
Affinity and Specificity for Binding to Glycosaminoglycans Can Be Tuned by Adapting Peptide Length and Sequence

 , , , , , and
, , , , , and
Abstract
:1. Introduction
2. Results
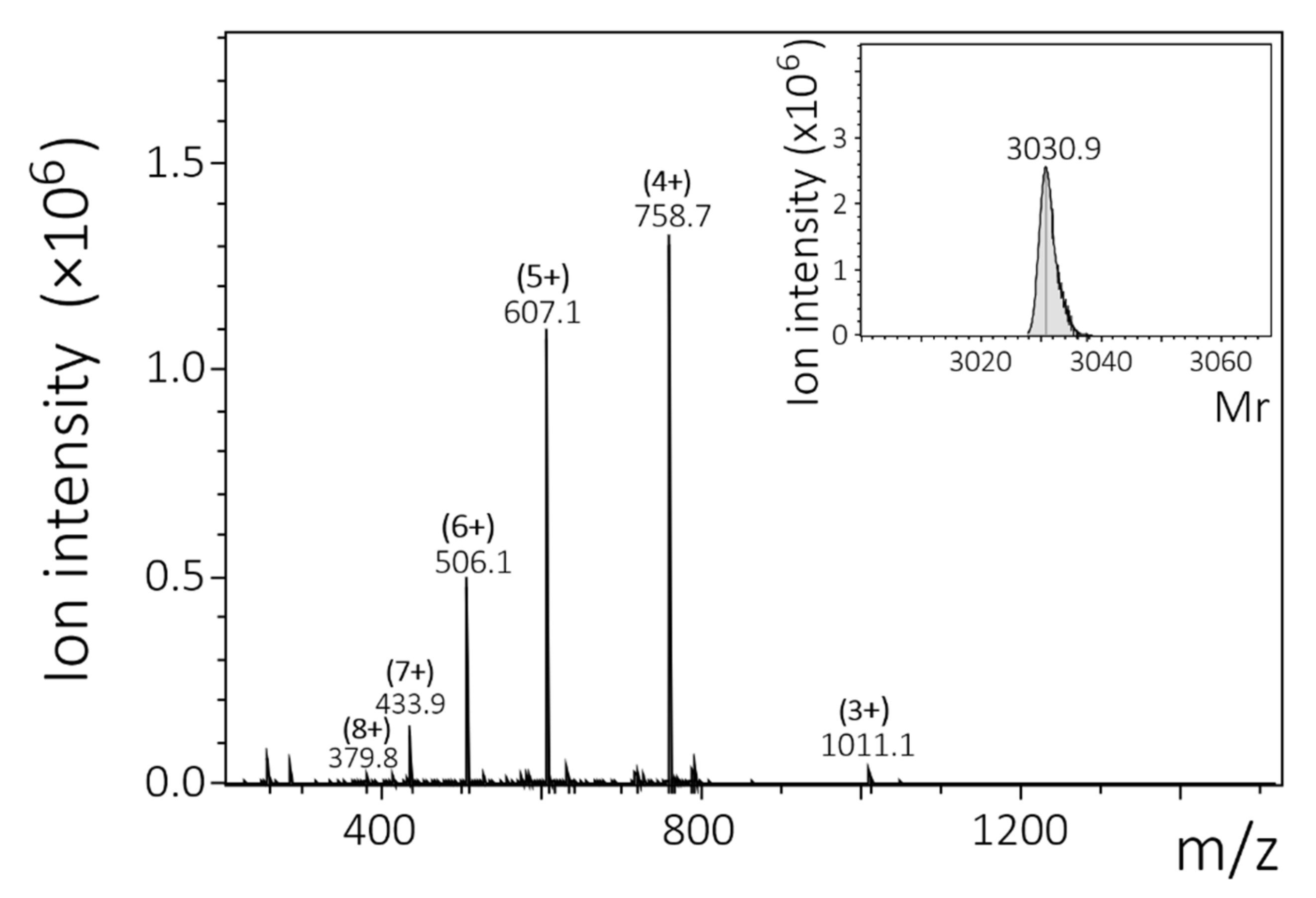
2.1. Chemical Synthesis and Fluorescent Labeling of Peptides
2.2. An Increased Number of GAG-Binding Motifs Results in Higher Affinity for HS
2.3. Gln Residues in Both GAG-Binding Motifs and Linker Sequences Increase the Affinity of Trimer Peptides for LMWH, dsLMWH and HS, but Not for HA
2.4. Dimer Peptides with Long versus Short GAG-Binding Motifs Are Characterized by Differential Affinity for LMWH and dsLMWH
2.5. Subcloning of a Mixed Population of pgsB-618 Cells Results in HS+ and HS− Subclones
2.6. Peptides Bind to Cellular GAGs with Differential Capacity
2.7. Binding of CXCL9(74-103) to Cellular GAGs Is Reduced by Adding Heparin or HS
3. Discussion
4. Materials and Methods
4.1. Solid-Phase Synthesis of Peptides
4.2. Production of Desulfated LMWH
4.3. Isothermal Fluorescence Titration (IFT)
4.4. Cells
4.5. Cellular Surface HS Expression
4.6. Subcloning of pgsB-618 Cells
4.7. Binding of Peptides to Cellular GAGs
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gandhi, N.S.; Mancera, R.L. The structure of glycosaminoglycans and their interactions with proteins. Chem. Biol. Drug Des. 2008, 72, 455–482. [Google Scholar] [CrossRef] [PubMed]
- Lindahl, U.; Couchman, J.; Kimata, K.; Esko, J.D. Chapter 17: Proteoglycans and sulfated glycosaminoglycans. In Essentials of Glycobiology, 3rd ed.; Varki, A., Cummings, R.D., Esko, J.D., Stanley, P., Hart, G.W., Aebi, M., Darvill, A.G., Kinoshita, T., Packer, N.H., Prestegard, J.H., Eds.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2015–2017; NBK453033. [Google Scholar]
- Vallet, S.D.; Clerc, O.; Ricard-Blum, S. Glycosaminoglycan-Protein Interactions: The First Draft of the Glycosaminoglycan Interactome. J. Histochem. Cytochem. 2021, 69, 93–104. [Google Scholar] [CrossRef] [PubMed]
- Shi, D.; Sheng, A.; Chi, L. Glycosaminoglycan-Protein Interactions and Their Roles in Human Disease. Front. Mol. Biosci. 2021, 8, 639666. [Google Scholar] [CrossRef]
- Esko, J.D.; Prestegard, J.H.; Linhardt, R.J. Chapter 38: Proteins that bind sulfated glycosaminoglycans. In Essentials of Glycobiology, 3rd ed.; Varki, A., Cummings, R.D., Esko, J.D., Stanley, P., Hart, G.W., Aebi, M., Darvill, A.G., Kinoshita, T., Packer, N.H., Prestegard, J.H., Eds.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2015–2017; NBK453026. [Google Scholar]
- Capila, I.; Linhardt, R.J. Heparin-protein interactions. Angew. Chem. Int. Ed. Engl. 2002, 41, 391–412. [Google Scholar] [CrossRef]
- Cardin, A.D.; Weintraub, H.J. Molecular modeling of protein-glycosaminoglycan interactions. Arteriosclerosis 1989, 9, 21–32. [Google Scholar] [CrossRef]
- Sobel, M.; Soler, D.F.; Kermode, J.C.; Harris, R.B. Localization and characterization of a heparin binding domain peptide of human von Willebrand factor. J. Biol. Chem. 1992, 267, 8857–8862. [Google Scholar] [CrossRef]
- Hileman, R.E.; Fromm, J.R.; Weiler, J.M.; Linhardt, R.J. Glycosaminoglycan-protein interactions: Definition of consensus sites in glycosaminoglycan binding proteins. Bioessays 1998, 20, 156–167. [Google Scholar] [CrossRef]
- Torrent, M.; Nogués, M.V.; Andreu, D.; Boix, E. The “CPC clip motif”: A conserved structural signature for heparin-binding proteins. PLoS ONE 2012, 7, e42692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rudd, T.R.; Preston, M.D.; Yates, E.A. The nature of the conserved basic amino acid sequences found among 437 heparin binding proteins determined by network analysis. Mol. Biosyst. 2017, 13, 852–865. [Google Scholar] [CrossRef] [Green Version]
- Jin, L.; Abrahams, J.P.; Skinner, R.; Petitou, M.; Pike, R.N.; Carrell, R.W. The anticoagulant activation of antithrombin by heparin. Proc. Natl. Acad. Sci. USA 1997, 94, 14683–14688. [Google Scholar] [CrossRef] [Green Version]
- Peterson, F.C.; Elgin, E.S.; Nelson, T.J.; Zhang, F.; Hoeger, T.J.; Linhardt, R.J.; Volkman, B.F. Identification and characterization of a glycosaminoglycan recognition element of the C chemokine lymphotactin. J. Biol. Chem. 2004, 279, 12598–12604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muñoz, E.M.; Linhardt, R.J. Heparin-binding domains in vascular biology. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 1549–1557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vanheule, V.; Janssens, R.; Boff, D.; Kitic, N.; Berghmans, N.; Ronsse, I.; Kungl, A.J.; Amaral, F.A.; Teixeira, M.M.; Van Damme, J.; et al. The Positively Charged COOH-terminal Glycosaminoglycan-binding CXCL9(74-103) Peptide Inhibits CXCL8-induced Neutrophil Extravasation and Monosodium Urate Crystal-induced Gout in Mice. J. Biol. Chem. 2015, 290, 21292–21304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vanheule, V.; Vervaeke, P.; Mortier, A.; Noppen, S.; Gouwy, M.; Snoeck, R.; Andrei, G.; Van Damme, J.; Liekens, S.; Proost, P. Basic chemokine-derived glycosaminoglycan binding peptides exert antiviral properties against dengue virus serotype 2, herpes simplex virus-1 and respiratory syncytial virus. Biochem. Pharmacol. 2016, 100, 73–85. [Google Scholar] [CrossRef]
- Vanheule, V.; Boff, D.; Mortier, A.; Janssens, R.; Petri, B.; Kolaczkowska, E.; Kubes, P.; Berghmans, N.; Struyf, S.; Kungl, A.J.; et al. CXCL9-derived peptides differentially inhibit neutrophil migration in vivo through interference with glycosaminoglycan interactions. Front. Immunol. 2017, 8, 530. [Google Scholar] [CrossRef] [Green Version]
- Vanheule, V.; Crijns, H.; Poosti, F.; Ruytinx, P.; Berghmans, N.; Gerlza, T.; Ronsse, I.; Pörtner, N.; Matthys, P.; Kungl, A.J.; et al. Anti-inflammatory effects of the GAG-binding CXCL9(74-103) peptide in dinitrofluorobenzene-induced contact hypersensitivity in mice. Clin. Exp. Allergy 2018, 48, 1333–1344. [Google Scholar] [CrossRef]
- Boff, D.; Crijns, H.; Janssens, R.; Vanheule, V.; Menezes, G.B.; Macari, S.; Silva, T.A.; Amaral, F.A.; Proost, P. The chemokine fragment CXCL9(74-103) diminishes neutrophil recruitment and joint inflammation in antigen-induced arthritis. J. Leukoc. Biol. 2018, 104, 413–422. [Google Scholar] [CrossRef]
- De Zutter, A.; Crijns, H.; Berghmans, N.; García-Caballero, M.; Vanbrabant, L.; Pörtner, N.; Vanheule, V.; Verscheure, P.; Siddiquei, M.M.; Abu El-Asrar, A.M.; et al. The Chemokine-Based Peptide, CXCL9(74-103), Inhibits Angiogenesis by Blocking Heparan Sulfate Proteoglycan-Mediated Signaling of Multiple Endothelial Growth Factors. Cancers 2021, 13, 5090. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://web.expasy.org/cellosaurus/CVCL_4590 (accessed on 18 October 2021).
- Esko, J.D.; Weinke, J.L.; Taylor, W.H.; Ekborg, G.; Rodén, L.; Anantharamaiah, G.; Gawish, A. Inhibition of chondroitin and heparan sulfate biosynthesis in Chinese hamster ovary cell mutants defective in galactosyltransferase I. J. Biol. Chem. 1987, 262, 12189–12195. [Google Scholar] [CrossRef]
- Almeida, R.; Levery, S.B.; Mandel, U.; Kresse, H.; Schwientek, T.; Bennett, E.P.; Clausen, H. Cloning and expression of a proteoglycan UDP-galactose:beta-xylose beta1,4-galactosyltransferase I. A seventh member of the human beta4-galactosyltransferase gene family. J. Biol. Chem. 1999, 274, 26165–26171. [Google Scholar] [CrossRef] [Green Version]
- Kufareva, I.; Salanga, C.L.; Handel, T.M. Chemokine and chemokine receptor structure and interactions: Implications for therapeutic strategies. Immunol. Cell. Biol. 2015, 93, 372–383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Proudfoot, A.E.I.; Johnson, Z.; Bonvin, P.; Handel, T.M. Glycosaminoglycan Interactions with Chemokines Add Complexity to a Complex System. Pharmaceuticals 2017, 10, 70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Middleton, J.; Patterson, A.M.; Gardner, L.; Schmutz, C.; Ashton, B.A. Leukocyte extravasation: Chemokine transport and presentation by the endothelium. Blood 2002, 100, 3853–3860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crijns, H.; Vanheule, V.; Proost, P. Targeting Chemokine-Glycosaminoglycan Interactions to Inhibit Inflammation. Front. Immunol. 2020, 11, 483. [Google Scholar] [CrossRef] [PubMed]
- Fromm, J.R.; Hileman, R.E.; Caldwell, E.E.; Weiler, J.M.; Linhardt, R.J. Differences in the interaction of heparin with arginine and lysine and the importance of these basic amino acids in the binding of heparin to acidic fibroblast growth factor. Arch. Biochem. Biophys. 1995, 323, 279–287. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Esko, J.D. Demystifying heparan sulfate-protein interactions. Annu. Rev. Biochem. 2014, 83, 129–157. [Google Scholar] [CrossRef] [PubMed]
- Verrecchio, A.; Germann, M.W.; Schick, B.P.; Kung, B.; Twardowski, T.; San Antonio, J.D. Design of peptides with high affinities for heparin and endothelial cell proteoglycans. J. Biol. Chem. 2000, 275, 7701–7707. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.H.; Narimatsu, Y.; Clausen, T.M.; Gomes, C.; Karlsson, R.; Steentoft, C.; Spliid, C.B.; Gustavsson, T.; Salanti, A.; Persson, A.; et al. The GAGOme: A cell-based library of displayed glycosaminoglycans. Nat. Methods 2018, 15, 881–888. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Li, L.; Tian, F.; Zhang, L.; Xue, C.; Linhardt, R.J. Glycosaminoglycanomics of cultured cells using a rapid and sensitive LC-MS/MS approach. ACS Chem. Biol. 2015, 10, 1303–1310. [Google Scholar] [CrossRef] [PubMed]
- Piva, M.B.; Suarez, E.R.; Melo, C.M.; Cavalheiro, R.P.; Nader, H.B.; Pinhal, M.A. Glycosaminoglycans affect heparanase location in CHO cell lines. Glycobiology 2015, 25, 976–983. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esko, J.D.; Selleck, S.B. Order out of chaos: Assembly of ligand binding sites in heparan sulfate. Annu. Rev. Biochem. 2002, 71, 435–471. [Google Scholar] [CrossRef] [PubMed]
- Scarcelli, J.J.; Hone, M.; Beal, K.; Ortega, A.; Figueroa, B.; Starkey, J.A.; Anderson, K. Analytical subcloning of a clonal cell line demonstrates cellular heterogeneity that does not impact process consistency or robustness. Biotechnol. Prog. 2018, 34, 602–612. [Google Scholar] [CrossRef] [PubMed]
- Rueda, P.; Balabanian, K.; Lagane, B.; Staropoli, I.; Chow, K.; Levoye, A.; Laguri, C.; Sadir, R.; Delaunay, T.; Izquierdo, E.; et al. The CXCL12gamma chemokine displays unprecedented structural and functional properties that make it a paradigm of chemoattractant proteins. PLoS ONE 2008, 3, e2543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sandhoff, R.; Grieshaber, H.; Djafarzadeh, R.; Sijmonsma, T.P.; Proudfoot, A.E.; Handel, T.M.; Wiegandt, H.; Nelson, P.J.; Gröne, H.J. Chemokines bind to sulfatides as revealed by surface plasmon resonance. Biochim. Biophys. Acta 2005, 1687, 52–63. [Google Scholar] [CrossRef] [PubMed]
- Jørgensen, A.S.; Brandum, E.P.; Mikkelsen, J.M.; Orfin, K.A.; Boilesen, D.R.; Egerod, K.L.; Moussouras, N.A.; Vilhardt, F.; Kalinski, P.; Basse, P.; et al. The C-terminal peptide of CCL21 drastically augments CCL21 activity through the dendritic cell lymph node homing receptor CCR7 by interaction with the receptor N-terminus. Cell. Mol. Life Sci. 2021, 78, 6963–6978. [Google Scholar] [CrossRef]
- Loos, T.; Mortier, A.; Proost, P. Chapter 1. Isolation, identification, and production of posttranslationally modified chemokines. Methods Enzymol. 2009, 461, 3–29. [Google Scholar] [CrossRef]
- Fryer, A.; Huang, Y.C.; Rao, G.; Jacoby, D.; Mancilla, E.; Whorton, R.; Piantadosi, C.A.; Kennedy, T.; Hoidal, J. Selective O-desulfation produces nonanticoagulant heparin that retains pharmacological activity in the lung. J. Pharmacol. Exp. Ther. 1997, 282, 208–219. [Google Scholar]
- Derler, R.; Kitic, N.; Gerlza, T.; Kungl, A.J. Isolation and characterization of heparan sulfate from human lung tissues. Molecules 2021, 26, 5512. [Google Scholar] [CrossRef]
- Hamel, D.J.; Sielaff, I.; Proudfoot, A.E.I.; Handel, T.M. Chapter 4. Interactions of chemokines with glycosaminoglycans. Methods Enzymol. 2009, 461, 71–102. [Google Scholar] [CrossRef]
- Goger, B.; Halden, Y.; Rek, A.; Mösl, R.; Pye, D.; Gallagher, J.; Kungl, A.J. Different affinities of glycosaminoglycan oligosaccharides for monomeric and dimeric interleukin-8: A model for chemokine regulation at inflammatory sites. Biochemistry 2002, 41, 1640–1646. [Google Scholar] [CrossRef]
- Lau, E.K.; Paavola, C.D.; Johnson, Z.; Gaudry, J.P.; Geretti, E.; Borlat, F.; Kungl, A.J.; Proudfoot, A.E.; Handel, T.M. Identification of the glycosaminoglycan binding site of the CC chemokine, MCP-1: Implications for structure and function in vivo. J. Biol. Chem. 2004, 279, 22294–22305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerlza, T.; Hecher, B.; Jeremic, D.; Fuchs, T.; Gschwandtner, M.; Falsone, A.; Gesslbauer, B.; Kungl, A.J. A combinatorial approach to biophysically characterise chemokine-glycan binding affinities for drug development. Molecules 2014, 19, 10618–10634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McNaughton, E.F.; Eustace, A.D.; King, S.; Sessions, R.B.; Kay, A.; Farris, M.; Broadbridge, R.; Kehoe, O.; Kungl, A.J.; Middleton, J. Novel anti-inflammatory peptides based on chemokine-glycosaminoglycan interactions reduce leukocyte migration and disease severity in a model of rheumatoid arthritis. J. Immunol. 2018, 200, 3201–3217. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Peptide Name | Amino Acid Sequence | Theoretical Average Mr | Experimental Mr |
|---|---|---|---|
| TAMRA 1-KKAK-2mer | * KKAKASASKKAKTT | 1859.2 | 1859.7 |
| TAMRA-KKAK-3mer | * KKAKASASKKAKASASKKAKTT | 2631.1 | 2631.8 |
| TAMRA-KKQK-2mer | * KKQKQSQSKKQKTT | 2087.4 | 2087.3 |
| TAMRA-KKQK-3mer | * KKQKQSQSKKQKQSQSKKQKTT | 3030.4 | 3030.9 |
| TAMRA-KKQKASAS-2mer | * KKQKASASKKQKTT | 1973.3 | 1973.1 |
| TAMRA-KKQKASAS-3mer | * KKQKASASKKQKASASKKQKTT | 2802.2 | 2802.4 |
| TAMRA-KKKQQK-2mer | * KKKQQKQSQSKKKQQKTT | 2600.0 | 2599.3 |
| TAMRA-CXCL9(74-103) | * KKKQKNGKKHQKKKVLKVRKSQRSRQKKTT | 4072.8 | 4072.9 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Crijns, H.; Adyns, L.; Ganseman, E.; Cambier, S.; Vandekerckhove, E.; Pörtner, N.; Vanbrabant, L.; Struyf, S.; Gerlza, T.; Kungl, A.; et al. Affinity and Specificity for Binding to Glycosaminoglycans Can Be Tuned by Adapting Peptide Length and Sequence. Int. J. Mol. Sci. 2022, 23, 447. https://doi.org/10.3390/ijms23010447
Crijns H, Adyns L, Ganseman E, Cambier S, Vandekerckhove E, Pörtner N, Vanbrabant L, Struyf S, Gerlza T, Kungl A, et al. Affinity and Specificity for Binding to Glycosaminoglycans Can Be Tuned by Adapting Peptide Length and Sequence. International Journal of Molecular Sciences. 2022; 23(1):447. https://doi.org/10.3390/ijms23010447
Chicago/Turabian StyleCrijns, Helena, Lowie Adyns, Eva Ganseman, Seppe Cambier, Eline Vandekerckhove, Noëmie Pörtner, Lotte Vanbrabant, Sofie Struyf, Tanja Gerlza, Andreas Kungl, and et al. 2022. "Affinity and Specificity for Binding to Glycosaminoglycans Can Be Tuned by Adapting Peptide Length and Sequence" International Journal of Molecular Sciences 23, no. 1: 447. https://doi.org/10.3390/ijms23010447
APA StyleCrijns, H., Adyns, L., Ganseman, E., Cambier, S., Vandekerckhove, E., Pörtner, N., Vanbrabant, L., Struyf, S., Gerlza, T., Kungl, A., & Proost, P. (2022). Affinity and Specificity for Binding to Glycosaminoglycans Can Be Tuned by Adapting Peptide Length and Sequence. International Journal of Molecular Sciences, 23(1), 447. https://doi.org/10.3390/ijms23010447