1. Introduction
Chirality plays a crucial role in many issues, beginning from the fundamental problem of the origin of life on the Earth up to the development of new functional materials [
1]. Biomolecules such as proteins and DNA are known to consist of L-amino acids and D-sugars. Until recently, the homochirality of proteins was believed to be maintained during the lifespan of an organism. However, proteins with D-amino acids have been detected in various tissues, and their amounts have been shown to increase with organism aging. According to recent studies, the replacement of L-amino acids by D-analogs during aging is one of the causes of Alzheimer’s and Parkinson’s [
1,
2]. It was found that optical isomers may undergo so-called chiral inversion—conversion of one optical configuration to another one under various conditions, e.g., change of temperature, solvent, under UV irradiation, action of enzymes and other chiral substances, etc. [
1]. On the other hand, the chirality of biomolecules also produces rigid requirements on the pharmaceutical industry, since the optical isomers of drugs (enantiomers) often have different and sometimes opposite therapeutic activities [
3]. Currently, more than half the drugs on the market are chiral compounds, and most of them are still produced in the form of racemates (equimolar mixtures of two enantiomers) [
3]. Numerous biochemical studies do not provide a final answer to the question of why enantiomers with identical physico-chemical properties demonstrate such differences in living organism [
4]. Thus, the difference in biological activity of enantiomers of both chiral drugs (xenobiotics) and amino acids (structural components of biomolecules) are challenging problems. Certain successes in this direction in the last decade are associated with the use of linked systems with two chiral centers [
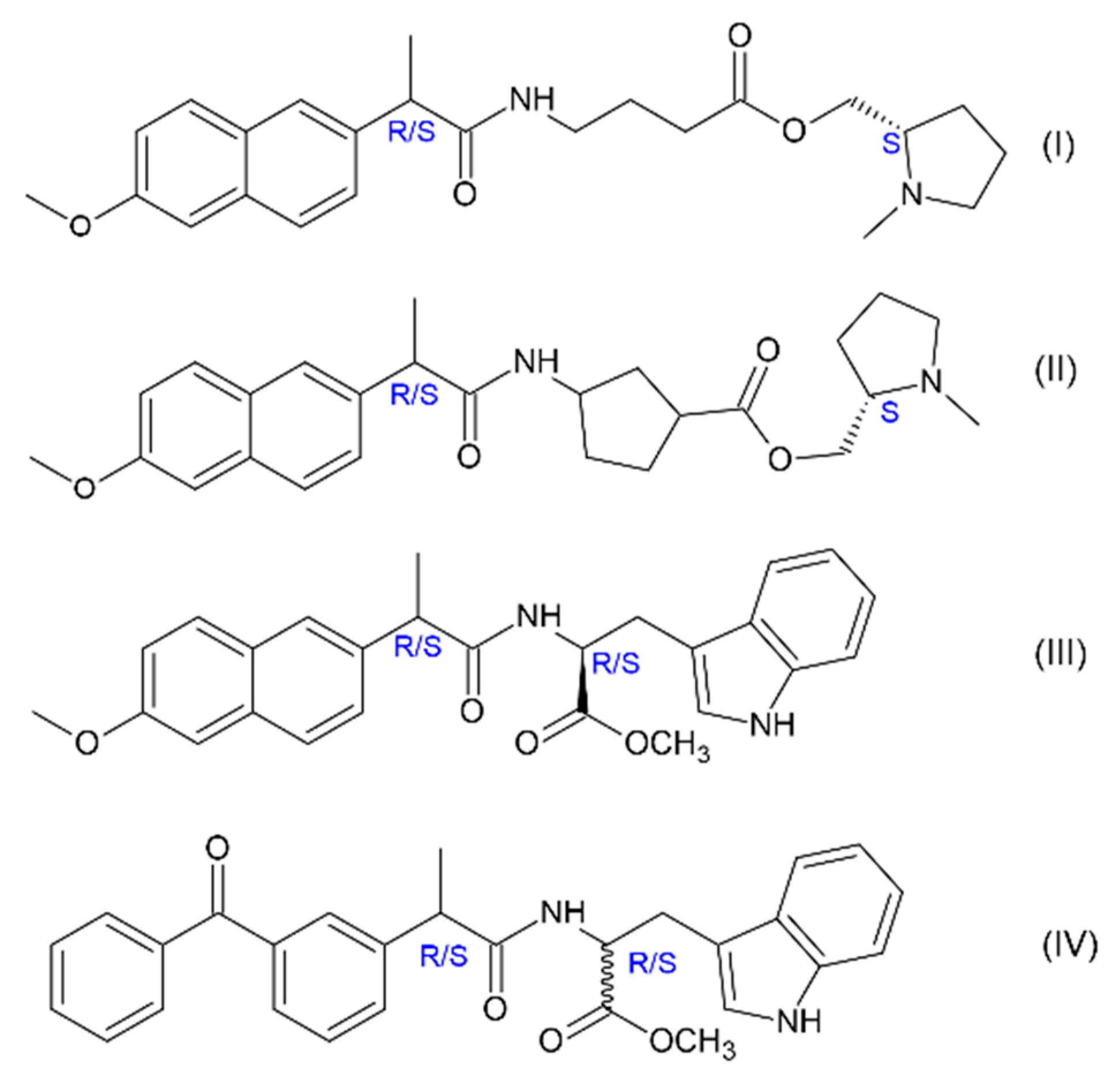
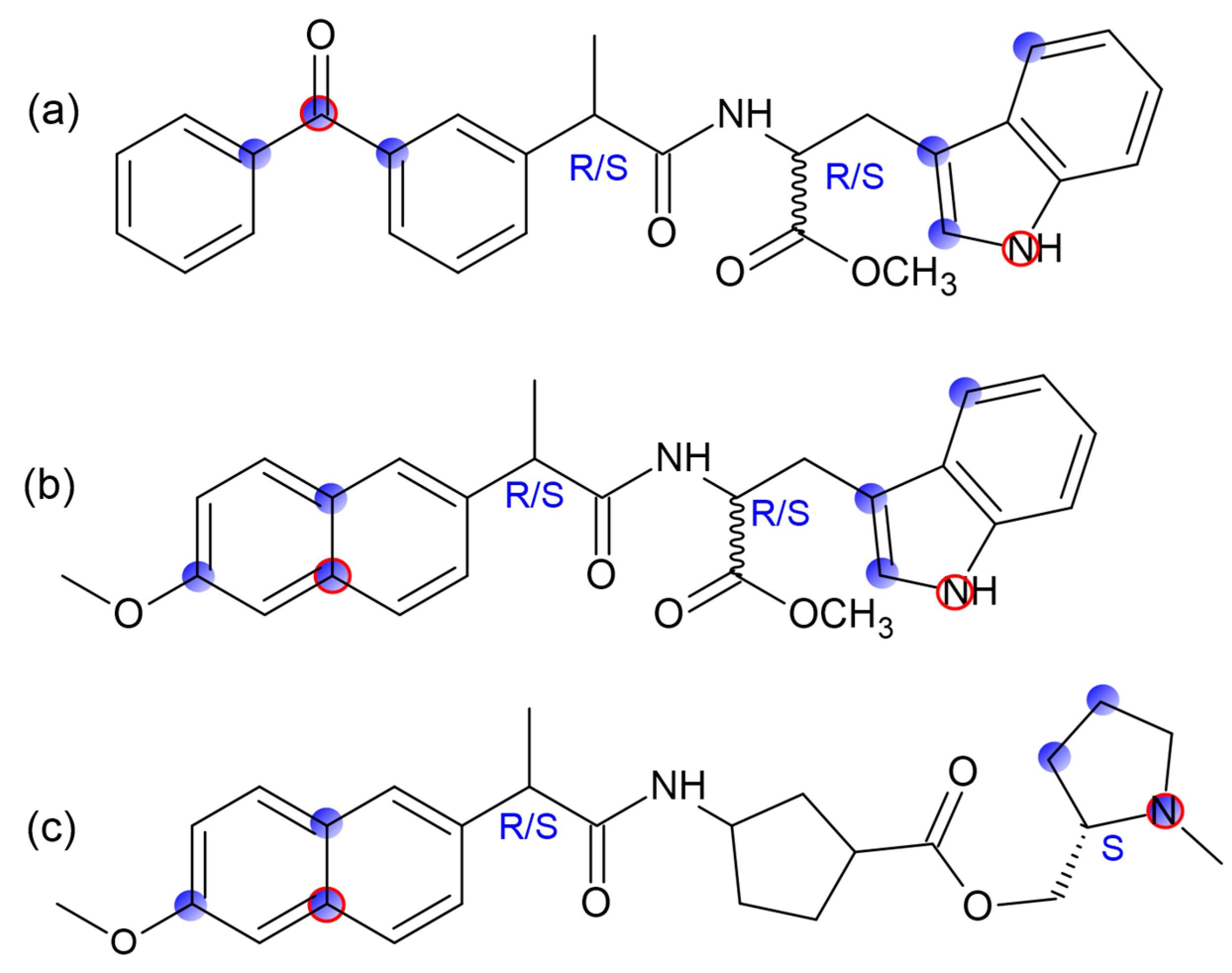
5]. Investigations of model systems—dyads where molecule of chiral drug linked with chiral electron donor seem to be promising to address this problem since elementary processes in such donor–acceptor dyads are believed to simulate binding of drug molecule with amino acid residue in active site of enzyme. For example, spin chemistry and photochemistry studies of dyads involving naproxen enantiomers linked with (
S)-N-methylpyrrolidine (I, II) and (
S)-tryptophan (III) (
Figure 1) revealed several peculiarities of electron transfer (ET)—stereo- and spin selectivity [
6]. Recent studies have also shown spin selectivity in dyads where enantiomers of naproxen and ketoprofen linked with (
R)-tryptophan (III and IV).
It is worth noting that representatives of non-steroidal anti-inflammatory drugs (NSAIDs), derivatives of propionic acid, especially naproxen, are among the most frequently prescribed medicines that exist in two enantiomeric forms with different therapeutic effects [
3]. Stereo-selectivity of ET processes in dyads with (
R)- and (
S)-naproxen and another NSAID, ketoprofen, and its relevance to biochemical studies have been described in detail [
6,
7,
8,
9].
This report aims to discuss the other peculiarity of chiral systems—spin selectivity—and highlight the relationship between this phenomenon and optical configuration of the dyad. Spin selectivity is the difference in CIDNP coefficients of diastereomers [
10]. The importance of this phenomenon is that the observed difference was explained by the difference in the distribution of spin density in paramagnetic precursors of diastereomers [
10,
11]. The latter also suggests a difference in the distribution of electron density in diastereomers. This is an important conclusion, in light of the abovementioned assumption, that processes occurring in diastereomers can simulate interaction of a chiral drug with chiral amino acid residues in the active sites of enzymes and receptors. If we consider the situation in this direction, then the difference in the distribution of electron density can be one of the real reasons for the differences in the therapeutic activity of drug enantiomers. It is well known that interactions between drug and amino acids at active sites involve charge redistribution.
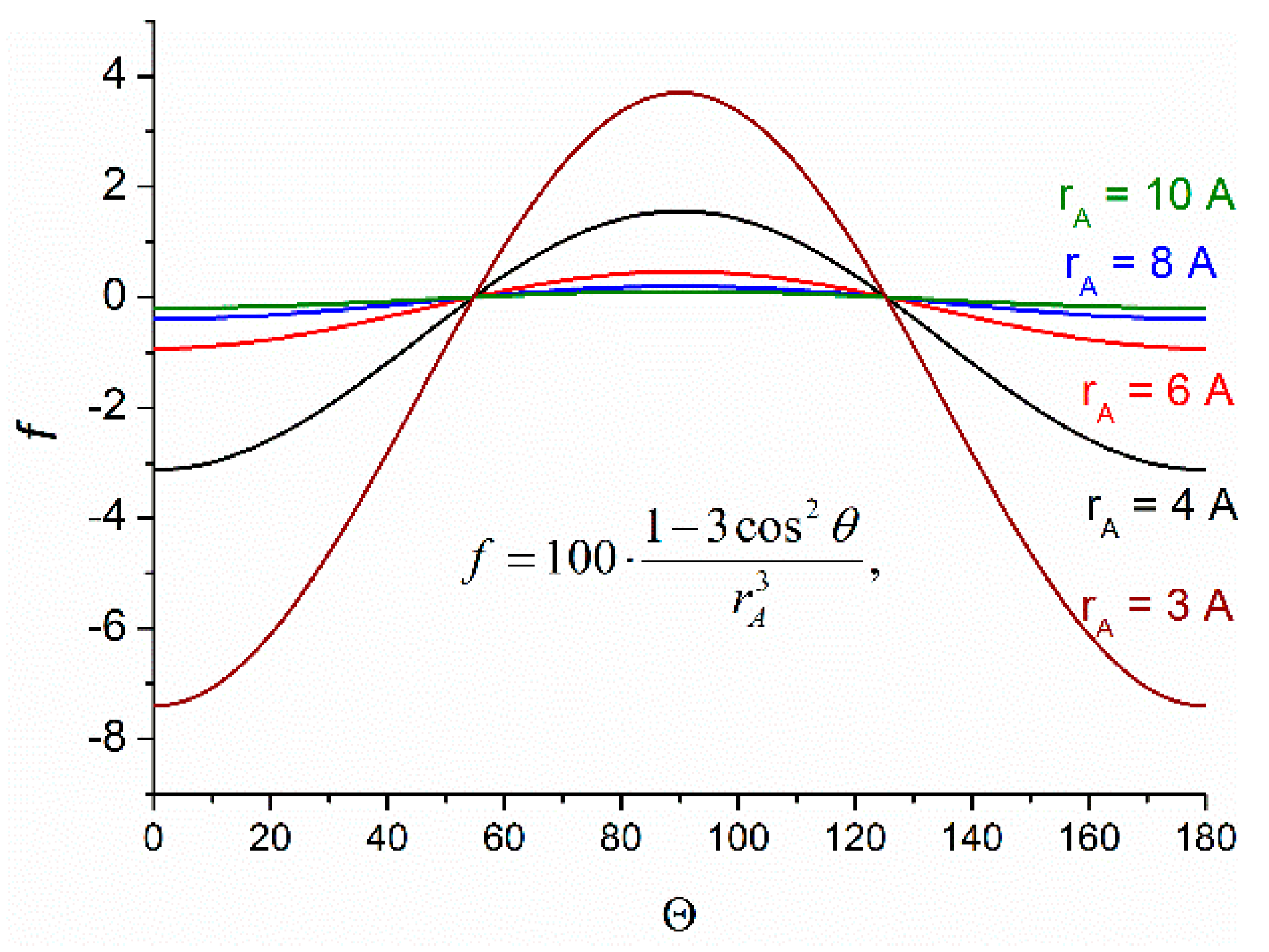
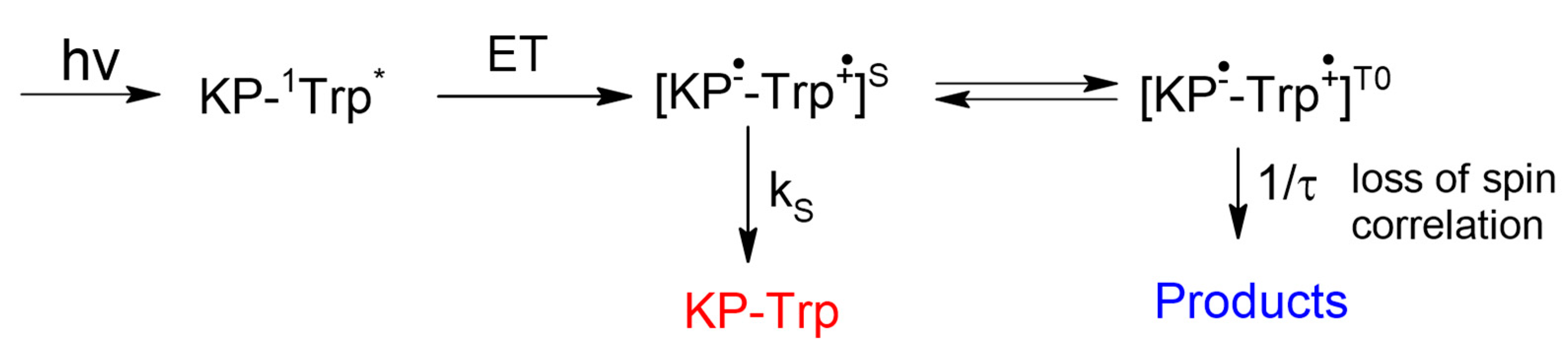
Since the spin selectivity phenomenon belongs to the area of spin effects, we should give a brief overview of chemically induced dynamic nuclear polarization (CIDNP). The CIDNP is the manifestation in NMR spectra of the products of radical reactions as signals of nuclei with the spin state populations different from Boltzmann (hyperpolarization). The phases (emission or enhanced absorption) and intensities of these signals depend on certain parameters, such as hyperfine interaction (HFI) constants, the difference in g-factors of partners in a radical pair (RP) or biradical, the multiplicity of RP precursors, and a few more parameters [
12]. In essence, CIDNP effects reflect the difference in the recombination probability of RPs with α
N and β
N nuclear spin projections on the magnetic field direction. The source of this difference is the difference in the energy of electron–nuclear interaction in RPs and in the Larmor precession frequencies of radical partners. The spin Hamiltonian describing these interactions in high magnetic field is presented below:
where
g1 and
g2 are g-factors of electrons,
β is the Bohr magneton,
B is external magnetic field induction,
S1z,
S2z and
Iiz,
Ikz are electron and nuclear spins operators (projection on
B direction), and a is the HFI constant. As a result of the analysis of the CIDNP effects, information can be obtained on the radical stages of the process, in particular, on the spin density distribution in the radical precursors of the products. This information on the spin density distribution in the paramagnetic forms of drugs and amino acids can be practically significant for drug–receptor binding known to involve charge transfer processes. It should also be noted that the role of the external magnetic field in the processes of chiral enrichment has already been predicted [
13].
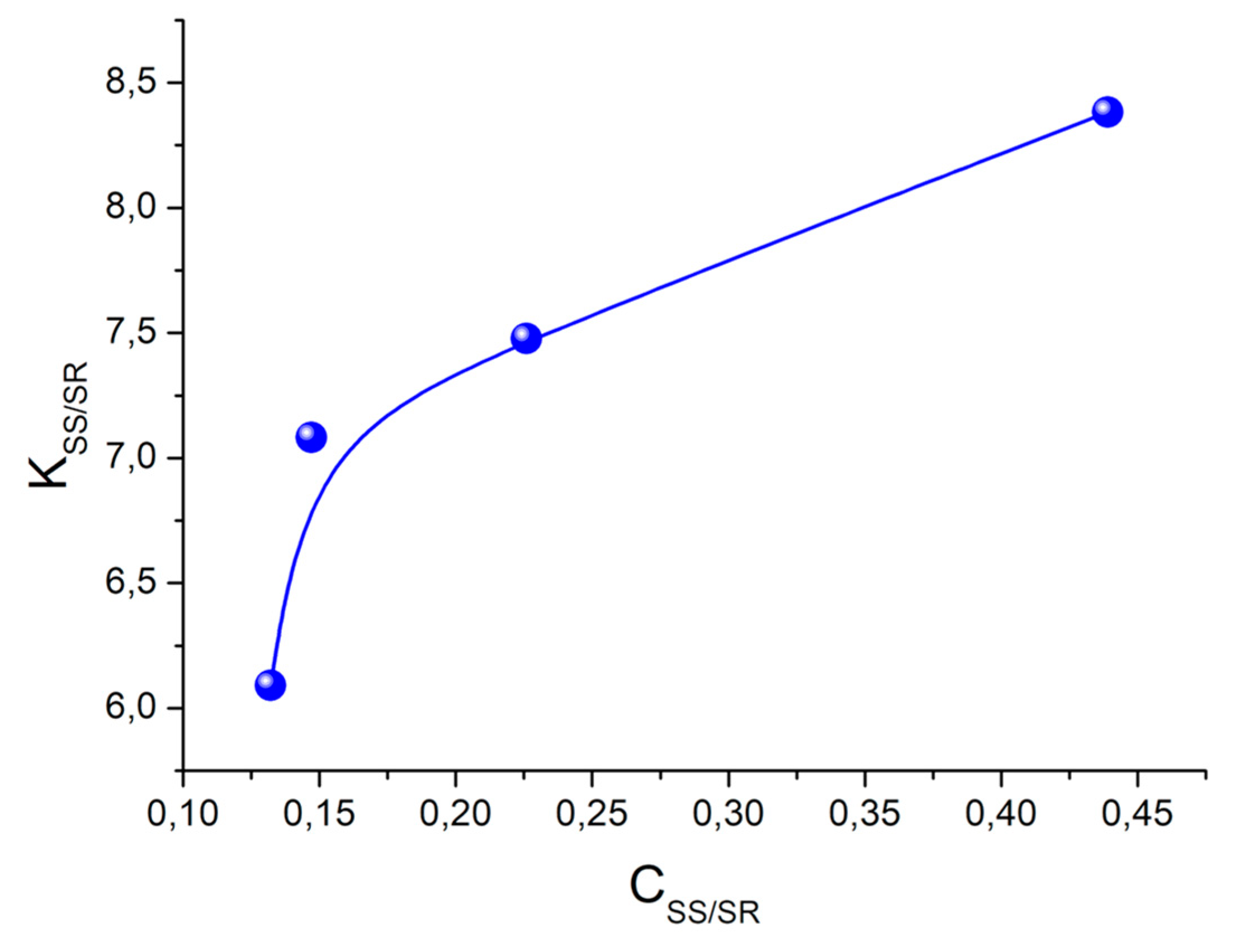
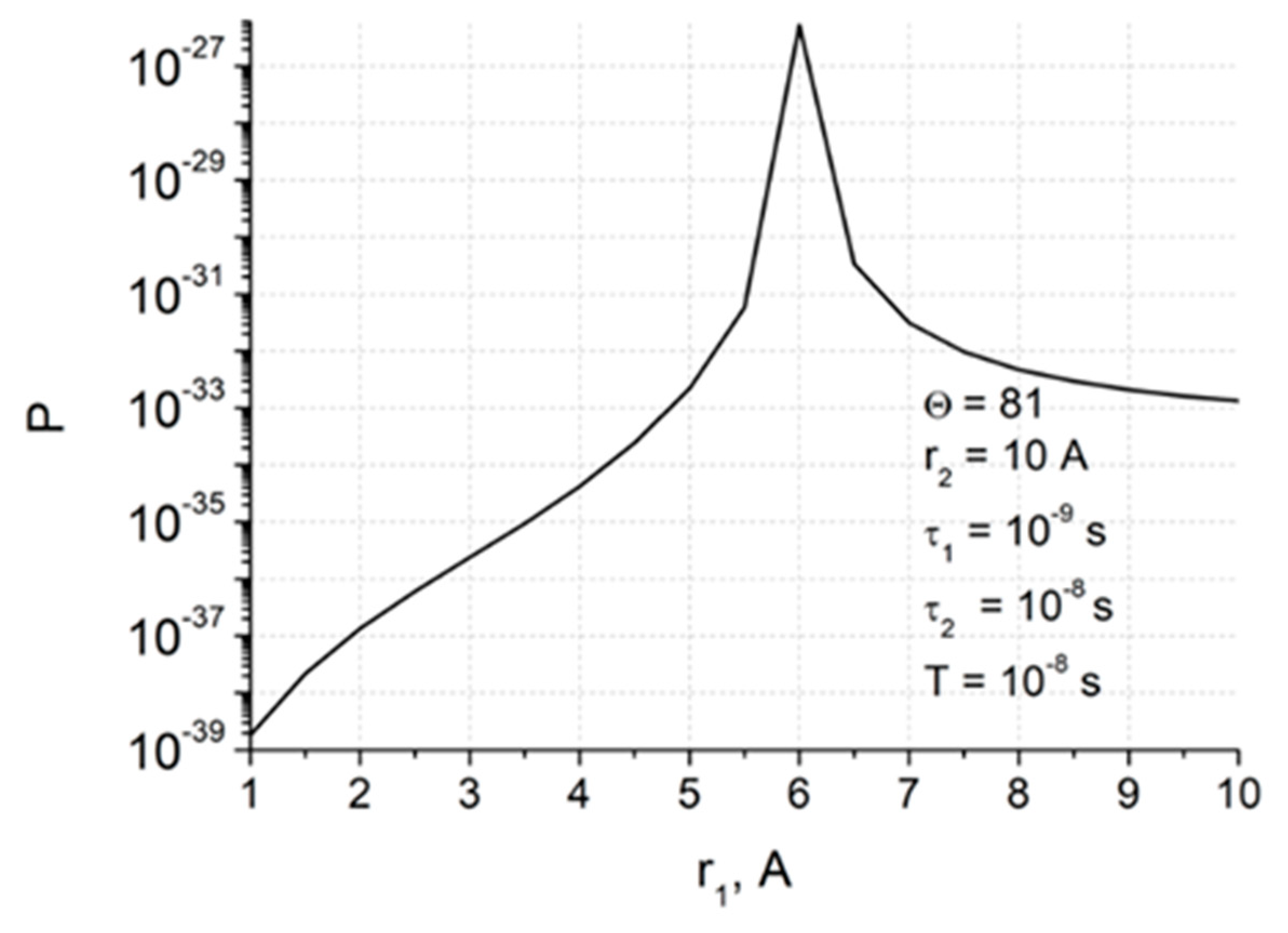
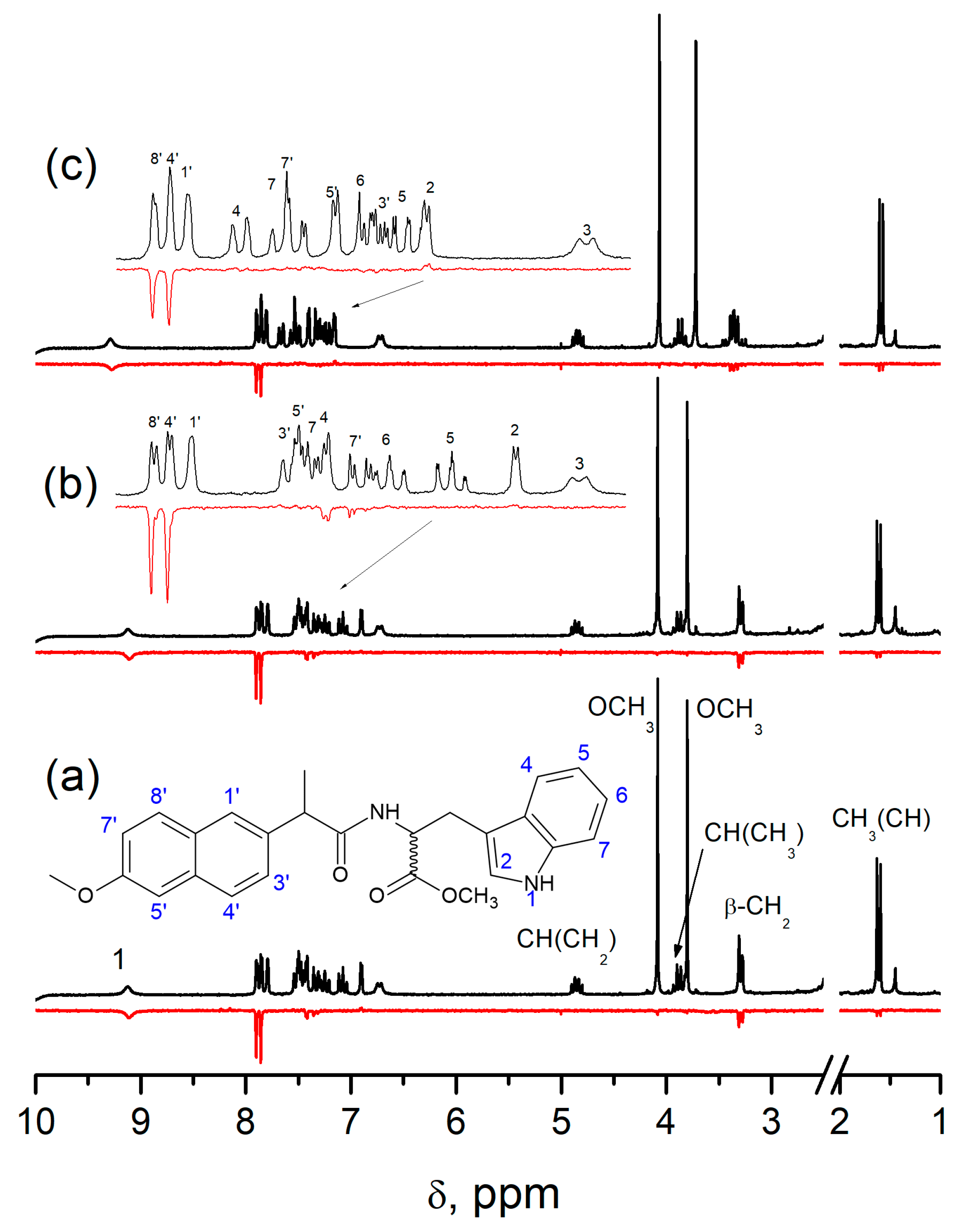
Spin selectivity is referred to as difference in the CIDNP enhancement coefficients calculated for one pair of radicals. In the case of studied dyads, it is a biradical-zwitterion (BZ). The CIDNP enhancement coefficient for a specific proton in dyad is equal to the intensity of the polarized proton signal (
Ipol) divided by the intensity of its signal in the equilibrium NMR spectrum (
Ieq) and the concentration of biradical-zwitterion (BZ) formed as result of intramolecular ET [
10]. For convenience, further, the ratio of CIDNP enhancement coefficients for diastereomers of dyads will be used:
This value
KRS/SS is the spin selectivity. The first systematic study of spin selectivity showed that the value of
KRS/SS in dyads II and III is about 2. Authors have listed several reasons of this phenomenon and have arrived at the conclusion that the origin of spin selectivity is the difference of HFI constants in biradical-zwitterions of diastereomers [
10,
11]. More recent studies of a model system involving enantiomers of another NSAID—ketoprofen linked with enantiomers of tryptophan (set of (
S,S)-, (
S,R)- and (
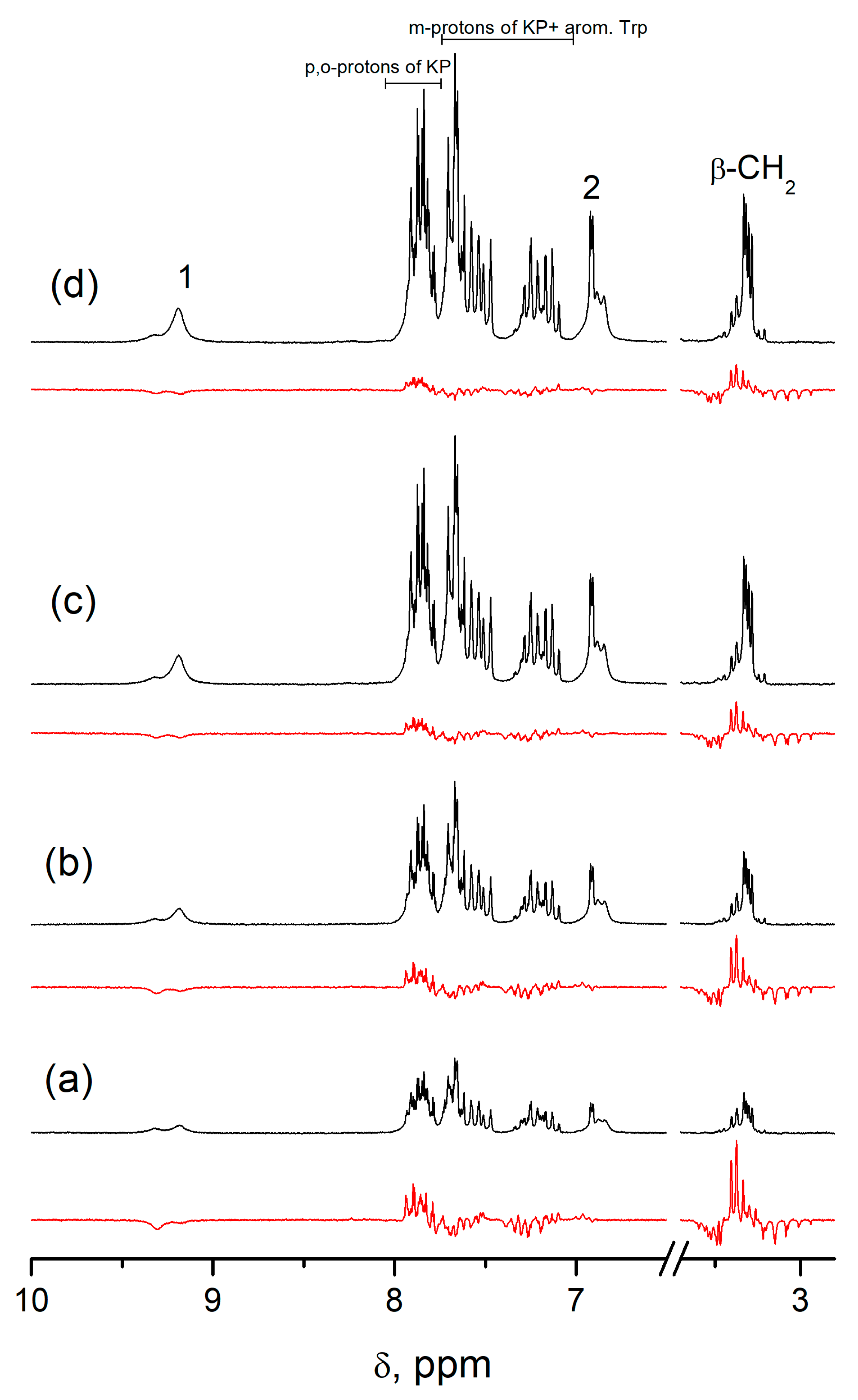
R,S)-optical configurations) IV—showed almost tenfold difference in CIDNP enhancement coefficients.
According to the literature data [
5,
12] the differences in the stereoselectivity of electron transfer in (
R,S)- and (
S,S)-diastereomers in similar systems vary within 25–40%. Taking into account that the CIDNP coefficient is associated with the manifestation of ET mechanism, it is difficult to imagine that the contributions of the ET mechanism can differ for (
S,S)- and (
R,S)-configurations by almost an order of magnitude. On the other hand, dyad IV is a system with two chromophores, and the prevailing mechanism of fluorescence quenching is resonance energy transfer (RET) [
13,
14]. According to [
13], the stereoselectivity of singlet–singlet energy transfer in diastereomers hardly reaches 2, and our own data for dyad IV gives a value of 1.6 [
14]. These differences in BZ concentrations in Equation (2), caused by RET stereoselectivity, could not provide an observable difference in the values of the CIDNP coefficients for the (
S,S)- and (
R,S)-configurations. Correspondingly, the difference in the CIDNP coefficients of the (
S,S)-, (
R,S)-, and (
S,R)-configurations of dyad IV is associated with anomalously low hyperpolarization of the (
R,S)- and (
S,R)-enantiomers. This reasoning leads to the conclusion that the interpretation of spin selectivity associated with the difference in HFI constants in BZ of diastereomers is not the only one.
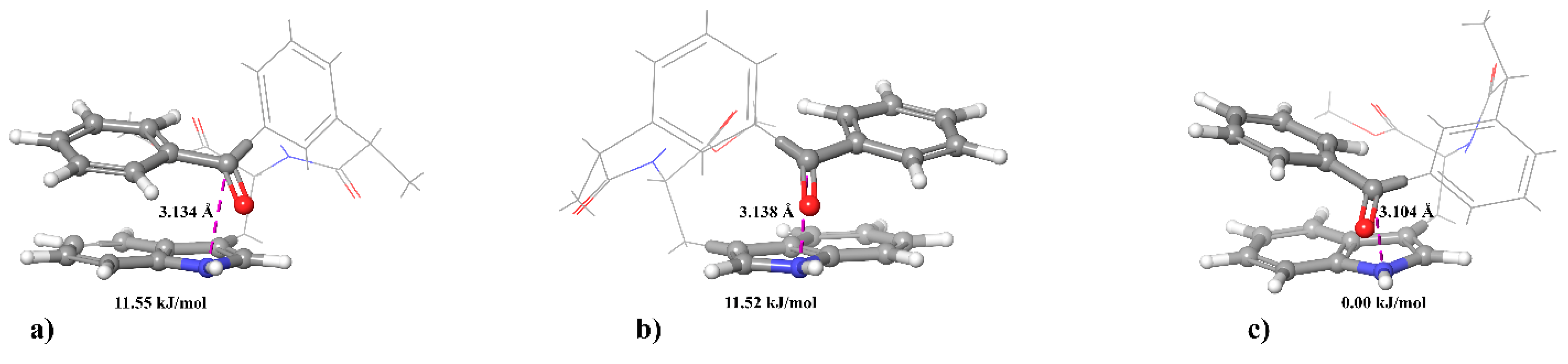
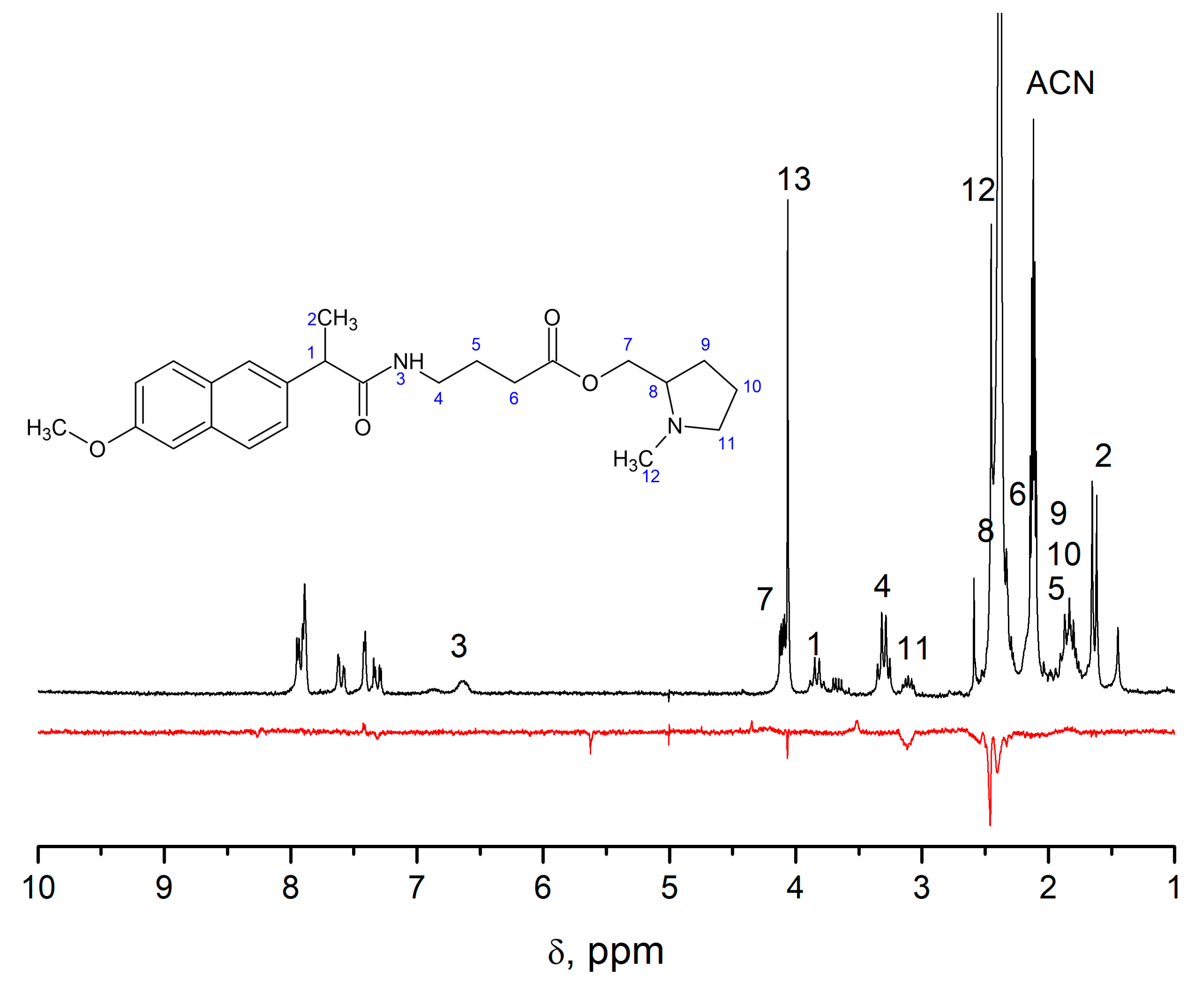
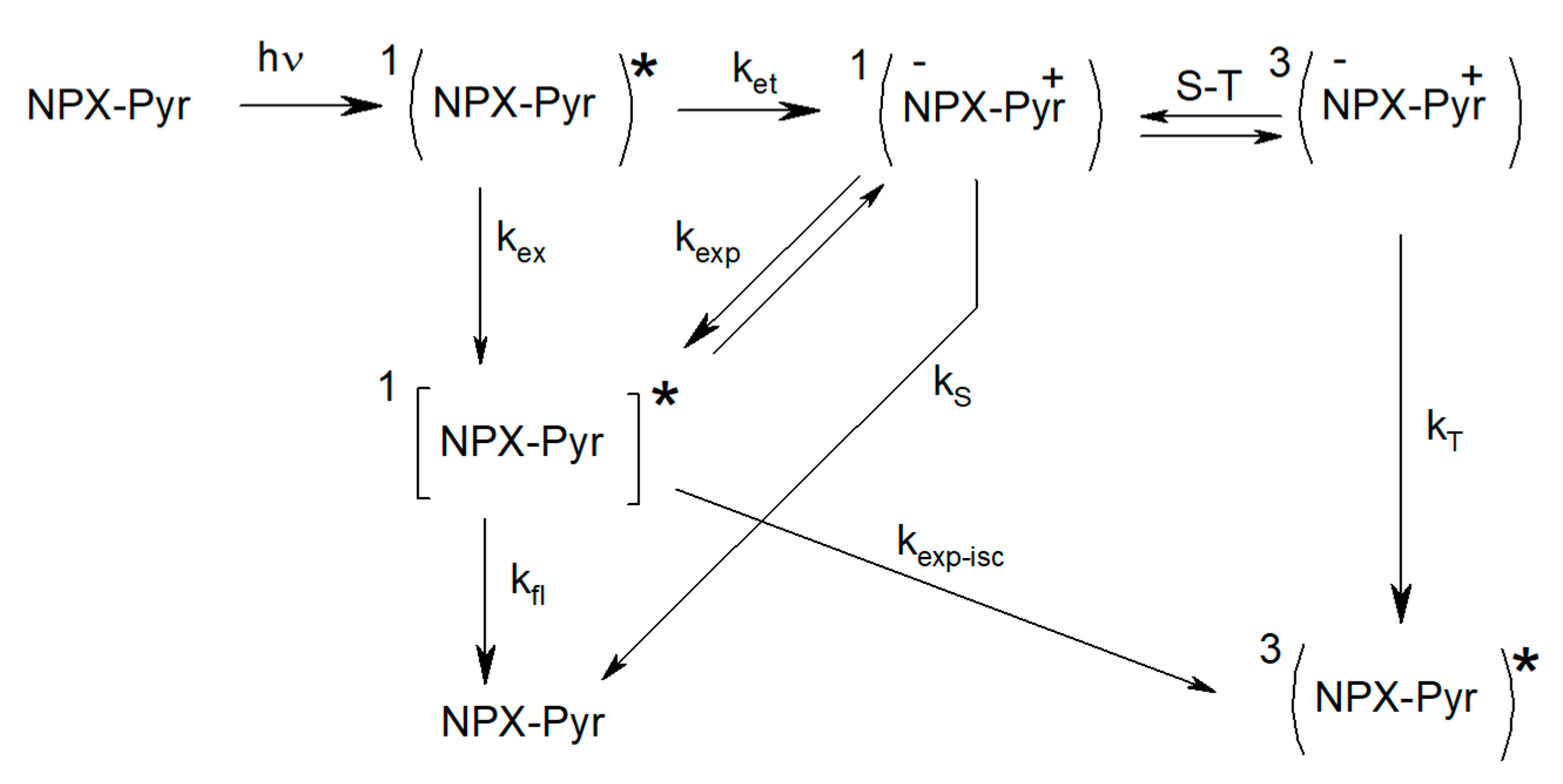
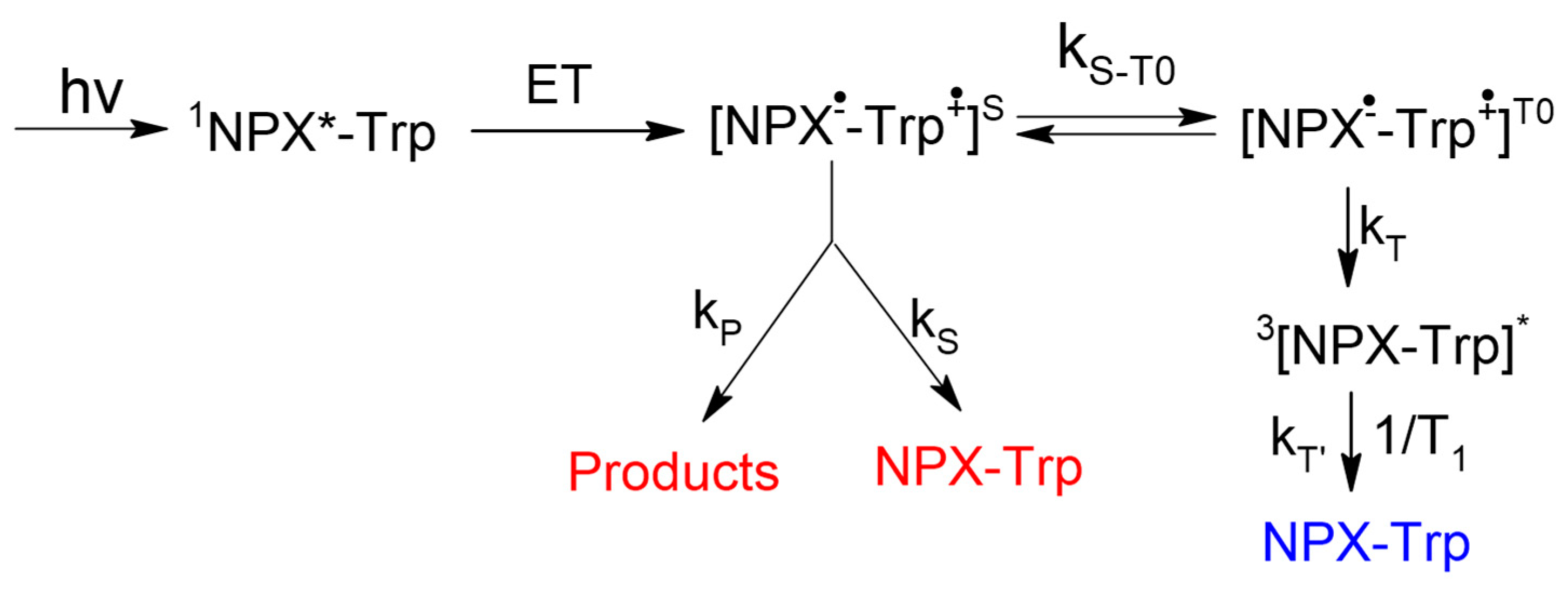
In order to trace some patterns in the abovementioned difference in the spin effects of optical isomers of dyads I–IV, a comprehensive analysis of CIDNP of all polarized protons and its spin selectivity will be carried out. This consideration involves the calculation of CIDNP in diastereomers of dyads in accordance with the S-T0 approximation using data from molecular dynamics and quantum chemical calculations.
In addition, an alternative explanation of the hyperpolarization will be given, involving magnetic dipole–dipole interaction of electrons in the spin Hamiltonian (1). An attempt to use the dipole–dipole electron–electron magnetic interaction to explain the difference in the CIDNP coefficients in the diastereomers of the dyad IV was already made in the work [
14], but it was the CIDNP calculation without taking into account BZ lifetime. In this article, the CIDNP formed in the biradical-zwitterion within the framework of the two-position model will be calculated taking into account the electron dipole–dipole interaction when BZ is in and out of the reaction zone.
Thus, consideration the different efficiency of spin effects in diastereomers of dyads I–IV together with comparison of experimental results with molecular simulating data seems to be promising to discover the influence of optical configuration on the structure and the reactivity of donor–acceptor dyads. In particular, the analysis of these data for diastereomers of dyads III and IV with D- and L- residues of tryptophan, including the abnormally weak hyperpolarization in the (R,S)- and (S,R)-enantiomers of dyad IV, can shed light on the nature of the differences between systems with optical isomers of tryptophan. The addressing to model systems in this case is especially important, since the structures of certain proteins and peptides found in living systems become highly disordered when L-amino acids are replaced by D-analogs, and they cannot be studied using high-resolution NMR and X-ray spectroscopy.


 ,
, 






















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}



