
B Lineage Cells in ANCA-Associated Vasculitis


{kind=link}
{kind=link}
Abstract
:1. Introduction
2. ANCA-Associated Vasculitis (AAV): Brief Background on Clinical Picture and Current Treatment Strategies
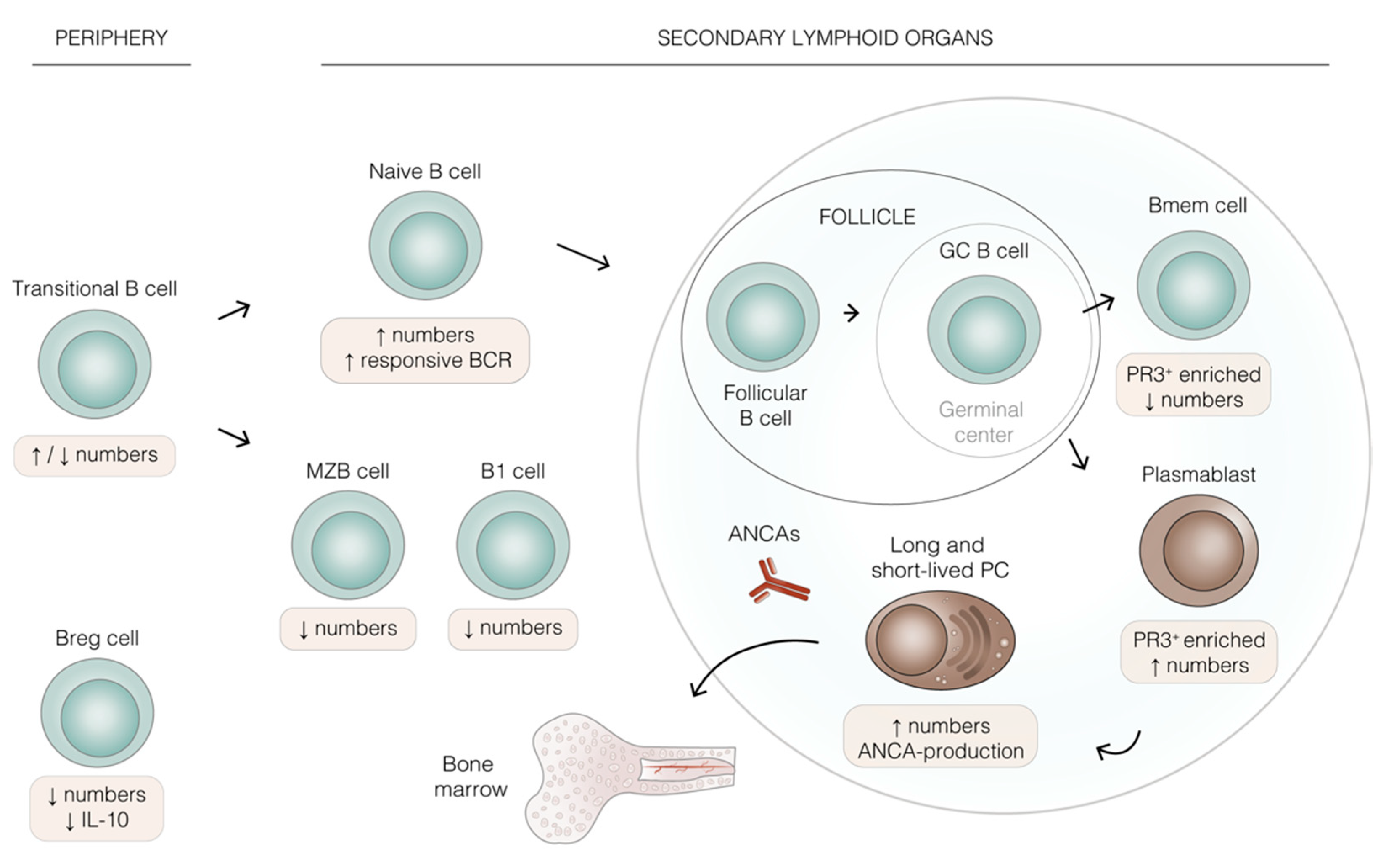
3. B Lineage Cells in AAV: Differentiation, Prevalence, and Function
4. Key Features of B Lineage Cells in AAV
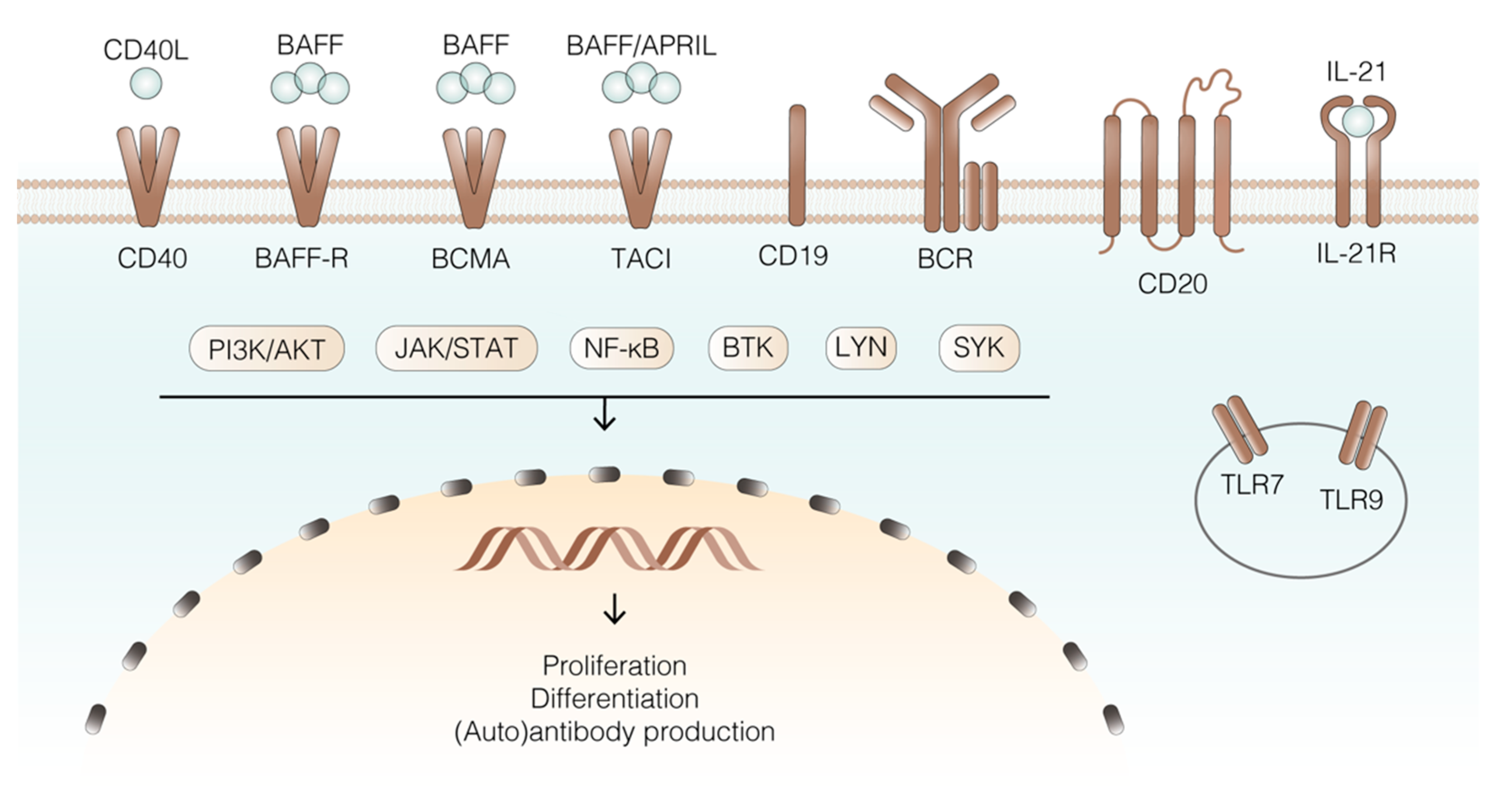
4.1. Receptors and Ligands
4.2. Intracellular Signalling Pathways
4.3. Cytokines and Chemokines
5. Concluding Remarks and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Nakazawa, D.; Masuda, S.; Tomaru, U.; Ishizu, A. Pathogenesis and Therapeutic Interventions for ANCA-Associated Vasculitis. Nat. Rev. Rheumatol. 2019, 15, 91–101. [Google Scholar] [CrossRef] [PubMed]
- Bugatti, S.; Codullo, V.; Caporali, R.; Montecucco, C. B Cells in Rheumatoid Arthritis. Autoimmun. Rev. 2007, 7, 137–142. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, K.; Clauder, A.K.; Manz, R.A. Targeting B Cells and Plasma Cells in Autoimmune Diseases. Front. Immunol. 2018, 9, 835. [Google Scholar] [CrossRef] [PubMed]
- Emery, P.; Gottenberg, J.E.; Rubbert-Roth, A.; Sarzi-Puttini, P.; Choquette, D.; Martínez Taboada, V.M.; Barile-Fabris, L.; Moots, R.J.; Ostor, A.; Andrianakos, A.; et al. Rituximab versus an Alternative TNF Inhibitor in Patients with Rheumatoid Arthritis Who Failed to Respond to a Single Previous TNF Inhibitor: SWITCH-RA, a Global, Observational, Comparative Effectiveness Study. Ann. Rheum. Dis. 2015, 74, 979–984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Emery, P.; Fleischmann, R.; Filipowicz-Sosnowska, A.; Schechtman, J.; Szczepanski, L.; Kavanaugh, A.; Racewicz, A.J.; Van Vollenhoven, R.F.; Li, N.F.; Agarwal, S.; et al. The Efficacy and Safety of Rituximab in Patients with Active Rheumatoid Arthritis despite Methotrexate Treatment: Results of a Phase IIb Randomized, Double-Blind, Placebo-Controlled, Dose-Ranging Trial. Arthritis Rheum. 2006, 54, 1390–1400. [Google Scholar] [CrossRef] [PubMed]
- Reddy, V.; Klein, C.; Isenberg, D.A.; Glennie, M.J.; Cambridge, G.; Cragg, M.S.; Leandro, M.J. Obinutuzumab Induces Superior B-Cell Cytotoxicity to Rituximab in Rheumatoid Arthritis and Systemic Lupus Erythematosus Patient Samples. Rheumatology 2017, 56, 1227–1237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sfikakis, P.P.; Boletis, J.N.; Lionaki, S.; Vigklis, V.; Fragiadaki, K.G.; Iniotaki, A.; Moutsopoulos, H.M. Remission of Proliferative Lupus Nephritis Following B Cell Depletion Therapy Is Preceded by Down-Regulation of the T Cell Costimulatory Molecule CD40 Ligand: An Open-Label Trial. Arthritis Rheum. 2005, 52, 501–513. [Google Scholar] [CrossRef] [PubMed]
- Tas, S.W.; Baeten, D.L.P. Recent Advances in the Treatment of Immune-Mediated Inflammatory Diseases. In Suppression and Regulation of Immune Responses: Methods and protocols; Cuturi, M.C., Anegon, I., Eds.; Springer: New York, NY, USA, 2016; Volume II, pp. 143–155. ISBN 978-1-4939-3139-2. [Google Scholar]
- Jennette, J.C.; Falk, R.J. B Cell-Mediated Pathogenesis of ANCA-Mediated Vasculitis. Semin. Immunopathol. 2014, 36, 327–338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stilmant, M.M.; Bolton, W.K.; Sturgill, B.C.; Schmitt, G.W.; Couser, W.G. Crescentic Glomerulonephritis without Immune Deposits: Clinicopathologic Features. Kidney Int. 1979, 15, 184–195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Paassen, P.; Cohen Tervaert, J.W.; Heeringa, P. Mechanisms of Vasculitis: How Pauci-Immune Is ANCA-Associated Renal Vasculitis? Nephron Exp. Nephrol. 2006, 105, e10–e16. [Google Scholar] [CrossRef] [PubMed]
- Roth, A.J.; Ooi, J.D.; Hess, J.J.; Van Timmeren, M.M.; Berg, E.A.; Poulton, C.E.; McGregor, J.A.; Burkart, M.; Hogan, S.L.; Hu, Y.; et al. Epitope Specificity Determines Pathogenicity and Detectability in Anca-Associated Vasculitis. J. Clin. Investig. 2013, 123, 1773–1783. [Google Scholar] [CrossRef] [Green Version]
- Shah, S.; Hruskova, Z.; Segelmark, M.; Morgan, M.D.; Hogan, J.; Lee, S.K.; Dale, J.; Harper, L.; Tesar, V.; Jayne, D.R.W.; et al. Treatment of Severe Renal Disease in ANCA Positive and Negative Small Vessel Vasculitis with Rituximab. Am. J. Nephrol. 2015, 41, 296–301. [Google Scholar] [CrossRef] [PubMed]
- Miloslavsky, E.M.; Lu, N.; Unizony, S.; Choi, H.K.; Merkel, P.A.; Seo, P.; Spiera, R.; Langford, C.A.; Hoffman, G.S.; Kallenberg, C.G.M.; et al. Myeloperoxidase–Antineutrophil Cytoplasmic Antibody (ANCA)–Positive and ANCA-Negative Patients with Granulomatosis with Polyangiitis (Wegener’s): Distinct Patient Subsets. Arthritis Rheumatol. 2016, 68, 2945–2952. [Google Scholar] [CrossRef]
- Iudici, M.; Pagnoux, C.; Courvoisier, D.S.; Cohen, P.; Hamidou, M.; Aouba, A.; Lifermann, F.; Ruivard, M.; Aumaître, O.; Bonnotte, B.; et al. Granulomatosis with Polyangiitis: Study of 795 Patients from the French Vasculitis Study Group Registry. Semin. Arthritis Rheum. 2021, 51, 339–346. [Google Scholar] [CrossRef] [PubMed]
- Jennette, J.C.; Falk, R.J. Pathogenesis of Antineutrophil Cytoplasmic Autoantibody-Mediated Disease. Nat. Rev. Rheumatol. 2014, 10, 463–473. [Google Scholar] [CrossRef] [PubMed]
- Lyons, P.A.; Rayner, T.F.; Trivedi, S.; Holle, J.U.; Watts, R.A.; Jayne, D.R.W.; Baslund, B.; Brenchley, P.; Bruchfeld, A.; Chaudhry, A.N.; et al. Genetically Distinct Subsets within ANCA-Associated Vasculitis. N. Engl. J. Med. 2012, 367, 214–223. [Google Scholar] [CrossRef] [Green Version]
- Rahmattulla, C.; Mooyaart, A.L.; Van Hooven, D.; Schoones, J.W.; Bruijn, J.A.; Dekkers, O.M.; Bajema, I.M. Genetic Variants in ANCA-Associated Vasculitis: A Meta-Analysis. Ann. Rheum. Dis. 2016, 75, 1687–1692. [Google Scholar] [CrossRef] [PubMed]
- Cornec, D.; Cornec-Le Gall, E.; Fervenza, F.C.; Specks, U. ANCA-Associated Vasculitis-Clinical Utility of Using ANCA Specificity to Classify Patients. Nat. Rev. Rheumatol. 2016, 12, 570–579. [Google Scholar] [CrossRef] [PubMed]
- Wallace, Z.S.; Stone, J.H. Personalized Medicine in ANCA-Associated Vasculitis ANCA Specificity as the Guide? Front. Immunol. 2019, 10, 2855. [Google Scholar] [CrossRef] [Green Version]
- Gou, S.J.; Yuan, J.; Chen, M.; Yu, F.; Zhao, M.H. Circulating Complement Activation in Patients with Anti-Neutrophil Cytoplasmic Antibody-Associated Vasculitis. Kidney Int. 2013, 83, 129–137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bansal, P.J.; Tobin, M.C. Neonatal Microscopic Polyangiitis Secondary to Transfer of Maternal Myeloperoxidase-Antineutrophil Cytoplasmic Antibody Resulting in Neonatal Pulmonary Hemorrhage and Renal Involvement. Ann. Allergy Asthma Immunol. 2004, 93, 398–401. [Google Scholar] [CrossRef]
- Stone, J.H.; Merkel, P.A.; Spiera, R.; Seo, P.; Langford, C.A.; Hoffman, G.S.; Kallenberg, C.G.M.; st. Clair, E.W.; Turkiewicz, A.; Tchao, N.K.; et al. Rituximab versus Cyclophosphamide for ANCA-Associated Vasculitis. N. Engl. J. Med. 2010, 363, 221–232. [Google Scholar] [CrossRef] [PubMed]
- Jones, R.B.; Cohen Tervaert, J.W.; Hauser, T.; Luqmani, R.; Morgan, M.D.; Peh, C.A.; Savage, C.O.; Segelmark, M.; Tesar, V.; van Paassen, P.; et al. Rituximab versus Cyclophosphamide in ANCA-Associated Renal Vasculitis. N. Engl. J. Med. 2010, 363, 211–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, R.B.; Furuta, S.; Tervaert, J.W.C.; Hauser, T.; Luqmani, R.; Morgan, M.D.; Peh, C.A.; Savage, C.O.; Segelmark, M.; Tesar, V.; et al. Rituximab versus Cyclophosphamide in ANCA-Associated Renal Vasculitis: 2-Year Results of a Randomised Trial. Ann. Rheum. Dis. 2015, 74, 1178–1182. [Google Scholar] [CrossRef] [PubMed]
- McClure, M.; Gopaluni, S.; Jayne, D.; Jones, R. B Cell Therapy in ANCA-Associated Vasculitis: Current and Emerging Treatment Options. Nat. Rev. Rheumatol. 2018, 14, 580–591. [Google Scholar] [CrossRef] [PubMed]
- Dumoitier, N.; Terrier, B.; London, J.; Lofek, S.; Mouthon, L. Implication of B Lymphocytes in the Pathogenesis of ANCA-Associated Vasculitides. Autoimmun. Rev. 2015, 14, 996–1004. [Google Scholar] [CrossRef]
- Guillevin, L.; Pagnoux, C.; Karras, A.; Khouatra, C.; Aumaître, O.; Cohen, P.; Maurier, F.; Decaux, O.; Ninet, J.; Gobert, P.; et al. Rituximab versus Azathioprine for Maintenance in ANCA-Associated Vasculitis. N. Engl. J. Med. 2014, 371, 1771–1780. [Google Scholar] [CrossRef] [Green Version]
- Cupps, T.R.; Edgar, L.C.; Fauci, A.S. Suppression of Human B Lymphocyte Function by Cyclophosphamide. J. Immunol. 1982, 128, 2453–2457. [Google Scholar] [PubMed]
- Reinhold-Keller, E.; Beuge, N.; Latza, U.; De Groot, K.; Rudert, H.; Nölle, B.; Heller, M.; Gross, W.L. An Interdisciplinary Approach to the Care of Patients with Wegener’s Granulomatosis: Long-Term Outcome in 155 Patients. Arthritis Rheum. 2000, 43, 1021–1032. [Google Scholar] [CrossRef]
- Hoffman, G.S.; Kerr, G.S.; Leavitt, R.Y.; Hallahan, C.W.; Lebovics, R.S.; Travis, W.D.; Rottem, M.; Fauci, A.S. Wegener Granulomatosis: An Analysis of 158 Patients. Ann. Intern. Med. 1992, 116, 488–498. [Google Scholar] [CrossRef] [PubMed]
- Pagnoux, C.; Mahr, A.; Hamidou, M.A.; Boffa, J.-J.; Ruivard, M.; Ducroix, J.-P.; Kyndt, X.; Lifermann, F.; Papo, T.; Lambert, M.; et al. Azathioprine or Methotrexate Maintenance for ANCA-Associated Vasculitis. N. Engl. J. Med. 2008, 359, 2790–2803. [Google Scholar] [CrossRef] [Green Version]
- Von Borstel, A.A.; Abdulahad, W.H.; Dekkema, G.; Rutgers, A.; Stegeman, C.A.; Veldman, J.; Heeringa, P.; Sanders, J.S. Mycophenolic Acid and 6-Mercaptopurine Both Inhibit B-Cell Proliferation in Granulomatosis with Polyangiitis Patients, Whereas Only Mycophenolic Acid Inhibits Bcell IL-6 Production. PLoS ONE 2020, 15, e0235743. [Google Scholar] [CrossRef] [PubMed]
- Terrier, B.; Pagnoux, C.; Perrodeau, É.; Karras, A.; Khouatra, C.; Aumaître, O.; Cohen, P.; Decaux, O.; Desmurs-Clavel, H.; Maurier, F.; et al. Long-Term Efficacy of Remission-Maintenance Regimens for ANCA-Associated Vasculitides. Ann. Rheum. Dis. 2018, 77, 1151–1157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melchers, F. Checkpoints That Control B Cell Development. J. Clin. Investig. 2015, 125, 2203–2210. [Google Scholar] [CrossRef] [Green Version]
- Brahim, F.; Osmond, D.G. Migration of Bone Marrow Lymphocytes Demonstrated by Selective Bone Marrow Labeling with Thymidine-H3. Anat. Rec. 1970, 168, 139–159. [Google Scholar] [CrossRef]
- Kurosaki, T.; Shinohara, H.; Baba, Y. B Cell Signalling and Fate Decision. Annu. Rev. Immunol. 2010, 28, 21–55. [Google Scholar] [CrossRef] [PubMed]
- Land, J.; Lintermans, L.L.; Stegeman, C.A.; Muñoz-Elías, E.J.; Tarcha, E.J.; Iadonato, S.P.; Heeringa, P.; Rutgers, A.; Abdulahad, W.H. Kv1.3 Channel Blockade Modulates the Effector Function of B Cells in Granulomatosis with Polyangiitis. Front. Immunol. 2017, 8, 1205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lepse, N.; Abdulahad, W.H.; Rutgers, A.; Kallenberg, C.G.M.; Stegeman, C.A.; Heeringa, P. Altered B Cell Balance, but Unaffected B Cell Capacity to Limit Monocyte Activation in Anti-Neutrophil Cytoplasmic Antibody-Associated Vasculitis in Remission. Rheumatology 2014, 53, 1683–1692. [Google Scholar] [CrossRef] [Green Version]
- Griffin, D.O.; Holodick, N.E.; Rothstein, T.L. Human B1 Cells in Umbilical Cord and Adult Peripheral Blood Express the Novel Phenotype CD20+CD27+CD43+CD70−. J. Exp. Med. 2011, 208, 67–80. [Google Scholar] [CrossRef]
- Appelgren, D.; Eriksson, P.; Ernerudh, J.; Segelmark, M. Marginal-Zone B-Cells Are Main Producers of IgM in Humans, and Are Reduced in Patients with Autoimmune Vasculitis. Front. Immunol. 2018, 9, 2242. [Google Scholar] [CrossRef] [Green Version]
- Lebien, T.W.; Tedder, T.F. B Lymphocytes: How They Develop and Function. Blood 2008, 112, 1570–1580. [Google Scholar] [CrossRef]
- Von Borstel, A.; Abdulahad, W.H.; Sanders, J.S.; Rip, J.; Neys, S.F.H.; Hendriks, R.W.; Stegeman, C.A.; Heeringa, P.; Rutgers, A.; Corneth, O.B.J. Evidence for Enhanced Bruton’s Tyrosine Kinase Activity in Transitional and Naïve B Cells of Patients with Granulomatosis with Polyangiitis. Rheumatology 2019, 58, 2230–2239. [Google Scholar] [CrossRef] [Green Version]
- Kurosaki, T.; Kometani, K.; Ise, W. Memory B Cells. Nat. Rev. Immunol. 2015, 15, 149–159. [Google Scholar] [CrossRef]
- Todd, S.K.; Pepper, R.J.; Draibe, J.; Tanna, A.; Pusey, C.D.; Mauri, C.; Salama, A.D. Regulatory B Cells Are Numerically but Not Functionally Deficient in Anti-Neutrophil Cytoplasm Antibody-Associated Vasculitis. Rheumatology 2014, 53, 1693–1703. [Google Scholar] [CrossRef] [Green Version]
- Cyster, J.G.; Allen, C.D.C. B Cell Responses: Cell Interaction Dynamics and Decisions. Cell 2019, 177, 524–540. [Google Scholar] [CrossRef] [Green Version]
- Brynjolfsson, S.F.; Mohaddes, M.; Kärrholm, J.; Wick, M.J. Long-Lived Plasma Cells in Human Bone Marrow Can Be Either CD191 or CD19−. Blood Adv. 2017, 1, 835–838. [Google Scholar] [CrossRef]
- Brynjolfsson, S.F.; Berg, L.P.; Ekerhult, T.O.; Rimkute, I.; Wick, M.J.; Martensson, I.L.; Grimsholm, O. Long-Lived Plasma Cells in Mice and Men. Front. Immunol. 2018, 9, 2673. [Google Scholar] [CrossRef]
- Voswinkel, J.; Mueller, A.; Kraemer, J.A.; Lamprecht, P.; Herlyn, K.; Holl-Ulrich, K.; Feller, A.C.; Pitann, S.; Gause, A.; Gross, W.L. B Lymphocyte Maturation in Wegener’s Granulomatosis: A Comparative Analysis of VH Genes from Endonasal Lesions. Ann. Rheum. Dis. 2006, 65, 859–864. [Google Scholar] [CrossRef] [Green Version]
- Mei, H.E.; Wirries, I.; Frölich, D.; Brisslert, M.; Giesecke, C.; Grün, J.R.; Alexander, T.; Schmidt, S.; Luda, K.; Kühl, A.A.; et al. A Unique Population of IgG-Expressing Plasma Cells Lacking CD19 Is Enriched in Human Bone Marrow. Blood 2015, 125, 1739–1748. [Google Scholar] [CrossRef]
- Wiik, A. Autoantibodies in Vasculitis. Arthritis Res. Ther. 2003, 5, 147–152. [Google Scholar] [CrossRef]
- Finkielman, J.D.; Merkel, P.A.; Schroeder, D.; Hoffman, G.S.; Spiera, R.; St. Clair, E.W.; Davis, J.C.; McCune, W.J.; Lears, A.K.; Ytterberg, S.R.; et al. Antiproteinase 3 Antineutrophil Cytoplasmic Antibodies and Disease Activity in Wegener Granulomatosis. Ann. Intern. Med. 2007, 147, 611–619. [Google Scholar] [CrossRef]
- Xu, P.C.; Cui, Z.; Chen, M.; Hellmark, T.; Zhao, M.H. Comparison of Characteristics of Natural Autoantibodies against Myeloperoxidase and Anti-Myeloperoxidase Autoantibodies from Patients with Microscopic Polyangiitis. Rheumatology 2011, 50, 1236–1243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keogh, K.A.; Wylam, M.E.; Stone, J.H.; Specks, U. Induction of Remission by B Lymphocyte Depletion in Eleven Patients with Refractory Antineutrophil Cytoplasmic Antibody-Associated Vasculitis. Arthritis Rheum. 2005, 52, 262–268. [Google Scholar] [CrossRef]
- Stasi, R.; Stipa, E.; Del Poeta, G.; Amadori, S.; Newland, A.C.; Provan, D. Long-Term Observation of Patients with Anti-Neutrophil Cytoplasmic Antibody-Associated Vasculitis Treated with Rituximab. Rheumatology 2006, 45, 1432–1436. [Google Scholar] [CrossRef] [Green Version]
- Chang, S.Y.; Keogh, K.A.; Lewis, J.E.; Ryu, J.H.; Cornell, L.D.; Garrity, J.A.; Yi, E.S. IgG4-Positive Plasma Cells in Granulomatosis with Polyangiitis (Wegener’s): A Clinicopathologic and Immunohistochemical Study on 43 Granulomatosis with Polyangiitis and 20 Control Cases. Hum. Pathol. 2013, 44, 2432–2437. [Google Scholar] [CrossRef] [PubMed]
- Perez Alamino, R.; Martínez, C.; Espinoza, L.R. IgG4-Associated Vasculitis. Curr. Rheumatol. Rep. 2013, 15, 348. [Google Scholar] [CrossRef]
- Al-Soudi, A.; Doorenspleet, M.E.; Esveldt, R.E.; Burgemeister, L.T.; Hak, A.E.; Van Den Born, B.J.H.; Tas, S.W.; Van Vollenhoven, R.F.; Klarenbeek, P.L.; De Vries, N. IgG4:IgG RNA Ratio Differentiates Active Disease from Remission in Granulomatosis with Polyangiitis: A New Disease Activity Marker? A Cross-Sectional and Longitudinal Study. Arthritis Res. Ther. 2019, 21, 43. [Google Scholar] [CrossRef] [Green Version]
- Berti, A.; Warner, R.; Johnson, K.; Cornec, D.; Schroeder, D.; Kabat, B.; Langford, C.A.; Hoffman, G.S.; Fervenza, F.C.; Kallenberg, C.G.M.; et al. Brief Report: Circulating Cytokine Profiles and Antineutrophil Cytoplasmic Antibody Specificity in Patients With Antineutrophil Cytoplasmic Antibody-Associated Vasculitis. Arthritis Rheumatol. 2018, 70, 1114–1121. [Google Scholar] [CrossRef] [PubMed]
- Ferraro, A.J.; Smith, S.W.; Neil, D.; Savage, C.O.S. Relapsed Wegener’s Granulomatosis after Rituximab Therapy—B Cells Are Present in New Pathological Lesions despite Persistent “depletion” of Peripheral Blood. Nephrol. Dial. Transplant. 2008, 23, 3030–3032. [Google Scholar] [CrossRef] [Green Version]
- Cornec, D.; Berti, A.; Hummel, A.; Peikert, T.; Pers, J.O.; Specks, U. Identification and Phenotyping of Circulating Autoreactive Proteinase 3-Specific B Cells in Patients with PR3-ANCA Associated Vasculitis and Healthy Controls. J. Autoimmun. 2017, 84, 122–131. [Google Scholar] [CrossRef]
- Steinmetz, O.M.; Velden, J.; Kneissler, U.; Marx, M.; Klein, A.; Helmchen, U.; Stahl, R.A.K.; Panzer, U. Analysis and Classification of B-Cell Infiltrates in Lupus and ANCA-Associated Nephritis. Kidney Int. 2008, 74, 448–457. [Google Scholar] [CrossRef] [Green Version]
- Wilde, B.; Thewissen, M.; Damoiseaux, J.; Knippenberg, S.; Hilhorst, M.; Van Paassen, P.; Witzke, O.; Cohen Tervaert, J.W. Regulatory B Cells in ANCA-Associated Vasculitis. Ann. Rheum. Dis. 2013, 72, 1416–1419. [Google Scholar] [CrossRef] [PubMed]
- Aybar, L.T.; Mcgregor, J.G.; Hogan, S.L.; Hu, Y.; Mendoza, C.E.; Brant, E.J.; Poulton, C.J.; Henderson, C.D.; Falk, R.J.; Bunch, D.O. Reduced CD5+CD24hiCD38hi and Interleukin-10+ Regulatory B Cells in Active Anti-Neutrophil Cytoplasmic Autoantibody-Associated Vasculitis Permit Increased Circulating Autoantibodies. Clin. Exp. Immunol. 2015, 180, 178–188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagai, M.; Hirayama, K.; Ebihara, I.; Shimohata, H.; Kobayashi, M.; Koyama, A. Serum Levels of BAFF and APRIL in Myeloperoxidase Anti-Neutrophil Cytoplasmic Autoantibody-Associated Renal Vasculitis: Association with Disease Activity. Nephron Clin. Pract. 2011, 118, 339–345. [Google Scholar] [CrossRef] [PubMed]
- Mackay, F.; Schneider, P. Cracking the BAFF Code. Nat. Rev. Immunol. 2009, 9, 491–502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vincent, F.B.; Saulep-Easton, D.; Figgett, W.A.; Fairfax, K.A.; Mackay, F. The BAFF/APRIL System: Emerging Functions beyond B Cell Biology and Autoimmunity. Cytokine Growth Factor Rev. 2013, 24, 203–215. [Google Scholar] [CrossRef] [PubMed]
- Salazar-Camarena, D.C.; Ortiz-Lazareno, P.C.; Cruz, A.; Oregon-Romero, E.; Machado-Contreras, J.R.; Muñoz-Valle, J.F.; Orozco-López, M.; Marín-Rosales, M.; Palafox-Sánchez, C.A. Association of BAFF, APRIL Serum Levels, BAFF-R, TACI and BCMA Expression on Peripheral B-Cell Subsets with Clinical Manifestations in Systemic Lupus Erythematosus. Lupus 2016, 25, 582–592. [Google Scholar] [CrossRef]
- Lenert, A.; Lenert, P. Current and Emerging Treatment Options for ANCA-Associated Vasculitis: Potential Role of Belimumab and Other BAFF/APRIL Targeting Agents. Drug Des. Dev. Ther. 2015, 9, 333–347. [Google Scholar] [CrossRef] [Green Version]
- Sanders, J.S.F.; Huitma, M.G.; Kallenberg, C.G.M.; Stegeman, C.A. Plasma Levels of Soluble Interleukin 2 Receptor, Soluble CD30, Interleukin 10 and B Cell Activator of the Tumour Necrosis Factor Family during Follow-up in Vasculitis Associated with Proteinase 3-Antineutrophil Cytoplasmic Antibodies: Associations with Di. Ann. Rheum. Dis. 2006, 65, 1484–1489. [Google Scholar] [CrossRef] [Green Version]
- Moore, P.A.; Belvedere, O.; Orr, A.; Pieri, K.; LaFleur, D.W.; Feng, P.; Soppet, D.; Charters, M.; Gentz, R.; Parmelee, D.; et al. BLyS: Member of the Tumor Necrosis Factor Family and B Lymphocyte Stimulator. Science 1999, 285, 260–263. [Google Scholar] [CrossRef] [PubMed]
- Schneider, P.; Mackay, F.; Steiner, V.; Hofmann, K.; Bodmer, J.L.; Holler, N.; Ambrose, C.; Lawton, P.; Bixler, S.; Acha-Orbea, H.; et al. BAFF, a Novel Ligand of the Tumour Necrosis Factor Family, Stimulates B Cell Growth. J. Exp. Med. 1999, 189, 1747–1756. [Google Scholar] [CrossRef] [PubMed]
- Holden, N.J.; Williams, J.M.; Morgan, M.D.; Challa, A.; Gordon, J.; Pepper, R.J.; Salama, A.D.; Harper, L.; Savage, C.O.S. ANCA-Stimulated Neutrophils Release BLyS and Promote B Cell Survival: A Clinically Relevant Cellular Process. Ann. Rheum. Dis. 2011, 70, 2229–2233. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Davidson, A. BAFF and Selection of Autoreactive B Cells. Trends Immunol. 2011, 32, 388–394. [Google Scholar] [CrossRef] [Green Version]
- Xin, G.; Su, Y.; Li, K.-S.; Chen, M.; Zhao, M.-H.; Xu, L.-X. Serum B-Cell Activating Factor in Myecloperoxiase-Antineutrophil Cytoplasmic Antibodies-Associated Vasculitis. Am. J. Med. Sci. 2014, 348, 25–29. [Google Scholar] [CrossRef] [PubMed]
- Bader, L.; Koldingsnes, W.; Nossent, J. B-Lymphocyte Activating Factor Levels Are Increased in Patients with Wegener’s Granulomatosis and Inversely Correlated with ANCA Titer. Clin. Rheumatol. 2010, 29, 1031–1035. [Google Scholar] [CrossRef]
- Baker, K.P.; Edwards, B.M.; Main, S.H.; Choi, G.H.; Wager, R.E.; Halpern, W.G.; Lappin, P.B.; Riccobene, T.; Abramian, D.; Sekut, L.; et al. Generation and Characterization of LymphoStat-B, a Human Monoclonal Antibody That Antagonizes the Bioactivities of B Lymphocyte Stimulator. Arthritis Rheum. 2003, 48, 3253–3265. [Google Scholar] [CrossRef] [PubMed]
- Navarra, S.V.; Guzmán, R.M.; Gallacher, A.E.; Hall, S.; Levy, R.A.; Jimenez, R.E.; Li, E.K.-M.; Thomas, M.; Kim, H.-Y.; León, M.G.; et al. Efficacy and Safety of Belimumab in Patients with Active Systemic Lupus Erythematosus: A Randomised, Placebo-Controlled, Phase 3 Trial. Lancet 2011, 377, 721–731. [Google Scholar] [CrossRef]
- Furie, R.; Petri, M.; Zamani, O.; Cervera, R.; Wallace, D.J.; Tegzová, D.; Sanchez-Guerrero, J.; Schwarting, A.; Merrill, J.T.; Chatham, W.W.; et al. A Phase III, Randomized, Placebo-Controlled Study of Belimumab, a Monoclonal Antibody That Inhibits B Lymphocyte Stimulator, in Patients with Systemic Lupus Erythematosus. Arthritis Rheum. 2011, 63, 3918–3930. [Google Scholar] [CrossRef] [PubMed]
- McClure, M.; Gopaluni, S.; Wason, J.; Rosen, A.N.; Henderson, R.; Salama, A.; Pusey, C.; Jayne, D.; Jones, R. 322. A randomised, double-blind, controlled, mechanistic study of rituximab and belimumab combination therapy in Pr3 ANCA-associated vasculitis (Combivas): Study protocol. Rheumatology 2019, 58, kez063.046. [Google Scholar] [CrossRef] [Green Version]
- Smulski, C.R.; Kury, P.; Seidel, L.M.; Staiger, H.S.; Edinger, A.K.; Willen, L.; Seidl, M.; Hess, H.; Salzer, U.; Rolink, A.G.; et al. BAFF- and TACI-Dependent Processing of BAFFR by ADAM Proteases Regulates the Survival of B Cells. Cell Rep. 2017, 18, 2189–2202. [Google Scholar] [CrossRef] [Green Version]
- Pieper, K.; Grimbacher, B.; Eibel, H. B-Cell Biology and Development. J. Allergy Clin. Immunol. 2013, 131, 959–971. [Google Scholar] [CrossRef] [PubMed]
- Thompson, J.S.; Bixler, S.A.; Qian, F.; Vora, K.; Scott, M.L.; Cachero, T.G.; Hession, C.; Schneider, P.; Sizing, I.D.; Mullen, C.; et al. BAFF-R, a Newly Identified TNF Receptor That Specifically Interacts with BAFF. Science 2001, 293, 2108–2111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rauch, M.; Tussiwand, R.; Bosco, N.; Rolink, A.G. Crucial Role for BAFF-BAFF-R Signalling in the Survival and Maintenance of Mature B Cells. PLoS ONE 2009, 4, e5456. [Google Scholar] [CrossRef] [Green Version]
- Lepse, N.; Land, J.; Rutgers, A.; Kallenberg, C.G.M.; Stegeman, C.A.; Abdulahad, W.H.; Heeringa, P. Toll-like Receptor 9 Activation Enhances B Cell Activating Factor and Interleukin-21 Induced Anti-Proteinase 3 Autoantibody Production in Vitro. Rheumatology 2016, 55, 162–172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sellam, J.; Miceli-Richard, C.; Gottenberg, J.E.; Ittah, M.; Lavie, F.; Lacabaratz, C.; Gestermann, N.; Proust, A.; Lambotte, O.; Mariette, X. Decreased B Cell Activating Factor Receptor Expression on Peripheral Lymphocytes Associated with Increased Disease Activity in Primary Sjögren’s Syndrome and Systemic Lupus Erythematosus. Ann. Rheum. Dis. 2007, 66, 790–797. [Google Scholar] [CrossRef] [Green Version]
- Mueller, A.; Brieske, C.; Schinke, S.; Csernok, E.; Gross, W.L.; Hasselbacher, K.; Voswinkel, J.; Holl-ulrich, K. Plasma Cells within Granulomatous Inflammation Display Signs Pointing to Autoreactivity and Destruction in Granulomatosis with Polyangiitis. Arthritis Res. Ther. 2014, 16, R55. [Google Scholar] [CrossRef] [Green Version]
- Hatzoglou, A.; Roussel, J.; Bourgeade, M.-F.; Rogier, E.; Madry, C.; Inoue, J.; Devergne, O.; Tsapis, A. TNF Receptor Family Member BCMA (B Cell Maturation) Associates with TNF Receptor-Associated Factor (TRAF) 1, TRAF2, and TRAF3 and Activates NF-ΚB, Elk-1, c-Jun N-Terminal Kinase, and P38 Mitogen-Activated Protein Kinase. J. Immunol. 2000, 165, 1322–1330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Darce, J.R.; Arendt, B.K.; Wu, X.; Jelinek, D.F. Regulated Expression of BAFF-Binding Receptors during Human B Cell Differentiation. J. Immunol. 2007, 179, 7276–7286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Connor, B.P.; Raman, V.S.; Erickson, L.D.; Cook, W.J.; Weaver, L.K.; Ahonen, C.; Lin, L.L.; Mantchev, G.T.; Bram, R.J.; Noelle, R.J. BCMA Is Essential for the Survival of Long-Lived Bone Marrow Plasma Cells. J. Exp. Med. 2004, 199, 91–97. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.A.; Farrah, T.; Goodwin, R.G. The TNF Receptor Superfamily of Cellular and Viral Proteins: Activation, Costimulation, and Death. Cell 1994, 76, 959–962. [Google Scholar] [CrossRef]
- Karpusas, M.; Hsu, Y.M.; Wang, J.H.; Thompson, J.; Lederman, S.; Chess, L.; Thomas, D. 2 A Crystal Structure of an Extracellular Fragment of Human CD40 Ligand. Structure 1995, 3, 1426. [Google Scholar] [CrossRef] [Green Version]
- Banner, D.W.; D’Arcy, A.; Janes, W.; Gentz, R.; Schoenfeld, H.-J.; Broger, C.; Loetscher, H.; Lesslauer, W. Crystal Structure of the Soluble Human 55 Kd TNF Receptor-Human TNFβ Complex: Implications for TNF Receptor Activation. Cell 1993, 73, 431–445. [Google Scholar] [CrossRef]
- Kehry, M.R. CD40-Mediated Signalling in B Cells. Balancing Cell Survival, Growth, and Death. J. Immunol. 1996, 156, 2345–2348. [Google Scholar]
- Berberich, I.; Shu, G.L.; Clark, E.A. Cross-Linking CD40 on B Cells Rapidly Activates Nuclear Factor-Kappa B. J. Immunol. 1994, 153, 4357–4366. [Google Scholar] [PubMed]
- Hoffmann, J.C.; Patschan, D.; Dihazi, H.; Müller, C.; Schwarze, K.; Henze, E.; Ritter, O.; Müller, G.A.; Patschan, S. Cytokine Profiling in Anti Neutrophil Cytoplasmic Antibody-Associated Vasculitis: A Cross-Sectional Cohort Study. Rheumatol. Int. 2019, 39, 1907–1917. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Satterthwaite, A.B. Bruton’s Tyrosine Kinase, a Component of B Cell Signalling Pathways, Has Multiple Roles in the Pathogenesis of Lupus. Front. Immunol. 2018, 8, 1986. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maxwell, M.J.; Tsantikos, E.; Kong, A.M.; Vanhaesebroeck, B.; Tarlinton, D.M.; Hibbs, M.L. Attenuation of Phosphoinositide 3-Kinase δ Signalling Restrains Autoimmune Disease. J. Autoimmun. 2012, 38, 381–391. [Google Scholar] [CrossRef]
- Puri, K.D.; Di Paolo, J.A.; Gold, M.R. B-Cell Receptor Signalling Inhibitors for Treatment of Autoimmune Inflammatory Diseases and B-Cell Malignancies. Int. Rev. Immunol. 2013, 32, 397–427. [Google Scholar] [CrossRef] [PubMed]
- Stadanlick, J.E.; Kaileh, M.; Karnell, F.G.; Scholz, J.L.; Miller, J.P.; Quinn III, W.J.; Brezski, R.J.; Treml, L.S.; Jordan, K.A.; Monroe, J.G.; et al. Tonic B Cell Antigen Receptor Signals Supply an NF-ΚB Substrate for Prosurvival BLyS Signalling. Nat. Immunol. 2008, 9, 1379–1387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Culton, D.A.; Nicholas, M.W.; Bunch, D.O.; Zhen, Q.L.; Kepler, T.B.; Dooley, M.A.; Mohan, C.; Nachman, P.H.; Clarke, S.H. Similar CD19 Dysregulation in Two Autoantibody-Associated Autoimmune Diseases Suggests a Shared Mechanism of B-Cell Tolerance Loss. J. Clin. Immunol. 2007, 27, 53–68. [Google Scholar] [CrossRef]
- Kuek, L.E.; Leffler, M.; Mackay, G.A.; Hulett, M.D. The MS4A Family: Counting Past 1, 2 and 3. Immunol. Cell Biol. 2016, 94, 11–23. [Google Scholar] [CrossRef] [PubMed]
- Pavlasova, G.; Mraz, M. The Regulation and Function of CD20: An “Enigma” of B-Cell Biology and Targeted Therapy. Haematologica 2020, 105, 1494–1506. [Google Scholar] [CrossRef] [PubMed]
- Konforte, D.; Simard, N.; Paige, C.J. IL-21: An Executor of B Cell Fate. J. Immunol. 2009, 182, 1781–1787. [Google Scholar] [CrossRef]
- Parrish-Novak, J.; Dillon, S.R.; Nelson, A.; Hammond, A.; Sprecher, C.; Gross, J.A.; Johnston, J.; Madden, K.; Xu, W.; West, J.; et al. Interleukin 21 and Its Receptor Are Involved in NK Cell Expansion and Regulation of Lymphocyte Function. Nature 2000, 408, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Good, K.L.; Bryant, V.L.; Tangye, S.G. Kinetics of Human B Cell Behavior and Amplification of Proliferative Responses Following Stimulation with IL-21. J. Immunol. 2006, 177, 5236–5247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuchen, S.; Robbins, R.; Sims, G.P.; Sheng, C.; Phillips, T.M.; Lipsky, P.E.; Ettinger, R. Essential Role of IL-21 in B Cell Activation, Expansion, and Plasma Cell Generation during CD4 + T Cell-B Cell Collaboration. J. Immunol. 2007, 179, 5886–5896. [Google Scholar] [CrossRef] [Green Version]
- Bryant, V.L.; Ma, C.S.; Avery, D.T.; Li, Y.; Good, K.L.; Corcoran, L.M.; de Waal Malefyt, R.; Tangye, S.G. Cytokine-Mediated Regulation of Human B Cell Differentiation into Ig-Secreting Cells: Predominant Role of IL-21 Produced by CXCR5 + T Follicular Helper Cells. J. Immunol. 2007, 179, 8180–8190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Ji, Z.; Yu, W.; Wu, S.; Chen, H.; Ma, L.; Ding, Z.; Jiang, L. Tofacitinib for the Treatment of Antineutrophil Cytoplasm Antibody-Associated Vasculitis: A Pilot Study. Ann. Rheum. Dis. 2021, 80, 1631–1633. [Google Scholar] [CrossRef] [PubMed]
- Kawasaki, T.; Kawai, T. Toll-like Receptor Signalling Pathways. Front. Immunol. 2014, 5, 461. [Google Scholar] [CrossRef] [Green Version]
- Horton, C.G.; Pan, Z.J.; Farris, A.D. Targeting Toll-like Receptors for Treatment of SLE. Mediat. Inflamm. 2010, 2010, 498980. [Google Scholar] [CrossRef] [PubMed]
- Hurtado, P.R.; Jeffs, L.; Nitschke, J.; Patel, M.; Sarvestani, G.; Cassidy, J.; Hissaria, P.; Gillis, D.; Peh, C.A. CpG Oligodeoxynucleotide Stimulates Production of Anti-Neutrophil Cytoplasmic Antibodies in ANCA Associated Vasculitis. BMC Immunol. 2008, 9, 34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, C.; Deng, H.; Gong, Y.; You, R.; Chen, M.; Zhao, M.H. Effect of High Mobility Group Box 1 on Toll-like Receptor 9 in B Cells in Myeloperoxidase-ANCA-Associated Vasculitis. Autoimmunity 2020, 53, 28–34. [Google Scholar] [CrossRef] [PubMed]
- Brodie, E.J.; Infantino, S.; Low, M.S.Y.; Tarlinton, D.M. Lyn, Lupus, and (B) Lymphocytes, a Lesson on the Critical Balance of Kinase Signalling in Immunity. Front. Immunol. 2018, 9, 401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saijo, K.; Schmedt, C.; Su, I.; Karasuyama, H.; Lowell, C.A.; Reth, M.; Adachi, T.; Patke, A.; Santana, A.; Tarakhovsky, A. Essential Role of Src-Family Protein Tyrosine Kinases in NF-ΚB Activation during B Cell Development. Nat. Immunol. 2003, 4, 274–279. [Google Scholar] [CrossRef]
- Stepanek, O.; Draber, P.; Drobek, A.; Horejsi, V.; Brdicka, T. Nonredundant Roles of Src-Family Kinases and Syk in the Initiation of B-Cell Antigen Receptor Signalling. J. Immunol. 2013, 190, 1807–1818. [Google Scholar] [CrossRef] [Green Version]
- Nishizumi, H.; Horikawa, K.; Mlinaric-Rascan, I.; Yamamoto, T. A Double-Edged Kinase Lyn: A Positive and Negative Regulator for Antigen Receptor-Mediated Signals. J. Exp. Med. 1998, 187, 1343–1348. [Google Scholar] [CrossRef] [Green Version]
- Ma, H.; Yankee, T.M.; Hu, J.; Asai, D.J.; Harrison, M.L.; Geahlen, R.L. Visualization of Syk-Antigen Receptor Interactions Using Green Fluorescent Protein: Differential Roles for Syk and Lyn in the Regulation of Receptor Capping and Internalization. J. Immunol. 2001, 166, 1507–1516. [Google Scholar] [CrossRef] [Green Version]
- Lamagna, C.; Hu, Y.; DeFranco, A.L.; Lowell, C.A. B Cell-Specific Loss of Lyn Kinase Leads to Autoimmunity. J. Immunol. 2014, 192, 919–928. [Google Scholar] [CrossRef] [Green Version]
- Flores-Borja, F.; Kabouridis, P.S.; Jury, E.C.; Isenberg, D.A.; Mageed, R.A. Decreased Lyn Expression and Translocation to Lipid Raft Signalling Domains in B Lymphocytes from Patients with Systemic Lupus Erythematosus. Arthritis Rheum. 2005, 52, 3955–3965. [Google Scholar] [CrossRef]
- Liossis, S.N.C.; Solomou, E.E.; Dimopoulos, M.A.; Panayiotidis, P.; Mavrikakis, M.M.; Sfikakis, P.P. B-Cell Kinase Lyn Deficiency in Patients with Systemic Lupus Erythematosus. J. Investig. Med. 2001, 49, 157–165. [Google Scholar] [CrossRef]
- Barr, P.M.; Wei, C.; Roger, J.; Schaefer-Cutillo, J.; Kelly, J.L.; Rosenberg, A.F.; Jung, J.; Sanz, I.; Friedberg, J.W. Syk Inhibition with Fostamatinib Leads to Transitional B Lymphocyte Depletion. Clin. Immunol. 2012, 142, 237–242. [Google Scholar] [CrossRef] [Green Version]
- McAdoo, S.P.; Prendecki, M.; Tanna, A.; Bhatt, T.; Bhangal, G.; McDaid, J.; Masuda, E.S.; Cook, H.T.; Tam, F.W.K.; Pusey, C.D. Spleen Tyrosine Kinase Inhibition Is an Effective Treatment for Established Vasculitis in a Pre-Clinical Model. Kidney Int. 2020, 97, 1196–1207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sadras, T.; Martin, M.; Kume, K.; Robinson, M.E.; Saravanakumar, S.; Lenz, G.; Chen, Z.; Song, J.Y.; Siddiqi, T.; Oksa, L.; et al. Developmental Partitioning of SYK and ZAP70 Prevents Autoimmunity and Cancer. Mol. Cell 2021, 81, 2094–2111.e9. [Google Scholar] [CrossRef] [PubMed]
- Yokozeki, T.; Adler, K.; Lankar, D.; Bonnerot, C. B Cell Receptor-Mediated Syk-Independent Activation of Phosphatidylinositol 3-Kinase, Ras, and Mitogen-Activated Protein Kinase Pathways. J. Immunol. 2003, 171, 1328–1335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Otero, D.C.; Omori, S.A.; Rickert, R.C. CD19-Dependent Activation of Akt Kinase in B-Lymphocytes. J. Biol. Chem. 2001, 276, 1474–1478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarbassov, D.D.; Guertin, D.A.; Ali, S.M.; Sabatini, D.M. Phosphorylation and Regulation of Akt/PKB by the Rictor-MTOR Complex. Science 2005, 307, 1098–1101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Werner, M.; Hobeika, E.; Jumaa, H. Role of PI3K in the Generation and Survival of B Cells. Immunol. Rev. 2010, 237, 55–71. [Google Scholar] [CrossRef]
- Bai, D.; Ueno, L.; Vogt, P.K. Akt-Mediated Regulation of NFκB and the Essentialness of NFκB for the Oncogenicity of PI3K and Akt. Int. J. Cancer 2009, 125, 2863–2870. [Google Scholar] [CrossRef] [Green Version]
- Bacalao, M.A.; Satterthwaite, A.B. Recent Advances in Lupus B Cell Biology: PI3K, IFNγ, and Chromatin. Front. Immunol. 2021, 11, 615673. [Google Scholar] [CrossRef]
- Donahue, A.C.; Fruman, D.A. PI3K Signalling Controls Cell Fate at Many Points in B Lymphocyte Development and Activation. Semin. Cell Dev. Biol. 2004, 15, 183–197. [Google Scholar] [CrossRef] [PubMed]
- Hodson, D.J.; Turner, M. The role of PI3K signalling in the B cell response to antigen. In Crossroads between Innate and Adaptive Immunity II; Schoenberger, S.P., Katsikis, P.D., Pulendran, B., Eds.; Springer: New York, NY, USA, 2009; pp. 43–53. ISBN 978-0-387-79311-5. [Google Scholar]
- Okkenhaug, K.; Vanhaesebroeck, B. PI3K in Lymphocyte Development, Differentiation and Activation. Nat. Rev. Immunol. 2003, 3, 317–330. [Google Scholar] [CrossRef]
- Corneth, O.B.J.; Klein Wolterink, R.G.J.; Hendriks, R.W. BTK Signalling in B Cell Differentiation and Autoimmunity. Curr. Top. Microbiol. Immunol. 2016, 393, 67–105. [Google Scholar] [CrossRef] [PubMed]
- Corneth, O.B.J.; Verstappen, G.M.P.; Paulissen, S.M.J.; de Bruijn, M.J.W.; Rip, J.; Lukkes, M.; van Hamburg, J.P.; Lubberts, E.; Bootsma, H.; Kroese, F.G.M.; et al. Enhanced Bruton’s Tyrosine Kinase Activity in Peripheral Blood B Lymphocytes from Patients with Autoimmune Disease. Arthritis Rheumatol. 2017, 69, 1313–1324. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, Y.; Iwai, K. Roles of the NF-κB Pathway in B-Lymphocyte Biology. In B Cell Receptor Signalling; Kurosaki, T., Wienands, J., Eds.; Springer International Publishing: Cham, Switzerland, 2016; pp. 177–209. ISBN 978-3-319-26133-1. [Google Scholar]
- Lim, K.-H.; Yang, Y.; Staudt, L.M. Pathogenetic Importance and Therapeutic Implications of NF-ΚB in Lymphoid Malignancies. Immunol. Rev. 2012, 246, 359–378. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.C. NF-ΚB Signalling in Inflammation. Signal Transduct. Target. Ther. 2017, 2, e17023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaileh, M.; Sen, R. NF-ΚB Function in B Lymphocytes. Immunol. Rev. 2012, 246, 254–271. [Google Scholar] [CrossRef]
- Müller, J.R.; Siebenlist, U. Lymphotoxin β Receptor Induces Sequential Activation of Distinct NF-ΚB Factors via Separate Signalling Pathways. J. Biol. Chem. 2003, 278, 12006–12012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shih, V.F.S.; Tsui, R.; Caldwell, A.; Hoffmann, A. A Single NFκB System for Both Canonical and Non-Canonical Signalling. Cell Res. 2011, 21, 86–102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brightbill, H.D.; Suto, E.; Blaquiere, N.; Ramamoorthi, N.; Sujatha-Bhaskar, S.; Gogol, E.B.; Castanedo, G.M.; Jackson, B.T.; Kwon, Y.C.; Haller, S.; et al. NF-ΚB Inducing Kinase Is a Therapeutic Target for Systemic Lupus Erythematosus. Nat. Commun. 2018, 9, 179. [Google Scholar] [CrossRef] [PubMed]
- Monach, P.A.; Warner, R.L.; Tomasson, G.; Specks, U.; Stone, J.H.; Ding, L.; Fervenza, F.C.; Fessler, B.J.; Hoffman, G.S.; Iklé, D.; et al. Serum Proteins Reflecting Inflammation, Injury and Repair as Biomarkers of Disease Activity in ANCA-Associated Vasculitis. Ann. Rheum. Dis. 2013, 72, 1342–1350. [Google Scholar] [CrossRef]
- Rieckmann, P.; Tuscano, J.M.; Kehrl, J.H. Tumour Necrosis Factor-α (TNF-α) and Interleukin-6 (IL-6) in B-Lymphocyte Function. Methods 1997, 11, 128–132. [Google Scholar] [CrossRef] [PubMed]
- Jónasdóttir, O.; Petersen, J.; Bendtzen, K. Tumour Necrosis Factor-α (TNF), Lymphotoxin and TNF Receptor Levels in Serum from Patients with Wegener’s Granulomatosis. Apmis 2001, 109, 781–786. [Google Scholar] [CrossRef] [PubMed]
- Eriksson, P.; Sandell, C.; Backteman, K.; Ernerudh, J. Expansions of CD4+CD28- and CD8+CD28- T Cells in Granulomatosis with Polyangiitis and Microscopic Polyangiitis Are Associated with Cytomegalovirus Infection but Not with Disease Activity. J. Rheumatol. 2012, 39, 1840–1843. [Google Scholar] [CrossRef] [PubMed]
- Bertram, A.; Lovric, S.; Engel, A.; Beese, M.; Wyss, K.; Hertel, B.; Park, J.K.; Becker, J.U.; Kegel, J.; Haller, H.; et al. Circulating ADAM17 Level Reflects Disease Activity in Proteinase-3 ANCA-Associated Vasculitis. J. Am. Soc. Nephrol. 2015, 26, 2860–2870. [Google Scholar] [CrossRef] [Green Version]
- Ostendorf, L.; Burns, M.; Durek, P.; Heinz, G.A.; Heinrich, F.; Garantziotis, P.; Enghard, P.; Richter, U.; Biesen, R.; Schneider, U.; et al. Targeting CD38 with Daratumumab in Refractory Systemic Lupus Erythematosus. N. Engl. J. Med. 2020, 383, 1149–1155. [Google Scholar] [CrossRef] [PubMed]
- Mougiakakos, D.; Krönke, G.; Völkl, S.; Kretschmann, S.; Aigner, M.; Kharboutli, S.; Böltz, S.; Manger, B.; Mackensen, A.; Schett, G. CD19-Targeted CAR T Cells in Refractory Systemic Lupus Erythematosus. N. Engl. J. Med. 2021, 385, 567–569. [Google Scholar] [CrossRef] [PubMed]
- Bontscho, J.; Schreiber, A.; Manz, R.A.; Schneider, W.; Luft, F.C.; Kettritz, R. Myeloperoxidase-Specific Plasma Cell Depletion by Bortezomib Protects from Anti-Neutrophil Cytoplasmic Autoantibodies-Induced Glomerulonephritis. J. Am. Soc. Nephrol. 2011, 22, 336–348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Novikov, P.; Moiseev, S.; Bulanov, N.; Shchegoleva, E. Bortezomib in Refractory ANCA-Associated Vasculitis: A New Option? Ann. Rheum. Dis. 2016, 75, e9. [Google Scholar] [CrossRef] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Merino-Vico, A.; van Hamburg, J.P.; Tas, S.W. B Lineage Cells in ANCA-Associated Vasculitis. Int. J. Mol. Sci. 2022, 23, 387. https://doi.org/10.3390/ijms23010387
Merino-Vico A, van Hamburg JP, Tas SW. B Lineage Cells in ANCA-Associated Vasculitis. International Journal of Molecular Sciences. 2022; 23(1):387. https://doi.org/10.3390/ijms23010387
Chicago/Turabian StyleMerino-Vico, Ana, Jan Piet van Hamburg, and Sander W. Tas. 2022. "B Lineage Cells in ANCA-Associated Vasculitis" International Journal of Molecular Sciences 23, no. 1: 387. https://doi.org/10.3390/ijms23010387
APA StyleMerino-Vico, A., van Hamburg, J. P., & Tas, S. W. (2022). B Lineage Cells in ANCA-Associated Vasculitis. International Journal of Molecular Sciences, 23(1), 387. https://doi.org/10.3390/ijms23010387