
Two Novel Dimorphism-Related Virulence Factors of Zymoseptoria tritici Identified Using Agrobacterium-Mediated Insertional Mutagenesis

 ,
, {kind=link}
{kind=link}
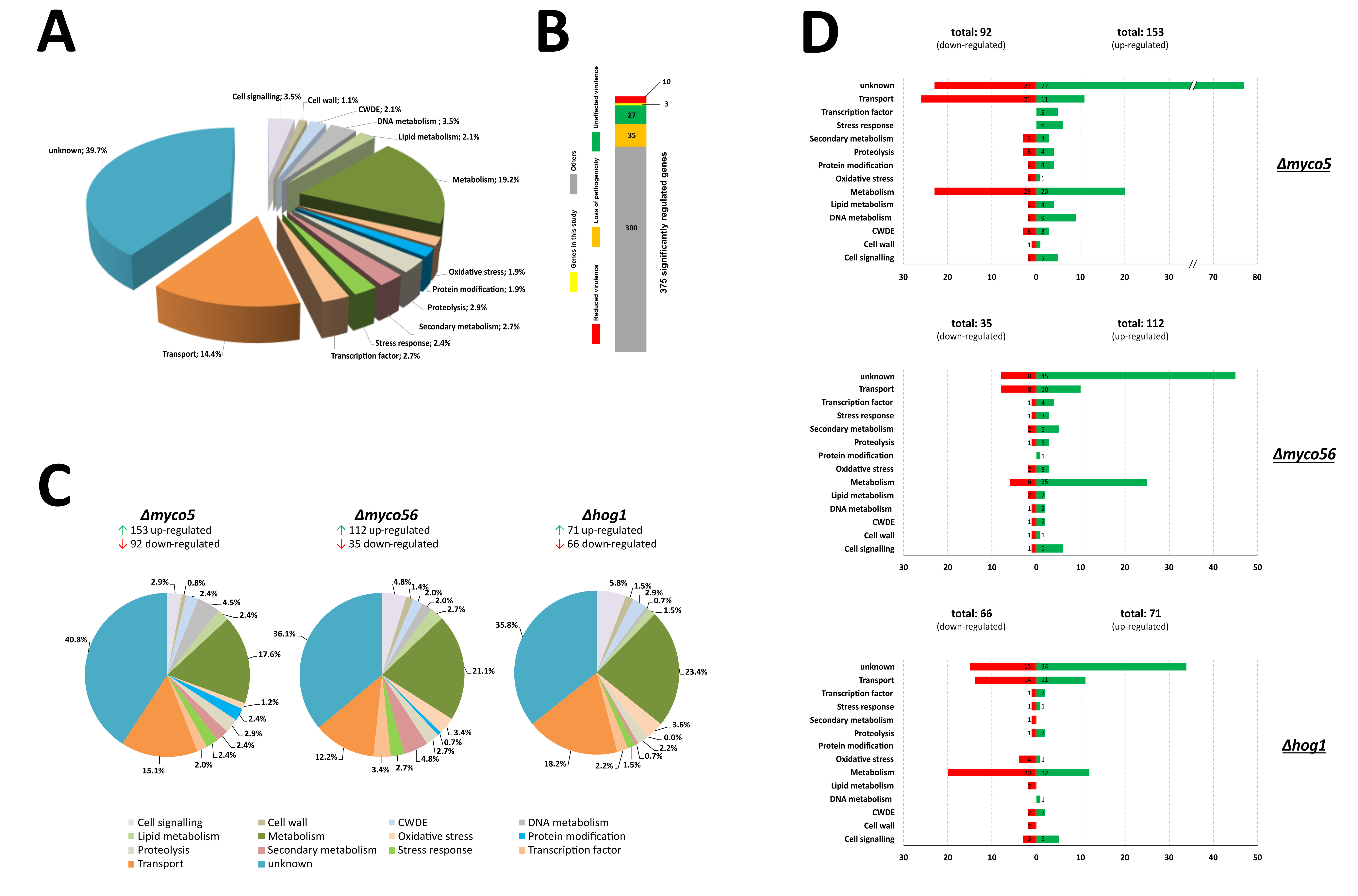{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
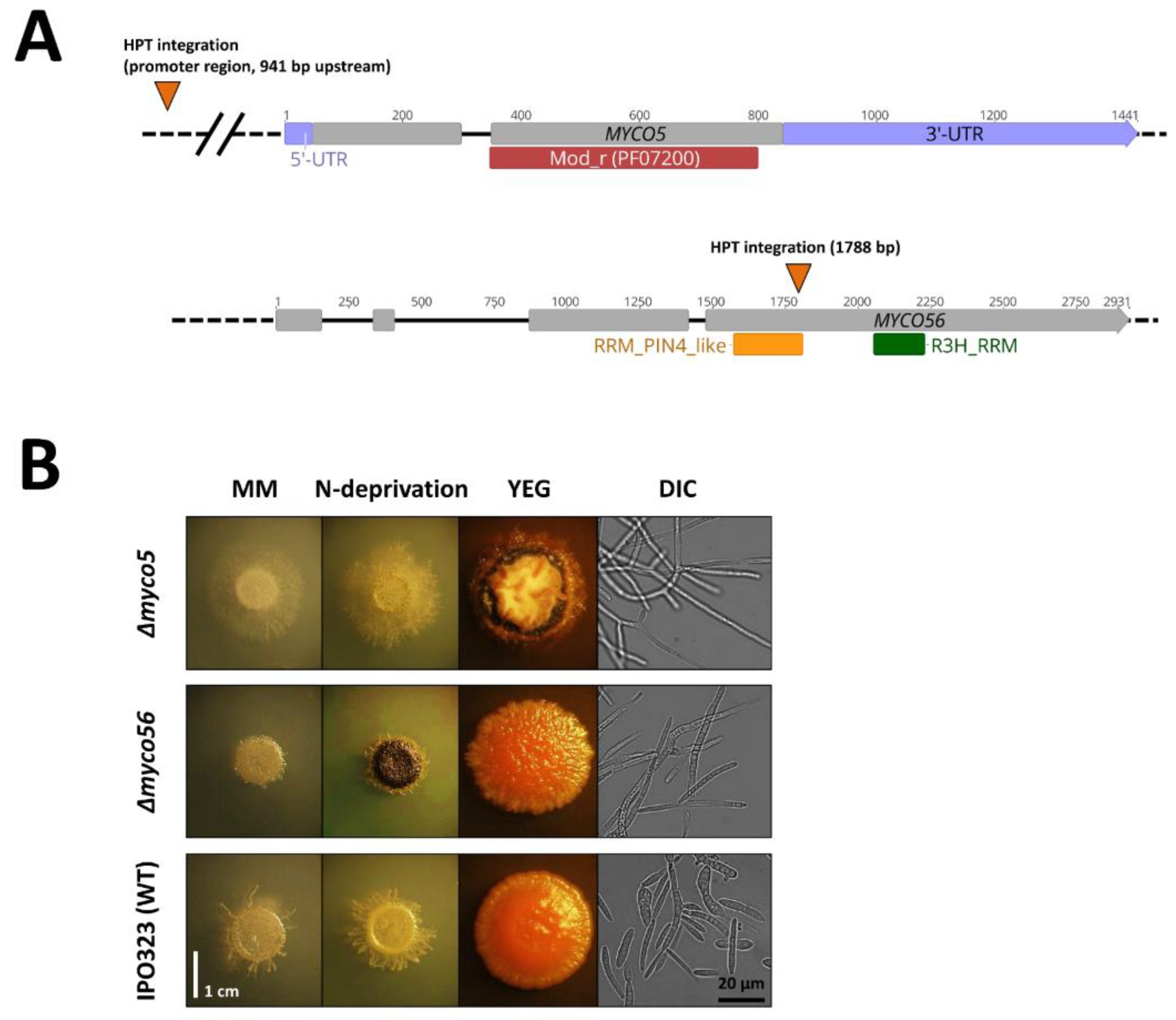
2.1. Identification of T-DNA Insertion Sites in myco#5 and myco#56 Mutant Strains and Gene Sequence Analysis
2.2. Targeted Gene Deletion Corroborates the Role in the Dimorphic Switch and Virulence for the MYCO5 and MYCO56 Genes
2.3. Transcriptome Analysis of Selected Strains during the Dimorphic Transition on Artificial Medium
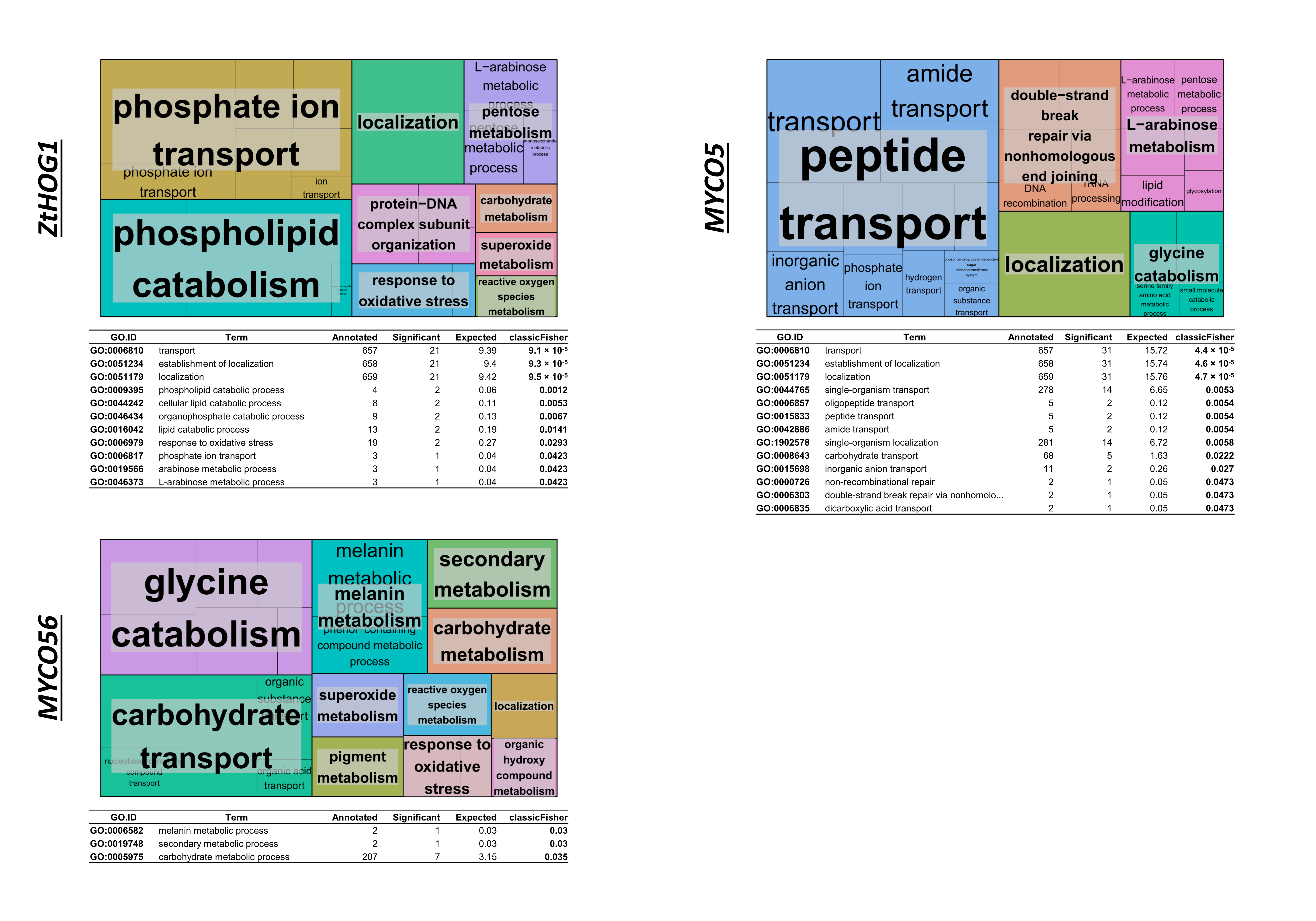
2.3.1. Genome-Wide Transcriptional Effects of MYCO5 Perturbation
2.3.2. Genome-Wide Transcriptional Effects of MYCO56 Perturbation
2.3.3. Genome-Wide Transcriptional Effects of ZtHOG1 Perturbation
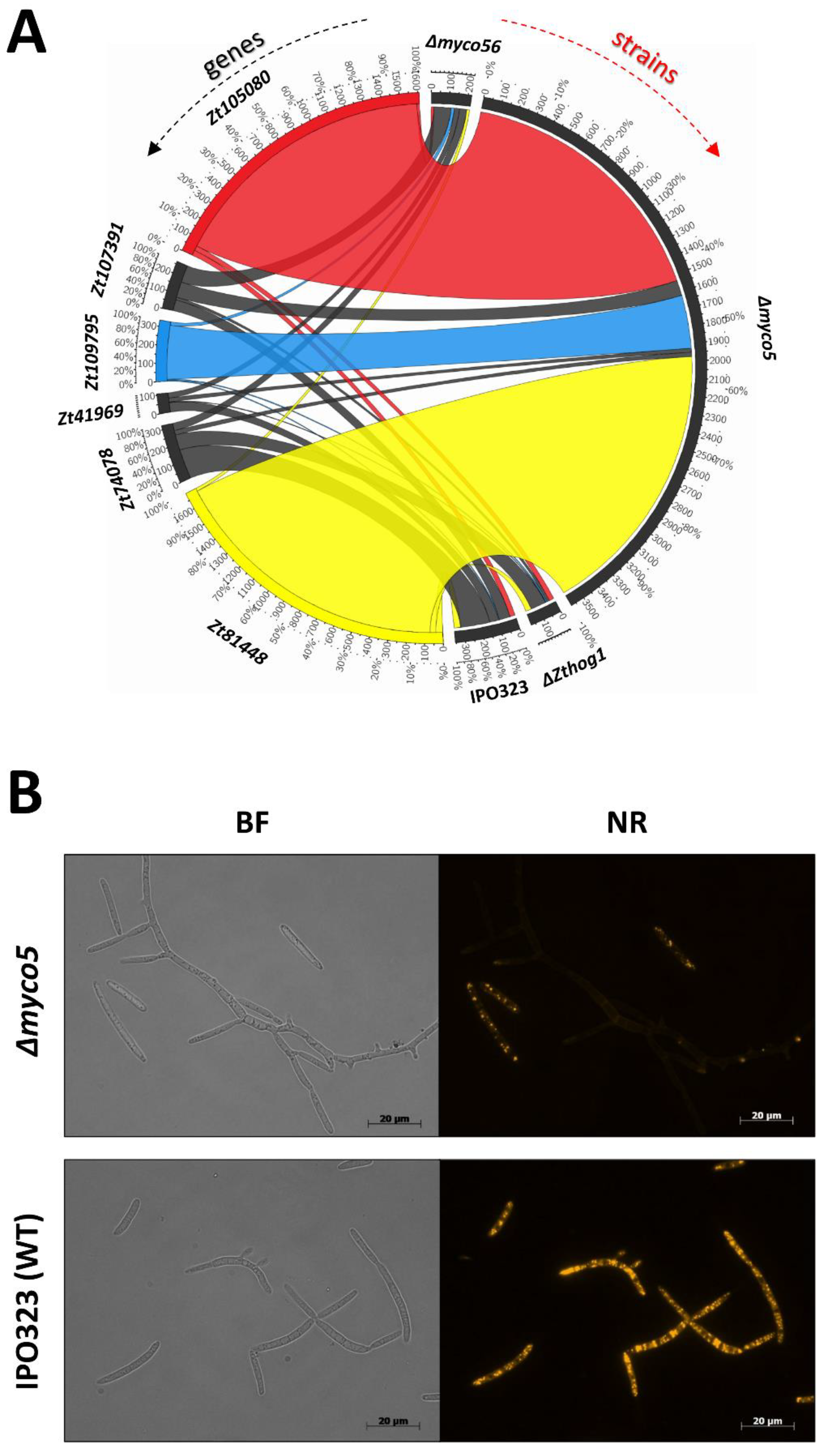
2.4. There Are Genes Unique to Zymoseptoria tritici among the DE Datasets
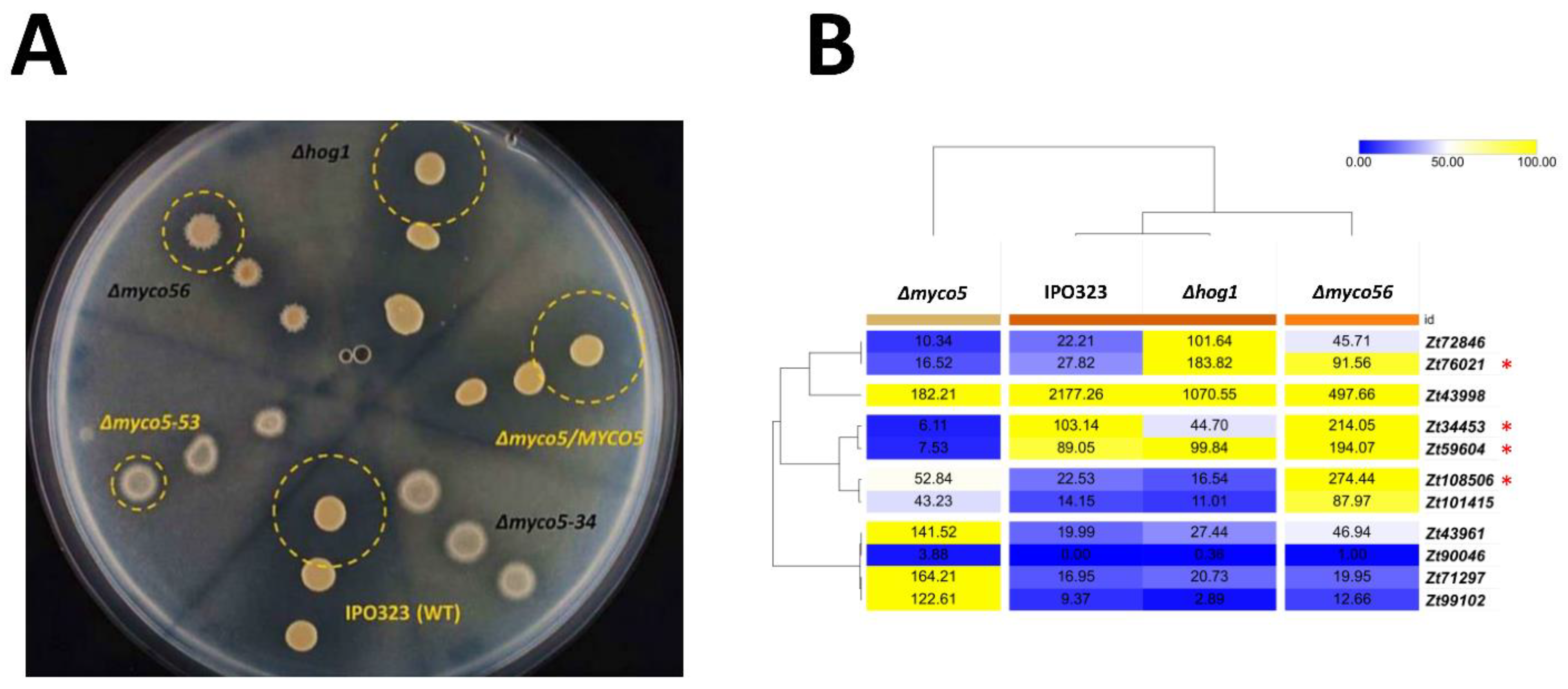
2.5. Δmyco56 Has Increased Pigmentation Following Growth on a Nutrient-Depleted Medium and When Grown as a Submerged Culture, while Δmyco5 Exhibits Thermo-Sensitivity
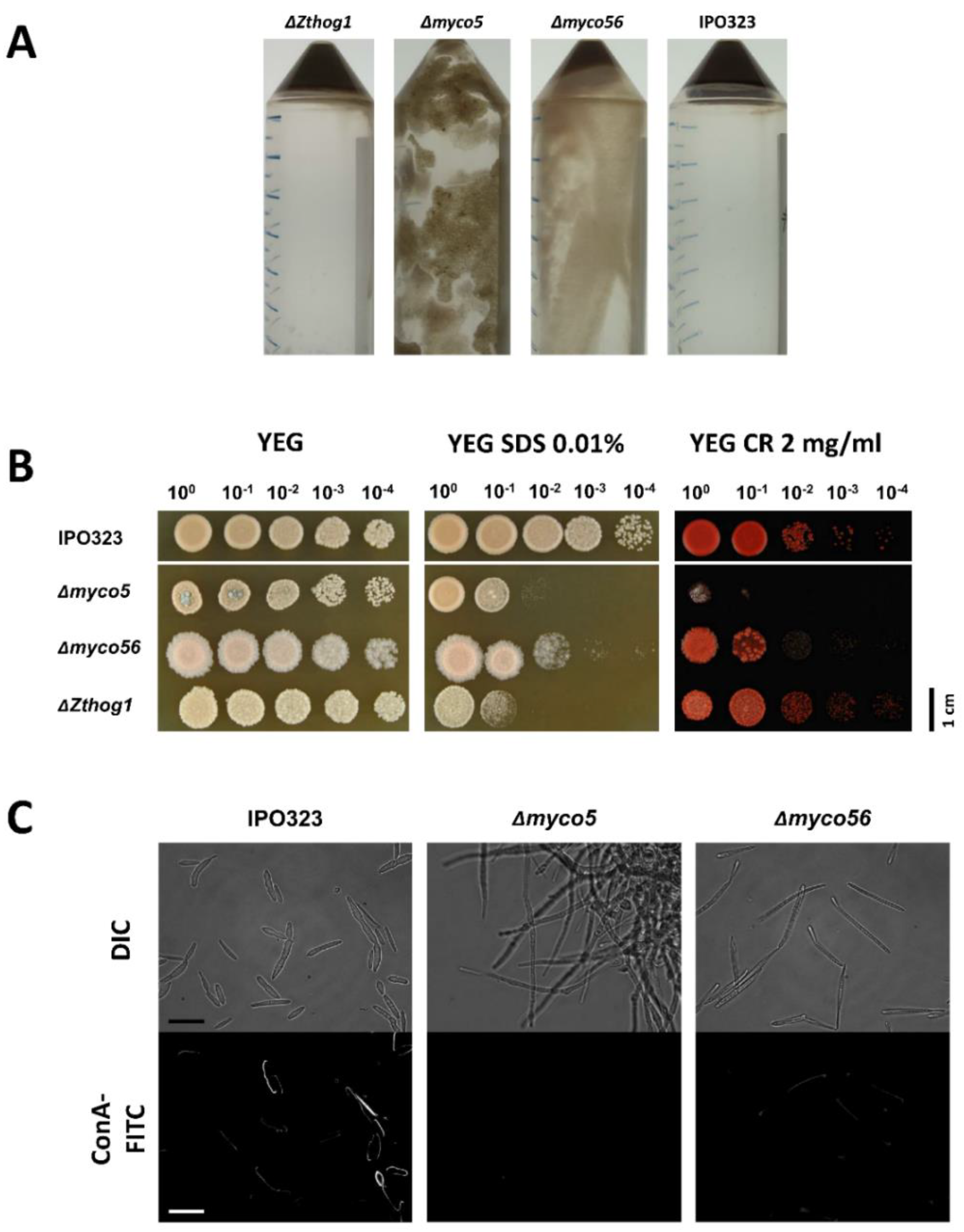
2.6. Vegetative Growth and Stress Response of the Δmyco5, Δmyco56 and ΔZthog1 Mutants
2.7. MYCO5 Plays a Role in the Oxidative Stress Response
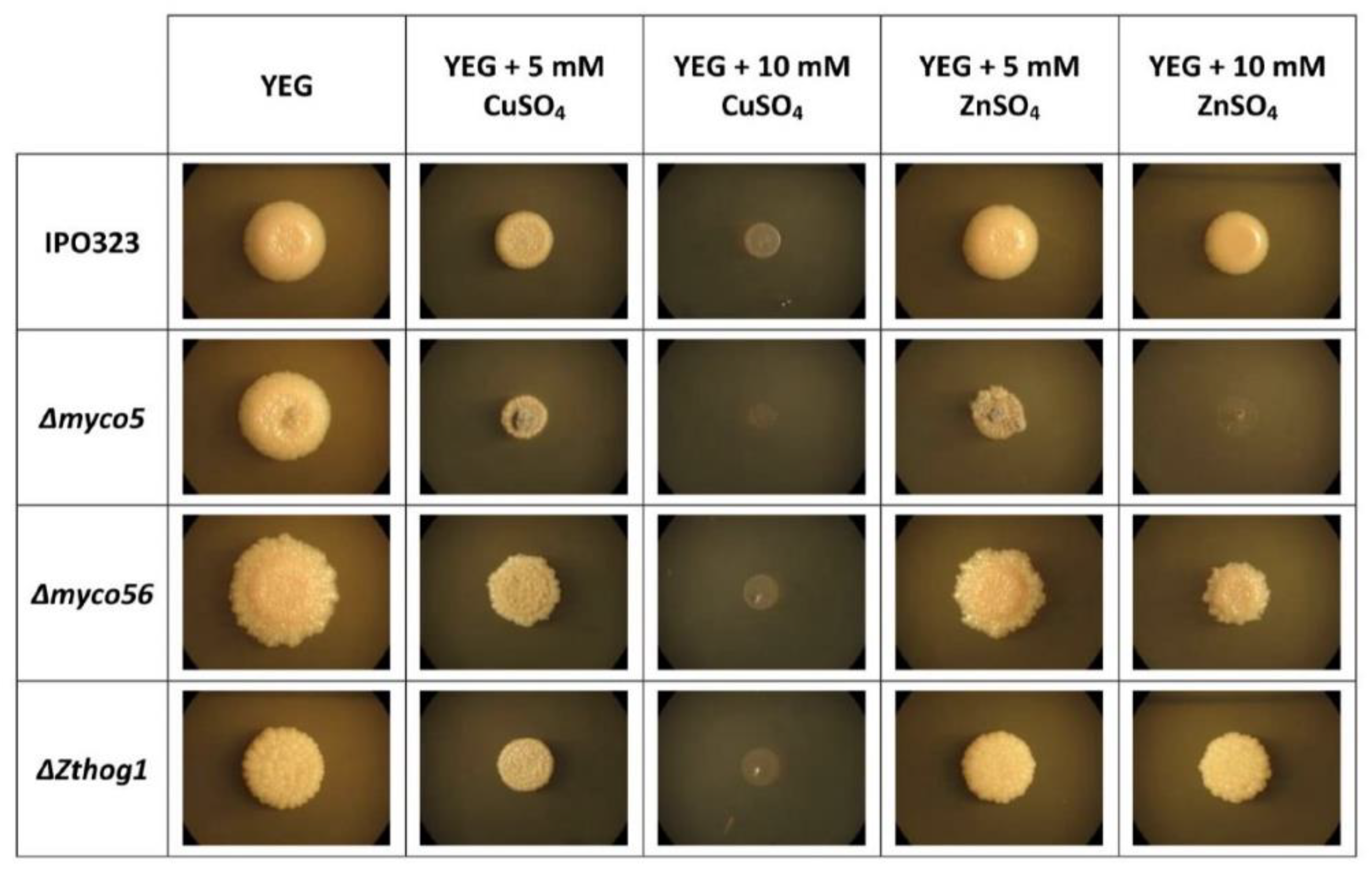
2.8. MYCO5 Is Involved in the Detoxification of Transient Metals
2.9. MYCO5 Has a Role in Maintaining Cellular Lipid Status
2.10. The Δmyco5 Mutant Exhibits Reduced Proteolytic Activity
2.11. Altered Cell-Wall Composition in the Δmyco5 and Δmyco56 Mutants
3. Discussion
3.1. MYCO5 Negatively Influences the Dimorphic Switch and Is Involved in Diverse Biological Processes
3.2. MYCO56 Encodes RNA-Binding Protein with a Role in Cell Branching, the Dimorphic Switch and Melanin Biosynthesis
3.3. Transcriptome Analysis Provides Crucial Insights into the Regulation of the Dimorphic Switch
4. Conclusions and Outlook
5. Material and Methods
5.1. Strains, Growth Conditions and Oligonucleotides
5.2. Nucleic Acid Manipulations
5.3. Pathogenicity Assays
5.4. Multiple Alignment and Phylogenetic Study of Zymoseptoria tritici Amino Acid Sequences
5.5. RNA-Seq Analysis
5.6. qRT-PCR Analysis of (Chloro) Peroxidases and Dismutases Encoding Genes
5.7. RNA-Seq Data Analysis of the Transcription of Known Pathogenicity Related Genes
5.8. Additional Analyses/Gene Ontology Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kettles, G.J.; Kanyuka, K. Dissecting the molecular interactions between wheat and the fungal pathogen Zymoseptoria tritici. Front. Plant Sci. 2016, 7, 508. [Google Scholar] [CrossRef] [Green Version]
- Orton, E.S.; Deller, S.; Brown, J.K. Mycosphaerella graminicola: From genomics to disease control. Mol. Plant Pathol. 2011, 12, 413–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rudd, J.J. Previous bottlenecks and future solutions to dissecting the Zymoseptoria tritici–wheat host-pathogen interaction. Fungal Genet. Biol. 2015, 79, 24–28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, F.; Li, W.; Jørgensen, H.J. Transcriptional reprogramming of wheat and the hemibiotrophic pathogen Septoria tritici during two phases of the compatible interaction. PLoS ONE 2013, 8, e81606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brunner, P.C.; Torriani, S.F.; Croll, D.; Stukenbrock, E.H.; McDonald, B.A. Coevolution and life cycle specialization of plant cell wall degrading enzymes in a hemibiotrophic pathogen. Mol. Biol. Evol. 2013, 30, 1337–1347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kellner, R.; Bhattacharyya, A.; Poppe, S.; Hsu, T.Y.; Brem, R.B.; Stukenbrock, E.H. Expression profiling of the wheat pathogen Zymoseptoria tritici reveals genomic patterns of transcription and host-specific regulatory programs. Genome Biol. Evol. 2014, 6, 1353–1365. [Google Scholar] [CrossRef] [PubMed]
- Rudd, J.J.; Kanyuka, K.; Hassani-Pak, K.; Derbyshire, M.; Andongabo, A.; Devonshire, J.; Hooper, J. Transcriptome and metabolite profiling of the infection cycle of Zymoseptoria tritici on wheat reveals a biphasic interaction with plant immunity involving differential pathogen chromosomal contributions and a variation on the hemibiotrophic lifestyle definition. Plant Physiol. 2015, 167, 1158–1185. [Google Scholar] [PubMed] [Green Version]
- Klein, B.S.; Tebbets, B. Dimorphism and virulence in fungi. Curr. Opin. Microbiol. 2007, 10, 314–319. [Google Scholar] [CrossRef] [Green Version]
- Boyce, K.J.; Andrianopoulos, A. Fungal dimorphism: The switch from hyphae to yeast is a specialized morphogenetic adaptation allowing colonization of a host. FEMS Microbiol. Rev. 2015, 39, 797–811. [Google Scholar] [CrossRef] [Green Version]
- Gauthier, G.M. Dimorphism in fungal pathogens of mammals, plants, and insects. PLoS Pathog 2015, 11, e1004608. [Google Scholar] [CrossRef] [PubMed]
- Mehrabi, R.; Zwiers, L.H.; de Waard, M.A.; Kema, G.H. MgHog1 regulates dimorphism and pathogenicity in the fungal wheat pathogen Mycosphaerella graminicola. Mol. Plant-Microbe Interact. 2006, 19, 1262–1269. [Google Scholar] [CrossRef] [Green Version]
- Mehrabi, R.; van der Lee, T.; Waalwijk, C.; Kema, G.H. MgSlt2, a cellular integrity MAP kinase gene of the fungal wheat pathogen Mycosphaerella graminicola, is dispensable for penetration but essential for invasive growth. Mol. Plant-Microbe Interact. 2006, 19, 389–398. [Google Scholar] [CrossRef] [Green Version]
- Choi, Y.E.; Goodwin, S.B. Gene encoding a c-type cyclin in Mycosphaerella graminicola is involved in aerial mycelium formation, filamentous growth, hyphal swelling, melanin biosynthesis, stress response, and pathogenicity. Mol. Plant-Microbe Interact. 2011, 24, 469–477. [Google Scholar] [CrossRef] [Green Version]
- Choi, Y.E.; Goodwin, S.B. MVE1, encoding the velvet gene product homolog in Mycosphaerella graminicola, is associated with aerial mycelium formation, melanin biosynthesis, hyphal swelling, and light signaling. Appl. Environ. Microbiol. 2011, 77, 942–953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perez-Nadales, E.; Nogueira, M.F.A.; Baldin, C.; Castanheira, S.; El Ghalid, M.; Grund, E.; Naik, V. Fungal model systems and the elucidation of pathogenicity determinants. Fungal Genet. Biol. 2014, 70, 42–67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Motteram, J.; Lovegrove, A.; Pirie, E.; Marsh, J.; Devonshire, J.; van de Meene, A.; Rudd, J.J. Aberrant protein N-glycosylation impacts upon infection-related growth transitions of the haploid plant-pathogenic fungus Mycosphaerella graminicola. Mol. Microbiol. 2011, 81, 415–433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mirzadi Gohari, A.; Mehrabi, R.; Robert, O.; Ince, I.A.; Boeren, S.; Schuster, M.; Kema, G.H. Molecular characterization and functional analyses of ZtWor1, a transcriptional regulator of the fungal wheat pathogen Zymoseptoria tritici. Mol. Plant Pathol. 2014, 15, 394–405. [Google Scholar] [CrossRef] [PubMed]
- Mohammadi, N.; Mehrabi, R.; Gohari, A.M.; Goltapeh, E.M.; Safaie, N.; Kema, G.H. The ZtVf1 transcription factor regulates development and virulence in the foliar wheat pathogen Zymoseptoria tritici. Fungal Genet. Biol. 2017, 109, 26–35. [Google Scholar] [CrossRef]
- Mohammadi, N.; Mehrabi, R.; Mirzadi Gohari, A.; Roostaei, M.; Mohammadi Goltapeh, E.; Safaie, N.; Kema, G.H. MADS-Box Transcription Factor ZtRlm1 Is Responsible for Virulence and Development of the Fungal Wheat Pathogen Zymoseptoria tritici. Front. Microbiol. 2020, 11, 1976. [Google Scholar] [CrossRef]
- Habig, M.; Bahena-Garrido, S.M.; Barkmann, F.; Haueisen, J.; Stukenbrock, E.H. The transcription factor Zt107320 affects the dimorphic switch, growth and virulence of the fungal wheat pathogen Zymoseptoria tritici. Mol. Plant Pathol. 2020, 21, 124–138. [Google Scholar] [CrossRef] [Green Version]
- Francisco, C.S.; Ma, X.; Zwyssig, M.M.; McDonald, B.A.; Palma-Guerrero, J. Morphological changes in response to environmental stresses in the fungal plant pathogen Zymoseptoria tritici. Sci. Rep. 2019, 9, 9642. [Google Scholar]
- Yemelin, A.; Brauchler, A.; Jacob, S.; Laufer, J.; Heck, L.; Foster, A.J.; Thines, E. Identification of factors involved in dimorphism and pathogenicity of Zymoseptoria tritici. PLoS ONE 2017, 12, 915. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Gurr, S.J. Walking into the unknown: A ‘step down’PCR-based technique leading to the direct sequence analysis of flanking genomic DNA. Gene 2000, 253, 145–150. [Google Scholar] [CrossRef]
- Hurley, J.H.; Hanson, P.I. Membrane budding and scission by the ESCRT machinery: It’s all in the neck. Nat. Rev. Mol. Cell Biol. 2010, 11, 556–566. [Google Scholar] [CrossRef]
- Deo, R.C.; Bonanno, J.B.; Sonenberg, N.; Burley, S.K. Recognition of polyadenylate RNA by the poly (A)-binding protein. Cell 1999, 98, 835–845. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez-Gabriel, M.A.; Burns, G.; McDonald, W.H.; Martín, V.; Yates, J.R.; Bähler, J.; Russell, P. RNA-binding protein Csx1 mediates global control of gene expression in response to oxidative stress. EMBO J. 2003, 22, 6256–6266. [Google Scholar] [CrossRef] [Green Version]
- Martin, V.; Rodríguez-Gabriel, M.A.; McDonald, W.H.; Watt, S.; Yates, J.R.; Bähler, J.; Russell, P. Cip1 and Cip2 are novel RNA-recognition-motif proteins that counteract Csx1 function during oxidative stress. Mol. Biol. Cell 2006, 17, 1176–1183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grishin, N.V. The R3H motif: A domain that binds single-stranded nucleic acids. Trends Biochem. Sci. 1998, 23, 329–330. [Google Scholar] [CrossRef]
- DeZwaan, T.M.; Carroll, A.M.; Valent, B.; Sweigard, J.A. Magnaporthe grisea pth11p is a novel plasma membrane protein that mediates appressorium differentiation in response to inductive substrate cues. Plant Cell 1999, 11, 2013–2030. [Google Scholar] [CrossRef] [Green Version]
- Hung, C.Y.; Seshan, K.R.; Yu, J.J.; Schaller, R.; Xue, J.; Basrur, V.; Cole, G.T. A metalloproteinase of Coccidioides posadasii contributes to evasion of host detection. Infect. Immun. 2005, 73, 6689–6703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kubo, Y.; Takano, Y.; Endo, N.; Yasuda, N.; Tajima, S.; Furusawa, I. Cloning and structural analysis of the melanin biosynthesis gene SCD1 encoding scytalone dehydratase in Colletotrichum lagenarium. Appl. Environ. Microbiol. 1996, 62, 4340–4344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yemelin, A. Making Visualization Work for You: Deriving Valuable Insights from Omics Data. In Magnaporthe Oryzae; Humana: New York, NY, USA, 2021; pp. 129–148. [Google Scholar]
- Schweizer, A.; Rupp, S.; Taylor, B.N.; Röllinghoff, M.; Schröppel, K. The TEA/ATTS transcription factor CaTec1p regulates hyphal development and virulence in Candida albicans. Mol. Microbiol. 2000, 38, 435–445. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.F.; Li, M.; Li, Y.Q.; Chen, X.D.; Gao, X.D. The TEA/ATTS transcription factor YlTec1p represses the yeast-to-hypha transition in the dimorphic yeast Yarrowia lipolytica. FEMS Yeast Res. 2013, 13, 50–61. [Google Scholar] [CrossRef] [PubMed]
- Andaluz, E.; Calderone, R.; Reyes, G.; Larriba, G. Phenotypic analysis and virulence of Candida albicans LIG4 mutants. Infect. Immun. 2001, 69, 137–147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mouyna, I.; Morelle, W.; Vai, M.; Monod, M.; Léchenne, B.; Fontaine, T.; Latgé, J.P. Deletion of GEL2 encoding for a β (1–3) glucanosyltransferase affects morphogenesis and virulence in Aspergillus fumigatus. Mol. Microbiol. 2005, 56, 1675–1688. [Google Scholar] [CrossRef] [PubMed]
- Brennwald, P.; Rossi, G. Spatial regulation of exocytosis and cell polarity: Yeast as a model for animal cells. FEBS Lett. 2007, 581, 2119–2124. [Google Scholar] [CrossRef] [Green Version]
- Takano, Y.; Kubo, Y.; Shimizu, K.; Mise, K.; Okuno, T.; Furusawa, I. Structural analysis of PKS1, a polyketide synthase gene involved in melanin biosynthesis in Colletotrichum lagenarium. Mol. Gen. Genet. MGG 1995, 249, 162–167. [Google Scholar] [CrossRef]
- Tsuji, G.; Sugahara, T.; Fujii, I.; Mori, Y.; Ebizuka, Y.; Shiraishi, T.; Kubo, Y. Evidence for involvement of two naphthol reductases in the first reduction step of melanin biosynthesis pathway of Colletotrichum lagenarium. Mycol. Res. 2003, 107, 854–860. [Google Scholar] [CrossRef]
- Perpetua, N.S.; Kubo, Y.; Yasuda, N.; Takano, Y.; Furusawa, I. Cloning and characterization of a melanin biosynthetic THR1 reductase gene essential for appressorial penetration of Colletotrichum lagenarium. Mol. Plant-Microbe Interact. 1996, 9, 323–329. [Google Scholar] [CrossRef]
- Tsuji, G.; Kenmochi, Y.; Takano, Y.; Sweigard, J.; Farrall, L.; Furusawa, I.; Kubo, Y. Novel fungal transcriptional activators, Cmr1p of Colletotrichum lagenarium and Pig1p of Magnaporthe grisea, contain Cys2His2 zinc finger and Zn (II) 2Cys6 binuclear cluster DNA-binding motifs and regulate transcription of melanin biosynthesis genes in a developmentally specific manner. Mol. Microbiol. 2000, 38, 940–954. [Google Scholar]
- Matheis, S.; Yemelin, A.; Scheps, D.; Andresen, K.; Jacob, S.; Thines, E.; Foster, A.J. Functions of the Magnaporthe oryzae Flb3p and Flb4p transcription factors in the regulation of conidiation. Microbiol. Res. 2017, 196, 106–117. [Google Scholar] [CrossRef] [PubMed]
- Leidlich, S.D.; Ibrahim, A.S.; Fu, Y.; Koul, A.; Jessup, C.; Vitullo, J.; Ghannoum, M.A. Cloning and disruption of caPLB1, a phospholipase B gene involved in the pathogenicity of Candida albicans. J. Biol. Chem. 1998, 273, 26078–26086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greenspan, P.; Mayer, E.P.; Fowler, S.D. Nile red: A selective fluorescent stain for intracellular lipid droplets. J. Cell Biol. 1985, 100, 965–973. [Google Scholar] [CrossRef] [Green Version]
- Baenziger, J.U.; Fiete, D. Structural determinants of concanavalin a specificity for oligosaccharides. J. Biol. Chem. 1979, 254, 2400–2407. [Google Scholar] [CrossRef]
- Korn, M.; Schmidpeter, J.; Dahl, M.; Müller, S.; Voll, L.M.; Koch, C. A genetic screen for pathogenicity genes in the hemibiotrophic fungus Colletotrichum higginsianum identifies the plasma membrane proton pump Pma2 required for host penetration. PLoS ONE 2015, 10, e0125960. [Google Scholar]
- Cousin, A.; Mehrabi, R.; Guilleroux, M.; Dufresne, M.; Van der Lee, T.; Waalwijk, C.; Kema, G.H. The MAP kinase-encoding gene MgFus3 of the non-appressorium phytopathogen Mycosphaerella graminicola is required for penetration and in vitro pycnidia formation. Mol. Plant Pathol. 2006, 7, 269–278. [Google Scholar] [CrossRef] [PubMed]
- Urban, M.; Bhargava, T.; Hamer, J.E. An ATP driven efflux pump is a novel pathogenicity factor in rice blast disease. EMBO J. 1999, 18, 512–521. [Google Scholar] [CrossRef] [Green Version]
- Bazan, N.G. Cellular and molecular events mediated by docosahexaenoic acid-derived neuroprotectin D1 signaling in photoreceptor cell survival and brain protection. Prostaglandins Leukot. Essent. Fat. Acids 2009, 81, 205–211. [Google Scholar] [CrossRef] [Green Version]
- Chumley, F.G.; Valent, B. Genetic analysis of melanin-deficient, nonpathogenic mutants of Magnaporthe grisea. Mol. Plant-Microbe Interact 1990, 3, 135–143. [Google Scholar] [CrossRef]
- Fan, R.; Klosterman, S.J.; Wang, C.; Subbarao, K.V.; Xu, X.; Shang, W.; Hu, X. Vayg1 is required for microsclerotium formation and melanin production in Verticillium dahliae. Fungal Genet. Biol. 2017, 98, 1–11. [Google Scholar] [CrossRef]
- Taborda, C.P.; Da Silva, M.B.; Nosanchuk, J.D.; Travassos, L.R. Melanin as a virulence factor of Paracoccidioides brasiliensis and other dimorphic pathogenic fungi: A minireview. Mycopathologia 2008, 165, 331–339. [Google Scholar] [CrossRef] [Green Version]
- Howard, R.J.; Ferrari, M.A. Role of melanin in appressorium function. Exp. Mycol. 1989, 13, 403–418. [Google Scholar] [CrossRef]
- Kramer, B.; Thines, E.; Foster, A.J. MAP kinase signalling pathway components and targets conserved between the distantly related plant pathogenic fungi Mycosphaerella graminicola and Magnaporthe grisea. Fungal Genet. Biol. 2009, 46, 667–681. [Google Scholar] [CrossRef]
- Sambrook JRussell, D.W. Molecular Cloning: A Laboratory Manual; Cold Spring laboratory Press: New York, NY, USA, 2001. [Google Scholar]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Thierer, T. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef] [PubMed]
- Edgar Robert, C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trapnell, C.; Roberts, A.; Goff, L.; Pertea, G.; Kim, D.; Kelley, D.R.; Pachter, L. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 2012, 7, 562–578. [Google Scholar] [CrossRef] [Green Version]
- Trapnell, C.; Pachter, L.; Salzberg, S.L. TopHat: Discovering splice junctions with RNA-Seq. Bioinformatics 2009, 25, 1105–1111. [Google Scholar] [CrossRef] [PubMed]
- Robinson, J.T.; Thorvaldsdóttir, H.; Winckler, W.; Guttman, M.; Lander, E.S.; Getz, G.; Mesirov, J.P. Integrative genomics viewer. Nat. Biotechnol. 2011, 29, 24–26. [Google Scholar] [CrossRef] [Green Version]
- Thorvaldsdóttir, H.; Robinson, J.T.; Mesirov, J.P. Integrative Genomics Viewer (IGV): High-performance genomics data visualization and exploration. Brief. Bioinform. 2013, 14, 178–192. [Google Scholar] [CrossRef] [Green Version]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Pfaffl, M.W. A new mathematical model for relative quantification in real-time RT–PCR. Nucleic Acids Res. 2001, 29, e45. [Google Scholar] [CrossRef]
- Urban, M.; Pant, R.; Raghunath, A.; Irvine, A.G.; Pedro, H.; Hammond-Kosack, K.E. The Pathogen-Host Interactions database (PHI-base): Additions and future developments. Nucleic Acids Res. 2014, 43, D645–D665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conesa, A.; Götz, S. Blast2GO: A comprehensive suite for functional analysis in plant genomics. Int. J. Plant Genom. 2008, 2008, 15. [Google Scholar] [CrossRef] [PubMed]
- Alexa, A.; Rahnenfuhrer, J. topGO: Enrichment Analysis for Gene Ontology, R Package Version 2.28.0, Cranio. Available online: https://www.bioconductor.org/packages/release/bioc/manuals/topGO/man/topGO.pdf (accessed on 10 October 2021).
- Supek, F.; Bošnjak, M.; Škunca, N.; Šmuc, T. REVIGO summarizes and visualizes long lists of gene ontology terms. PLoS ONE 2011, 6, e21800. [Google Scholar] [CrossRef] [PubMed] [Green Version]











Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yemelin, A.; Brauchler, A.; Jacob, S.; Foster, A.J.; Laufer, J.; Heck, L.; Antelo, L.; Andresen, K.; Thines, E. Two Novel Dimorphism-Related Virulence Factors of Zymoseptoria tritici Identified Using Agrobacterium-Mediated Insertional Mutagenesis. Int. J. Mol. Sci. 2022, 23, 400. https://doi.org/10.3390/ijms23010400
Yemelin A, Brauchler A, Jacob S, Foster AJ, Laufer J, Heck L, Antelo L, Andresen K, Thines E. Two Novel Dimorphism-Related Virulence Factors of Zymoseptoria tritici Identified Using Agrobacterium-Mediated Insertional Mutagenesis. International Journal of Molecular Sciences. 2022; 23(1):400. https://doi.org/10.3390/ijms23010400
Chicago/Turabian StyleYemelin, Alexander, Annamaria Brauchler, Stefan Jacob, Andrew J. Foster, Julian Laufer, Larissa Heck, Luis Antelo, Karsten Andresen, and Eckhard Thines. 2022. "Two Novel Dimorphism-Related Virulence Factors of Zymoseptoria tritici Identified Using Agrobacterium-Mediated Insertional Mutagenesis" International Journal of Molecular Sciences 23, no. 1: 400. https://doi.org/10.3390/ijms23010400
APA StyleYemelin, A., Brauchler, A., Jacob, S., Foster, A. J., Laufer, J., Heck, L., Antelo, L., Andresen, K., & Thines, E. (2022). Two Novel Dimorphism-Related Virulence Factors of Zymoseptoria tritici Identified Using Agrobacterium-Mediated Insertional Mutagenesis. International Journal of Molecular Sciences, 23(1), 400. https://doi.org/10.3390/ijms23010400