witek, H.A.; Ebata, T. Vacuum Ultraviolet Photoionization Induced Proton Migration and Formation of a New C−N Bond in Pyridine Clusters Revealed by Infrared Spectroscopy and Mass Spectrometry. J. Phys. Chem. Lett. 2021, 12, 4936–4943. [Google Scholar] [CrossRef]
- Rap, D.B.; Marimuthu, A.N.; Redlich, B.; Brünken, S. Stable Isomeric Structures of the Pyridine Cation (C5H5N•+) and Protonated Pyridine (C5H5NH•+) elucidated by Cold Ion Infrared Spectroscopy. J. Mol. Spectrosc. 2020, 373, 111357. [Google Scholar] [CrossRef]
- Mackie, J.C.; Colket, M.B.; Nelson, P.F. Shock Tube Pyrolysis of Pyridine. J. Phys. Chem. 1990, 94, 4099–4106. [Google Scholar] [CrossRef]
- Kiefer, J.H.; Zhang, Q.; Kern, R.D.; Yao, J.; Jursic, B. Pyrolyses of Aromatic Azines: Pyrazine, Pyrimidine, and Pyridine. J. Phys. Chem. A 1997, 101, 7061–7073. [Google Scholar] [CrossRef]
- Hore, N.R.; Russell, D.K. Radical Pathways in the Thermal Decomposition of Pyridine and Diazines: A Laser Pyrolysis and Semi-empirical Study. J. Chem. Soc. Perkin Trans. 2 1998, 2, 269–276. [Google Scholar] [CrossRef]
- Ni, C.-K.; Tseng, C.-M.; Lin, M.F.; Dyakov, Y.A. Photodissociation Dynamics of Small Aromatic Molecules Studied by Multimass Ion Imaging. J. Phys. Chem. B 2007, 111, 12631–12642. [Google Scholar] [CrossRef]
- Zhong, D.; Diau, E.W.-G.; Bernhardt, T.; De Feyter, S.; Roberts, J.D.; Zewail, A.H. Femtosecond dynamics of valence-bond isomers of azines: Transition states and conical intersections. Chem. Phys. Lett. 1998, 298, 129. [Google Scholar] [CrossRef]
- Lobastov, V.A.; Srinivasan, R.; Goodson, B.M.; Ruan, C.-Y.; Feenstra, J.S.; Zewail, A.H. Ultrafast Diffraction of Transient Molecular Structures in Radiationless Transitions. J. Phys. Chem. A 2001, 105, 11159. [Google Scholar] [CrossRef]
- Ehbrecht, A.; Kowalski, A.; Ottinger, C. Hot-atom chemiluminescence: A beam study of the reactions C(3P) + H2→CH (A2Δ, B2Σ−, C2Σ+) + H. Chem. Phys. Lett. 1998, 284, 205–213. [Google Scholar] [CrossRef]
- Drozdowski, R.; Kowalski, A. Luminescence cross sections in the low-energy collisions of H+, H2+, and H3+ ions with H2. Eur. Phys. J. D 2018, 72, 220. [Google Scholar] [CrossRef]
- Glenewinkel-Meyer, T.; Muller, B.; Ottinger, C.; Tischer, H. Measurement of the HCl+(A 2Σ+–X 2Π) electronic transition moment using quasiresonant charge transfer at low energy. J. Chem. Phys. 1988, 88, 3475. [Google Scholar] [CrossRef]
- Pyridine. Available online: https://www.sigmaaldrich.com/PL/pl/product/sial/270970 (accessed on 23 November 2021).








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Transition | H+ + C5H5N | H2+ + C5H5N | He+ + C5H5N | |||
|---|---|---|---|---|---|---|
| Tv [K] | TR [K] | Tv [K] | TR [K] | Tv [K] | TR [K] | |
| CH(A2Δ→X2Πr) Δν = 0 | 7900 | 4100 | 9500 | 4200 | 10,000 | 4800 |
| CH(B2Σ−→ X2Πr) Δν = 0 | 3000 | 3200 | 3800 | 3500 | 3900 | 3500 |
| CN(B2Σ+→ X2Σ+) Δν = 0 | 9000 | 4500 | 9500 | 5500 | 13,500 | 6000 |
| Fragment | RA (%) | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| H+ + C5H5N | H2+ + C5H5N | He+ + C5H5N † | H+ + C4H8O | H2+ + C4H8O | H3+ + C4H8O | |||||||
| Hβ | 43.6 (6.0) | 61.4 | 32.0 (3.7) | 45.9 | 5.1 (0.6) | 10.8 | 67.4 (5.2) | 88.8 | 57.4 (4.5) | 76.2 | 51.1 (3.0) | 67.3 |
| Hγ | 12.5 (2.0) | 9.6 (1.1) | 3.6 (0.5) | 15.0 (2.0) | 12.5 (2.2) | 11.2 (1.8) | ||||||
| Hδ | 3.6 (0.7) | 2.6 (0.3) | 2.1 (0.4) | 4.7 (1.0) | 4.7 (0.9) | 3.5 (0.7) | ||||||
| Hε | 1.6 (0.3) | 1.7 (0.2) | - | 1.7 (0.5) | 1.6 (0.5) | 1.5 (0.4) | ||||||
| CH(A2Δ→X2Πr) Δν = 0 | 19.9 (3.4) | 26.9 | 30.6 (3.7) | 40.4 | 28.2 (1.4) | 42.2 | 8.8 (2.0) | 11.2 | 18.9 (1.9) | 23.8 | 26.6 (2.3) | 32.7 |
| CH(B2Σ−→X2Πr) Δν = 0 | 4.1 (0.6) | 5.6 (0.7) | 11.7 (0.9) | 2.4 (0.9) | 4.9 (1.0) | 6.1 (0.8) | ||||||
| CH(C2Σ+→X2Πr) Δν = 0 | 2.9 (0.4) | 4.2 (0.6) | 2.3 (0.5) | - | - | - | ||||||
| CN(B2Σ+→ X2Σ+) Δν = 0 | 7.1 (1.3) | 9.2 | 7.9 (0.9) | 10.4 | 23.4 (1.3) | 27.8 | - | - | - | - | - | - |
| CN(B2Σ+→ X2Σ+) Δν = 1 | 2.1 (0.3) | 2.5 (0.3) | 4.4 (0.8) | - | - | - | ||||||
| NH(A3Π→X3Σ−) Δν = 0 | 1.0 (0.2) | 1.0 | 1.5 (0.4) | 1.5 | 1.0 (0.4) | 1.0 | - | - | - | - | - | - |
| C (2p3s 1P1→2p2 1D2) λ=193.1 nm | 1.1 (0.3) | 1.5 | 1.0 (0.2) | 1.8 | 2.4 (0.5) | 3.3 | - | - | - | - | - | - |
| C (2p3s 1P1→2p2 1S0) λ=247.9 nm | 0.4 (0.1) | 0.8 (0.4) | 0.9 (0.3) | - | - | - | ||||||
| C2 Δν = 0, 1 | - | - | - | - | 13.3 (1.0) | 13.3 | - | - | - | - | - | - |
| He | - | - | - | - | 1.6 (0.4) | 1.6 | - | - | - | - | - | - |
| No. | Reactants | Products | ETH [eV] | Collisional Process |
|---|---|---|---|---|
| 1. | H+ + C5H5N | H + C5H5N+ | −4.40 | CT |
| 2. | H(n = 4) + C5H5N+ | 8.35 | CT | |
| 3. | H + C5H4N+ + H(n = 4) | 13.13 | CT | |
| 4. | H + NCCHCHCHCH+ + H(n = 4) | 14.24 | CT | |
| 5. | H+ + C5H4N + H(n = 4) | 17.53 | DE | |
| 6. | H+ + NCCHCHCHCH + H(n = 4) | 18.64 | DE | |
| 7. | H+ + C5H4N+ + H(n = 4) | 27.75 | DI | |
| 8. | H + NCCHCH+ + CH(A2Δ)+ CH2 | 9.48 | CT | |
| 9. | H + NCCHCHCH+ + CH(A2Δ) + H | 9.61 | CT | |
| 10. | H+ + NCCHCH + CH(A2Δ)+ CH2 | 13.88 | DE | |
| 11. | H+ + NCCHCHCH + CH(A2Δ) + H | 14.01 | DE | |
| 12. | H+ + C3H3N+ + CH(A2Δ) + CH | 26.68 | DI | |
| 13. | H + CN(B2Σ+) + CHCHCHCH2+ | 5.17 | CT | |
| 14. | H + CN(B2Σ+) + H + CH2CCHCH+ | 7.00 | CT | |
| 15. | H+ + CN(B2Σ+) + CHCHCHCH2 | 9.54 | DE | |
| 16. | H+ + CN(B2Σ+) + H + CH2CCHCH | 11.40 | DE | |
| 17. | H+ + CN(B2Σ+) + H+C4H4+ | 22.61 | DI | |
| 18. | H2+ + C5H5N | H2 + C5H5N+ | −6.23 | CT |
| 19. | H + H(n = 4) + C5H5N+ | 9.17 | CT | |
| 20. | H2 + C5H4N+ + H(n = 4) | 11.30 | CT | |
| 21. | H2 + NCCHCHCHCH+ + H(n = 4) | 12.41 | CT | |
| 22. | H+ + H(n=4)+C5H5N | 17.23 | DP | |
| 23. | H2++C5H4N+H(n = 4) | 17.53 | DE | |
| 24. | H2+ + NCCHCHCHCH+H(n = 4) | 18.64 | DE | |
| 25. | H2+ + C5H4N+ + H(n = 4) | 27.75 | DI | |
| 26. | H2 + NCCHCH+ + CH(A2Δ) + CH2 | 7.65 | CT | |
| 27. | H2 + NCCHCHCH+ + CH(A2Δ) + H | 7.78 | CT | |
| 28. | H2+ + NCCHCH + CH(A2Δ) + CH2 | 13.88 | DE | |
| 29. | H2+ + NCCHCHCH + CH(A2Δ) + H | 14.01 | DE | |
| 30. | H2+ + C3H3N+ + CH(A2Δ)+CH | 26.68 | DI | |
| 31. | H2 + CN(B2Σ+) + CHCHCHCH2+ | 3.31 | CT | |
| 32. | H2 + CN(B2Σ+) + H + CH2CCHCH+ | 5.17 | CT | |
| 33. | H2+ + CN(B2Σ+) + CHCHCHCH2 | 9.54 | DE | |
| 34. | H2+ + CN(B2Σ+) + H + CH2CCHCH | 11.40 | DE | |
| 35. | H2+ + CN(B2Σ+) + H + C4H4+ | 22.61 | DI |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wasowicz, T.J. Neutral Dissociation of Pyridine Evoked by Irradiation of Ionized Atomic and Molecular Hydrogen Beams. Int. J. Mol. Sci. 2022, 23, 205. https://doi.org/10.3390/ijms23010205
Wasowicz TJ. Neutral Dissociation of Pyridine Evoked by Irradiation of Ionized Atomic and Molecular Hydrogen Beams. International Journal of Molecular Sciences. 2022; 23(1):205. https://doi.org/10.3390/ijms23010205
Chicago/Turabian StyleWasowicz, Tomasz J. 2022. "Neutral Dissociation of Pyridine Evoked by Irradiation of Ionized Atomic and Molecular Hydrogen Beams" International Journal of Molecular Sciences 23, no. 1: 205. https://doi.org/10.3390/ijms23010205
APA StyleWasowicz, T. J. (2022). Neutral Dissociation of Pyridine Evoked by Irradiation of Ionized Atomic and Molecular Hydrogen Beams. International Journal of Molecular Sciences, 23(1), 205. https://doi.org/10.3390/ijms23010205