
Mechanisms of Viral Degradation of Cellular Signal Transducer and Activator of Transcription 2

Abstract
:1. Introduction
2. STAT2 Degradation by the Ubiquitin–Proteasome System (UPS)
2.1. Basic Features of the Ubiquitin–Proteasome System
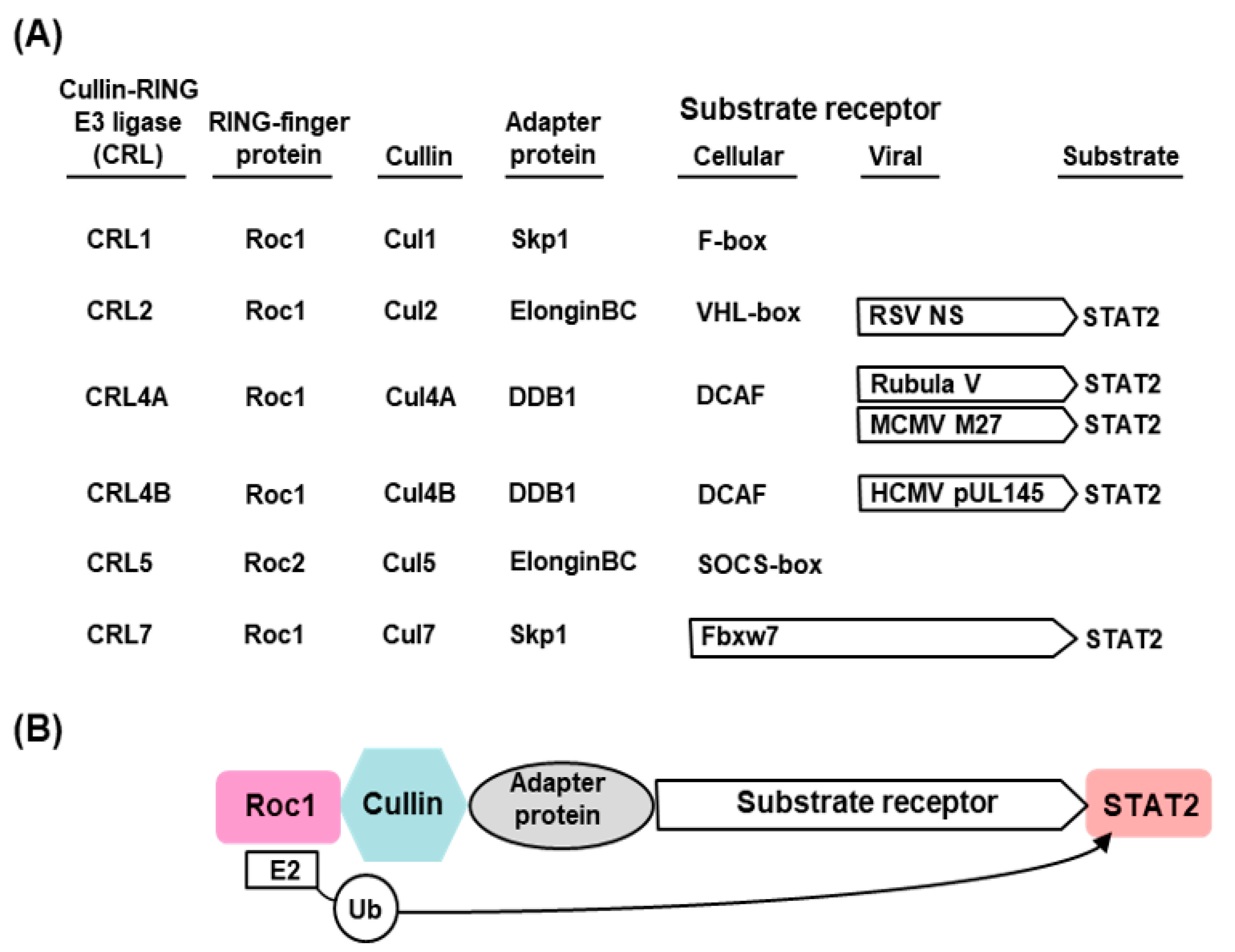
2.2. The E3 Ligases: Assembly and Function
2.3. Viruses and Viral Proteins That Promote STAT2 Degradation by the UPS
2.3.1. Flaviviridae
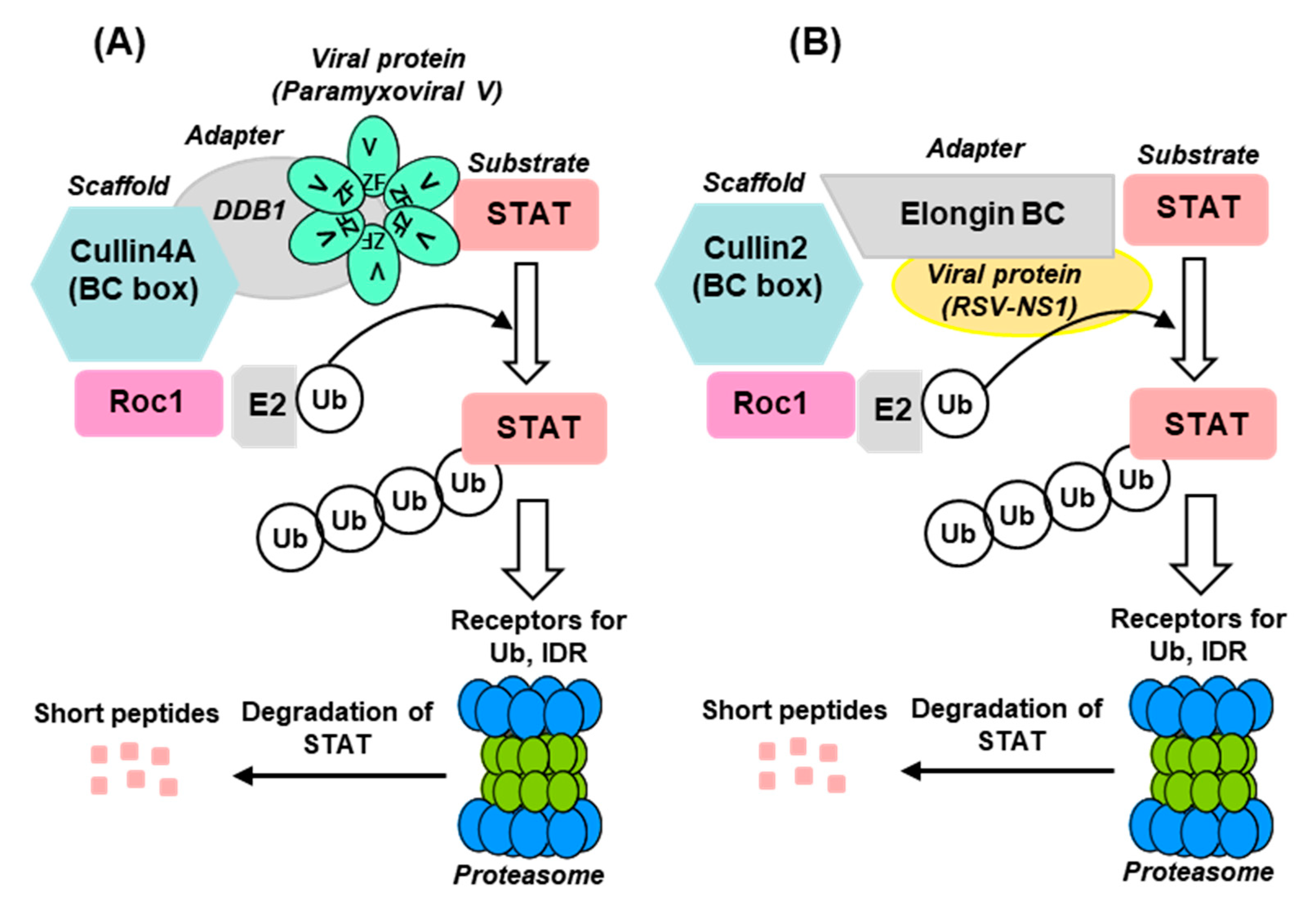
2.3.2. Paramyxoviridae
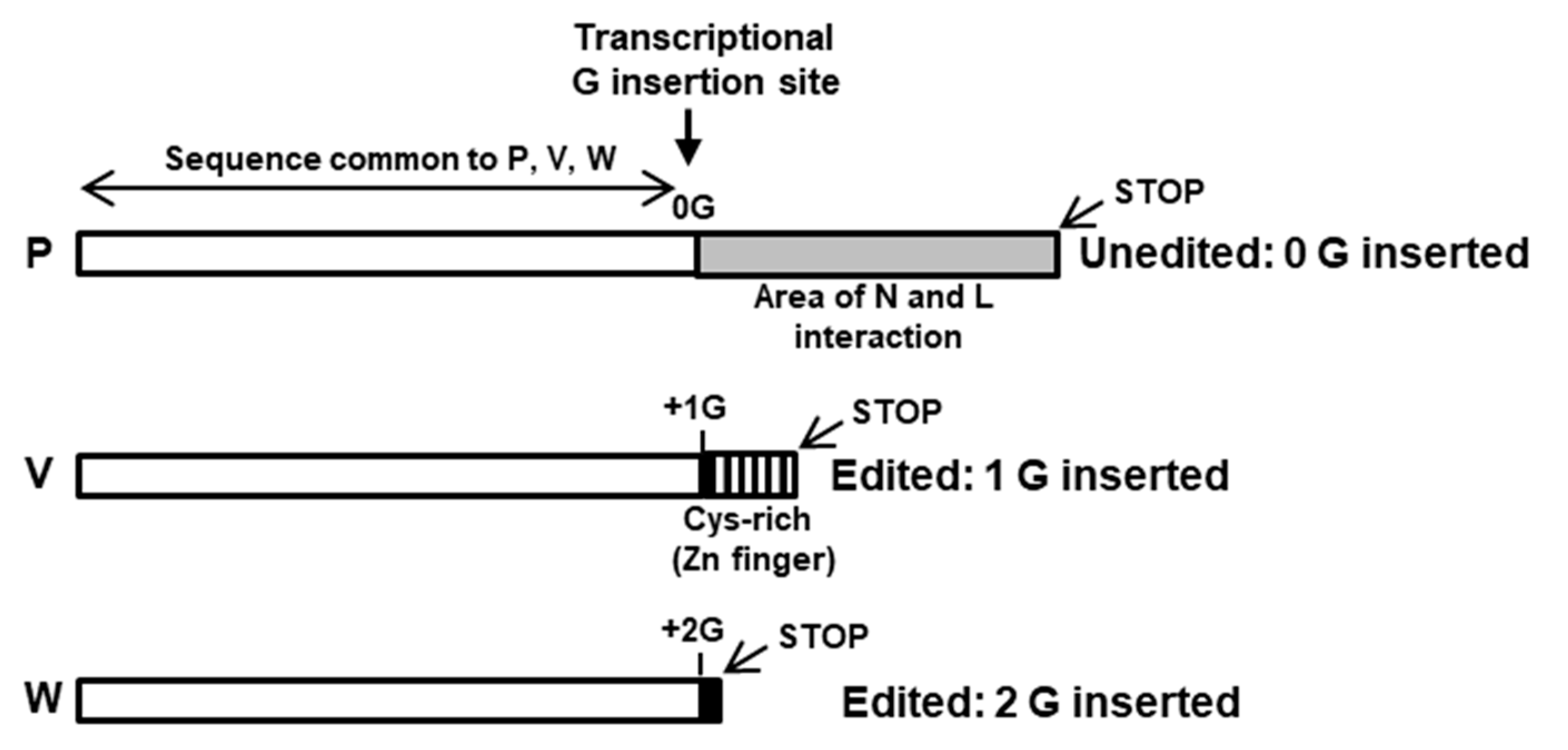
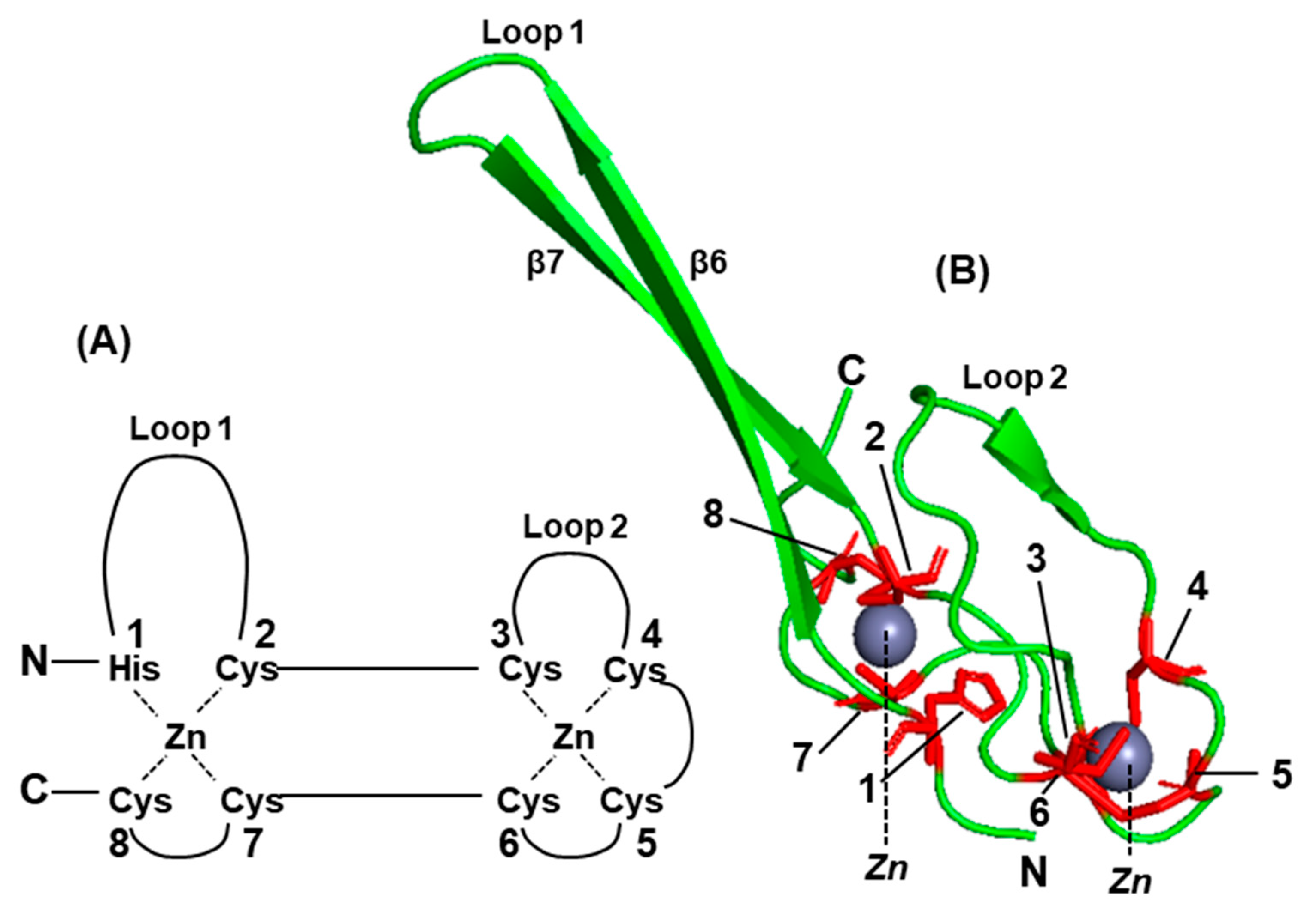
Transcriptional RNA Editing of the Viral P Gene Produces Multiple Immune Suppressor Proteins
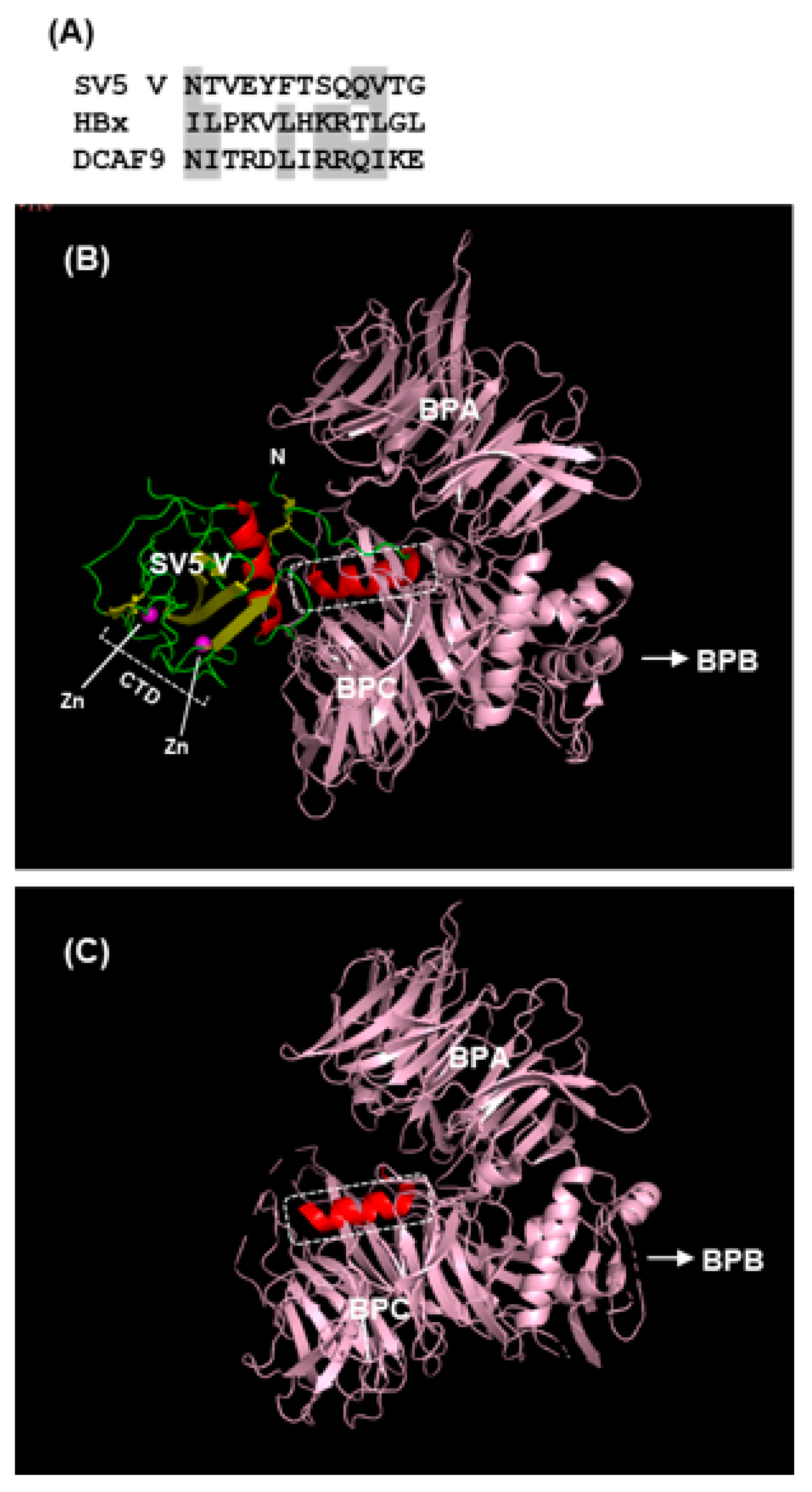
The Nature of the V-Dependent E3 Ligase Complex and Its Role in STAT Regulation
2.3.3. Pneumoviridae
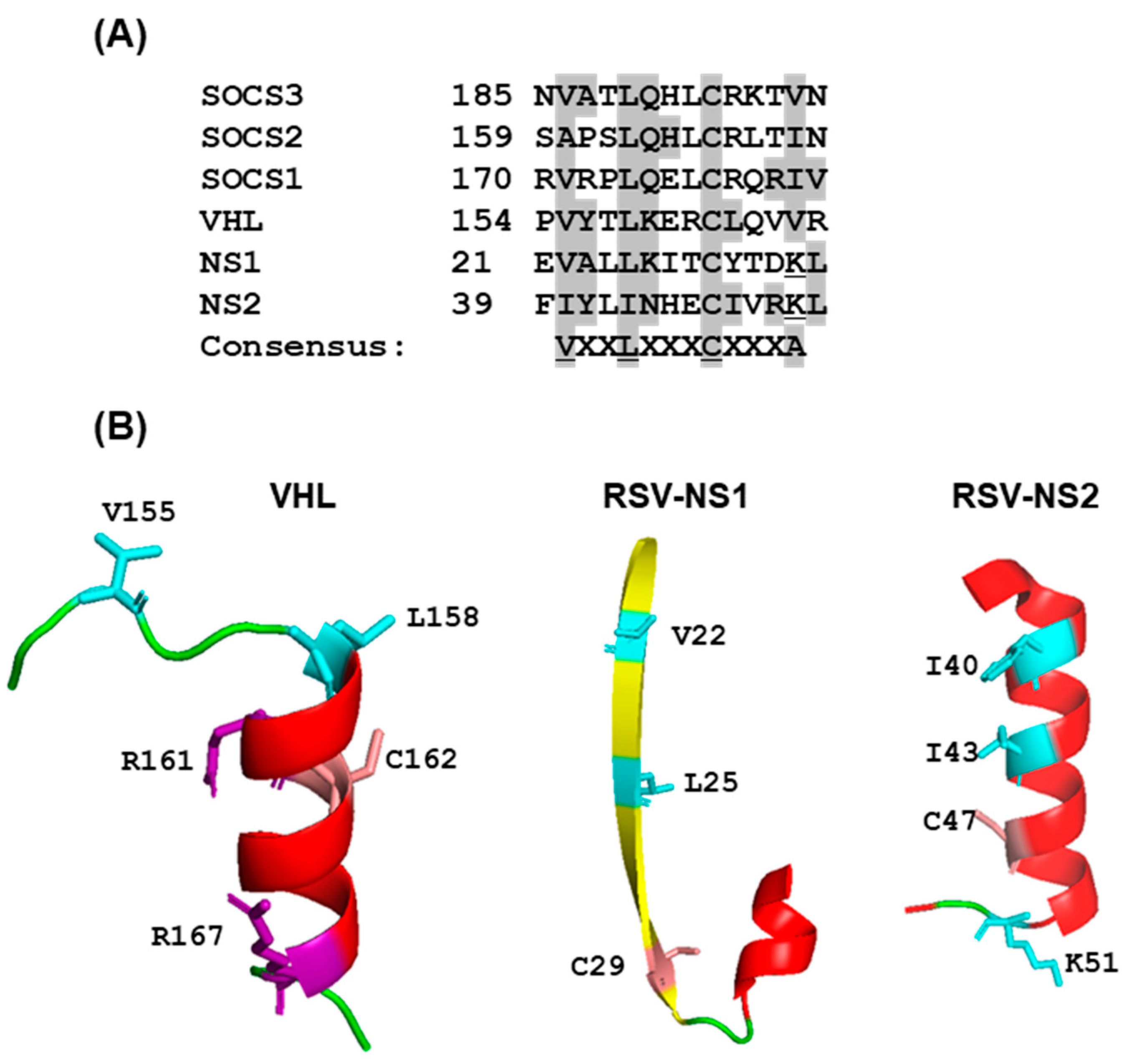
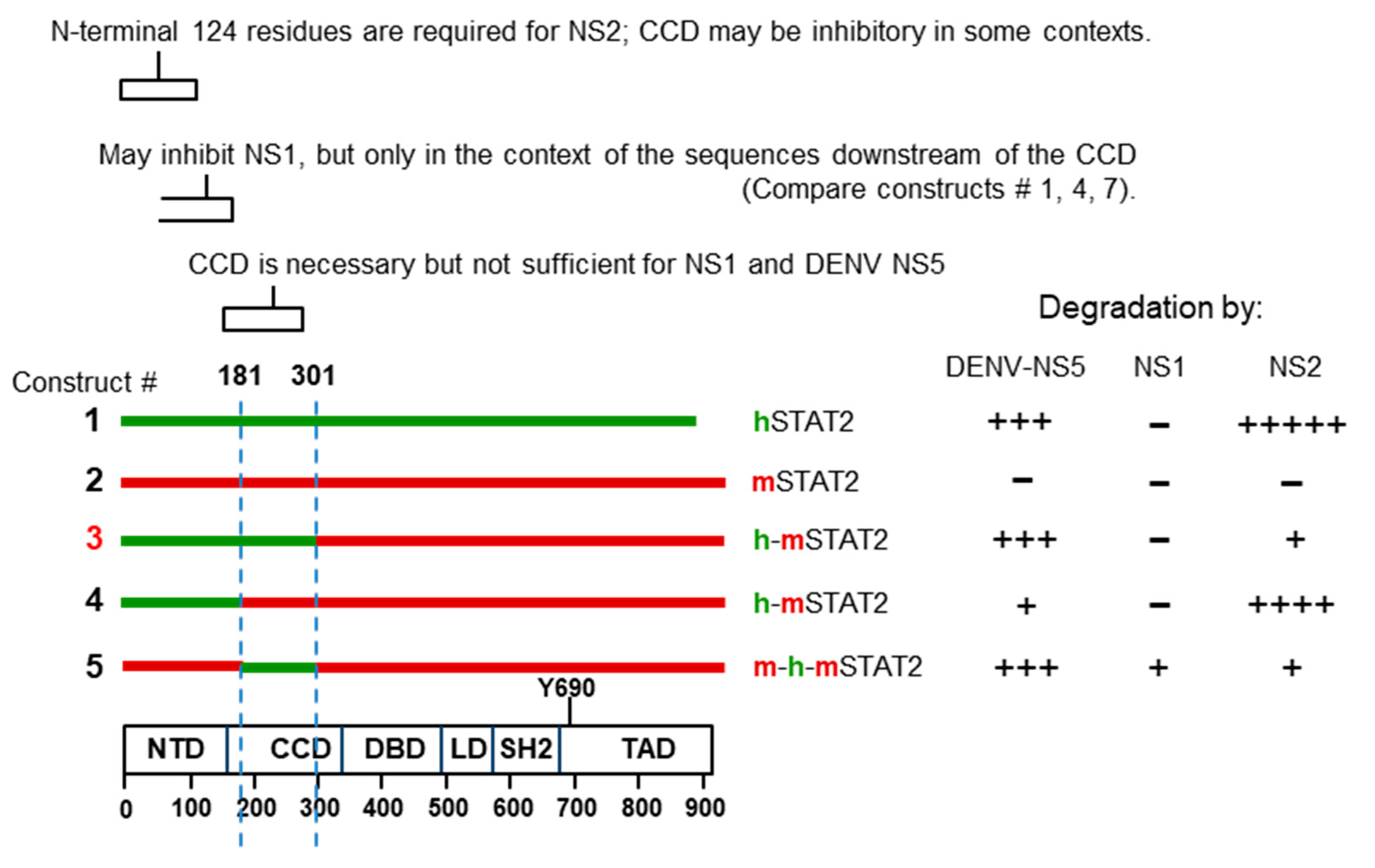
Viral Nonstructural Proteins, NS1 and NS2
Search for NS-Interacting Domains of STAT Proteins
2.3.4. Arteriviridae
2.3.5. Herpesviridae
3. Efficiency of Viral Suppression of IFN
4. Summary and Directions for Future Research
4.1. Diversity, Multiplicity, and Specificity
4.2. Directions for Future Research
Supplementary Materials
Funding
Acknowledgments
Conflicts of Interest
References
- Steen, H.C.; Gamero, A.M. STAT2 phosphorylation and signaling. Jak-Stat 2013, 2, e25790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Majoros, A.; Platanitis, E.; Kernbauer-Hölzl, E.; Rosebrock, F.; Müller, M.; Decker, T. Canonical and non-canonical aspects of JAK-STAT signaling: Lessons from interferons for cytokine responses. Front. Immunol. 2017, 8, 29. [Google Scholar] [CrossRef] [Green Version]
- Nan, Y.; Wu, C.; Zhang, Y.J. Signaling activated by type I interferons and viral antagonism. Front. Immunol. 2017, 8, 1758. [Google Scholar] [CrossRef] [PubMed]
- Fu, X.Y.; Kessler, D.S.; Veals, S.A.; Levy, D.E.; Darnell, J.E. ISGF3, the transcriptional activator induced by interferon alpha, consists of multiple interacting polypeptide chains. Proc. Natl. Acad. Sci. USA 1990, 87, 8555–8559. [Google Scholar] [CrossRef] [Green Version]
- Müller, M.; Laxton, C.; Briscoe, J.; Schindler, C.; Improta, T.; Darnell, J.E., Jr.; Stark, G.R.; Kerr, I.M. Complementation of a mutant cell line: Central role of the 91 kDa polypeptide of ISGF3 in the interferon-alpha and -gamma signal transduction pathways. EMBO J. 1993, 12, 4221–4228. [Google Scholar] [CrossRef]
- Ivashkiv, L.B.; Donlin, L.T. Regulation of type I interferon responses. Nat. Rev. Immunol. 2014, 14, 36–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kessler, D.S.; Veals, S.A.; Fu, X.Y.; Levy, D.E. Interferon-α regulates nuclear translocation and DNA-binding affinity of ISGF3, a multimeric transcriptional activator. Genes Dev. 1990, 4, 1753–1765. [Google Scholar] [CrossRef] [Green Version]
- Sadler, A.J.; Williams, B.R. Interferon-inducible antiviral effectors. Nat. Rev. Immunol. 2008, 8, 559–568. [Google Scholar] [CrossRef] [PubMed]
- Schoggins, J.W. Interferon-stimulated genes: Roles in viral pathogenesis. Curr. Opin. Virol. 2014, 6, 40–46. [Google Scholar] [CrossRef] [PubMed]
- Rabbani, M.A.; Ribaudo, M.; Guo, J.T.; Barik, S. Identification of interferon-stimulated gene proteins that inhibit human parainfluenza virus type 3. J. Virol. 2016, 90, 11145–11156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chowdhury, F.Z.; Farrar, J.D. STAT2: A shape-shifting anti-viral super STAT. Jak-Stat 2013, 2, e23633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reich, N.C. STAT dynamics. Cytokine Growth Factor Rev. 2007, 18, 511–518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Levy, D.E. Comparative evolutionary genomics of the STAT family of transcription factors. Jak-Stat 2012, 1, 23–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boudinot, P.; Bird, S.; Du Pasquier, L.; Collet, B. The repertoire of vertebrate STAT transcription factors: Origin and variations in fish. Dev. Comp. Immunol. 2021, 116, 103929. [Google Scholar] [CrossRef] [PubMed]
- Baris, S.; Alroqi, F.; Kiykim, A.; Karakoc-Aydiner, E.; Ogulur, I.; Ozen, A.; Charbonnier, L.M.; Bakır, M.; Boztug, K.; Chatila, T.A.; et al. Severe early-onset combined immunodeficiency due to heterozygous gain-of-function mutations in STAT1. J. Clin. Immunol. 2016, 36, 641–648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blaszczyk, K.; Olejnik, A.; Nowicka, H.; Ozgyin, L.; Chen, Y.L.; Chmielewski, S.; Kostyrko, K.; Wesoly, J.; Balint, B.L.; Lee, C.K.; et al. STAT2/IRF9 directs a prolonged ISGF3-like transcriptional response and antiviral activity in the absence of STAT1. Biochem. J. 2015, 466, 511–524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.; Yin, Y.; Xu, L.; Su, J.; Huang, F.; Wang, Y.; Boor, P.P.C.; Chen, K.; Wang, W.; Cao, W.; et al. Unphosphorylated ISGF3 drives constitutive expression of interferon-stimulated genes to protect against viral infections. Sci. Signal. 2017, 10, eaah4248. [Google Scholar] [CrossRef]
- Platanitis, E.; Demiroz, D.; Schneller, A.; Fischer, K.; Capelle, C.; Hartl, M.; Gossenreiter, T.; Müller, M.; Novatchkova, M.; Decker, T. A molecular switch from STAT2-IRF9 to ISGF3 underlies interferon-induced gene transcription. Nat. Commun. 2019, 10, 2921. [Google Scholar] [CrossRef] [PubMed]
- Sung, P.S.; Cheon, H.; Cho, C.H.; Hong, S.-H.; Park, D.Y.; Seo, H.-E.; Park, S.-H.; Yoon, S.K.; Stark, G.R.; Shin, E.-C. Roles of unphosphorylated ISGF3 in HCV infection and interferon responsiveness. Proc. Natl. Acad. Sci. USA 2015, 112, 10443––10448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheon, H.J.; Holvey-Bates, E.G.; Schoggins, J.W.; Forster, S.; Hertzog, P.; Imanaka, N.; Rice, C.M.; Jackson, M.W.; Junk, D.J.; Stark, G.R. IFNβ-dependent increases in STAT1, STAT2, and IRF9 mediate resistance to viruses and DNA damage. EMBO J. 2013, 32, 2751–2763. [Google Scholar] [CrossRef] [Green Version]
- Hahm, B.; Trifilo, M.J.; Zuniga, E.I.; Oldstone, M.B. Viruses evade the immune system through type I interferon-mediated STAT2-dependent, but STAT1-independent, signaling. Immunity 2005, 22, 247–257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brierley, M.M.; Marchington, K.L.; Jurisica, I.; Fish, E.N. Identification of GAS-dependent interferon-sensitive target genes whose transcription is STAT2-dependent but ISGF3-independent. FEBS J. 2006, 273, 1569–1581. [Google Scholar] [CrossRef]
- Matsumoto, M.; Tanaka, N.; Harada, H.; Kimura, T.; Yokochi, T.; Kitagawa, M.; Schindler, C.; Taniguchi, T. Activation of the transcription factor ISGF3 by interferon-gamma. Biol. Chem. 1999, 380, 699–703. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, A.; Trilling, M.; Wagner, M.; Wilborn, M.; Bubic, I.; Jonjic, S.; Koszinowski, U.; Hengel, H. A cytomegaloviral protein reveals a dual role for STAT2 in IFN-γ signaling and antiviral responses. J. Exp. Med. 2005, 201, 1543–1553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trilling, M.; Le, V.T.K.; Fiedler, M.; Zimmermann, A.; Bleifuss, E.; Hengel, H. Identification of DNA-damage DNA-binding protein 1 as a conditional essential factor for cytomegalovirus replication in interferon-γ-stimulated cells. PLoS Pathog. 2011, 7, e1002069. [Google Scholar] [CrossRef] [Green Version]
- Park, C.; Li, S.; Cha, E.; Schindler, C. Immune response in Stat2 knockout mice. Immunity 2000, 13, 795–804. [Google Scholar] [CrossRef] [Green Version]
- Hickerson, B.T.; Westover, J.B.; Wettere, A.J.V.; Rigas, J.D.; Miao, J.; Conrad, B.L.; Motter, N.E.; Wang, Z.; Gowen, B.B. Pathogenesis of Rift Valley fever virus aerosol infection in STAT2 knockout hamsters. Viruses 2018, 10, 651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ranadheera, C.; Valcourt, E.J.; Warner, B.M.; Poliquin, G.; Rosenke, K.; Frost, K.; Tierney, K.; Saturday, G.; Miao, J.; Westover, J.B.; et al. Characterization of a novel STAT2 knock-out hamster model of Crimean-Congo hemorrhagic fever virus pathogenesis. Sci. Rep. 2020, 10, 12378. [Google Scholar] [CrossRef] [PubMed]
- Boudewijns, R.; Thibaut, H.J.; Kaptein, S.J.F.; Li, R.; Vergote, V.; Seldeslachts, L.; Weyenbergh, J.V.; Keyzer, C.D.; Bervoets, L.; Sharma, S.; et al. STAT2 signaling restricts viral dissemination but drives severe pneumonia in SARS-CoV-2 infected hamsters. Nat. Commun. 2020, 11, 5838. [Google Scholar] [CrossRef]
- Freij, B.J.; Hanrath, A.T.; Chen, R.; Hambleton, S.; Duncan, C.J.A. Life-threatening influenza, hemophagocytic lymphohistiocytosis and probable vaccine-strain varicella in a novel case of homozygous STAT2 deficiency. Front. Immunol. 2021, 11, 624415. [Google Scholar] [CrossRef] [PubMed]
- Duncan, C.J.A.; Hambleton, S. Human disease phenotypes associated with loss and gain of function mutations in STAT2: Viral susceptibility and type I interferonopathy. J. Clin. Immunol. 2021, 41, 1446–1456. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.-K.; Bluyssen, H.A.R.; Levy, D.E. Regulation of interferon-α responsiveness by the duration of Janus kinase activity. J. Biol. Chem. 1997, 272, 21872–21877. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Didcock, L.; Young, D.F.; Goodbourn, S.; Randall, R.E. The V protein of simian virus 5 inhibits interferon signalling by targeting STAT1 for proteasome-mediated degradation. J. Virol. 1999, 73, 9928–9933. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parisien, J.P.; Lau, J.F.; Rodriguez, J.J.; Ulane, C.M.; Horvath, C.M. Selective STAT protein degradation induced by paramyxoviruses requires both STAT1 and STAT2 but is independent of alpha/beta interferon signal transduction. J. Virol. 2002, 76, 4190–4198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bochtler, M.; Ditzel, L.; Groll, M.; Hartmann, C.; Huber, R. The proteasome. Annu. Rev. Biophys. Biomol. Str. 1999, 28, 295–317. [Google Scholar] [CrossRef]
- Voges, D.; Zwickl, P.; Baumeister, W. The 26S proteasome: A molecular machine designed for controlled proteolysis. Annu. Rev. Biochem. 1999, 68, 1015–1068. [Google Scholar] [CrossRef]
- Wang, J.; Maldonado, M.A. The ubiquitin-proteasome system and its role in inflammatory and autoimmune diseases. Cell. Mol. Immunol. 2006, 3, 255–261. [Google Scholar]
- O’Connor, H.F.; Huibregtse, J.M. Enzyme-substrate relationships in the ubiquitin system: Approaches for identifying substrates of ubiquitin ligases. Cell. Mol. Life Sci. 2017, 74, 3363–3375. [Google Scholar] [CrossRef] [PubMed]
- Fenteany, G.; Standaert, R.F.; Lane, W.S.; Choi, S.; Corey, E.J.; Schreiber, S.L. Inhibition of proteasome activities and subunit-specific amino-terminal threonine modification by lactacystin. Science 1995, 268, 726–731. [Google Scholar] [CrossRef]
- Lee, D.H.; Goldberg, A.L. Proteasome inhibitors: Valuable new tools for cell biologists. Trends Cell. Biol. 1998, 8, 397–403. [Google Scholar] [CrossRef]
- Buetow, L.; Huang, D.T. Structural insights into the catalysis and regulation of E3 ubiquitin ligases. Nat. Rev. Mol. Cell. Biol. 2016, 17, 626–642. [Google Scholar] [CrossRef] [Green Version]
- Bhowmick, P.; Pancsa, R.; Guharoy, M.; Tompa, P. Functional diversity and structural disorder in the human ubiquitination pathway. PLoS ONE 2013, 8, e1725. [Google Scholar] [CrossRef] [Green Version]
- Stewart, M.D.; Ritterhoff, T.; Klevit, R.E.; Brzovic, P.S. E2 enzymes: More than just middle men. Cell Res. 2016, 26, 423–440. [Google Scholar] [CrossRef] [Green Version]
- Bhattacharyya, S.; Yu, H.; Mim, C.; Matouschek, A. Regulated protein turnover: Snapshots of the proteasome in action. Nat. Rev. Mol. Cell Biol. 2014, 15, 122–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, H.; Matouschek, A. Recognition of client proteins by the proteasome. Annu. Rev. Biophys. 2017, 46, 149–173. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Zhang, N.-Y.; Zurawel, A.; Hansen, K.C.; Liu, C.-W. Degradation of some polyubiquitinated proteins requires an intrinsic proteasomal binding element in the substrates. J. Biol. Chem. 2010, 285, 4771–4780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erales, J.; Coffino, P. Ubiquitin-independent proteasomal degradation. Biochim. Biophys. Acta. 2014, 1843, 216–221. [Google Scholar] [CrossRef] [Green Version]
- Laney, J.D.; Hochstrasser, M. Substrate targeting in the ubiquitin system. Cell 1999, 97, 427–430. [Google Scholar] [CrossRef] [Green Version]
- Jin, J.; Cardozo, T.; Lovering, R.C.; Elledge, S.J.; Pagano, M.; Harper, J.W. Systematic analysis and nomenclature of mammalian F-box proteins. Genes Dev. 2004, 18, 2573–2580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamura, T.; Sato, S.; Haque, D.; Liu, L.; Kaelin, W.G., Jr.; Conaway, R.C.; Conaway, J.W. The Elongin BC complex interacts with the conserved SOCS-box motif present in members of the SOCS, ras, WD-40 repeat, and ankyrin repeat families. Genes Dev. 1998, 12, 3872–3881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stebbins, C.E.; Kaelin, W.G., Jr.; Pavletich, N.P. Structure of the VHL-ElonginC-ElonginB complex: Implications for VHL tumor suppressor function. Science 1999, 284, 455–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Angers, S.; Li, T.; Yi, X.; MacCoss, M.J.; Moon, R.T.; Zheng, N. Molecular architecture and assembly of the DDB1-CUL4A ubiquitin ligase machinery. Nature 2006, 443, 590–593. [Google Scholar] [CrossRef]
- Higa, L.A.; Wu, M.; Ye, T.; Kobayashi, R.; Sun, H.; Zhang, H. CUL4-DDB1 ubiquitin ligase interacts with multiple WD40-repeat proteins and regulates histone methylation. Nat. Cell. Biol. 2006, 8, 1277–1283. [Google Scholar] [CrossRef] [PubMed]
- Chu, G.; Chang, E. Xeroderma pigmentosum group E cells lack a nuclear factor that binds to damaged DNA. Science 1988, 242, 564–567. [Google Scholar] [CrossRef] [PubMed]
- Bullock, A.N.; Debreczeni, J.W.; Edwards, A.M.; Sundström, M.; Knapp, S. Crystal structure of the SOCS2-elongin C-elongin B complex defines a prototypical SOCS box ubiquitin ligase. Proc. Natl. Acad. Sci. USA 2006, 103, 7637–7642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elliott, J.; Lynch, O.T.; Suessmuth, Y.; Qian, P.; Boyd, C.R.; Burrows, J.F.; Buick, R.; Stevenson, N.J.; Touzelet, O.; Gadina, M.; et al. Respiratory syncytial virus NS1 protein degrades STAT2 by using the Elongin-Cullin E3 ligase. J. Virol. 2007, 81, 3428–3436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Becker, T.; Le-Trilling, V.T.K.; Trilling, M. Cellular cullin RING ubiquitin ligases: Druggable host dependency factors of cytomegaloviruses. Int. J. Mol. Sci. 2019, 20, 1636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, K.; Deshaies, R.J.; Liu, X. Assembly and regulation of CRL ubiquitin ligases. Adv. Exp. Med. Biol. 2020, 1217, 33–46. [Google Scholar] [CrossRef] [PubMed]
- Sarikas, A.; Hartmann, T.; Pan, Z.-Q. The cullin protein family. Genome Biol. 2011, 12, 220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kadeppagari, R.-K.; Sanchez, R.L.; Foster, T.P. HSV-2 inhibits type-I interferon signaling via multiple complementary and compensatory STAT2-associated mechanisms. Virus Res. 2012, 167, 273–284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, M.; Fu, M.; Li, M.; Hu, H.; Gong, S.; Hu, Q. Herpes simplex virus type 2 inhibits type I IFN signaling mediated by the novel E3 ubiquitin protein ligase activity of viral protein ICP22. J. Immunol. 2020, 205, 1281–1292. [Google Scholar] [CrossRef] [PubMed]
- Ulane, C.M.; Horvath, C.M. Paramyxoviruses SV5 and HPIV2 assemble STAT protein ubiquitin ligase complexes from cellular components. Virology 2002, 304, 160–166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mackenzie, J.S.; Gubler, D.J.; Petersen, L.R. Emerging flaviviruses: The spread and resurgence of Japanese encephalitis, West Nile and dengue viruses. Nat. Med. 2004, 10, S98–S109. [Google Scholar] [CrossRef]
- Pierson, T.C.; Diamond, T.C. The continued threat of emerging flaviviruses. Nat. Microbiol. 2020, 5, 796–812. [Google Scholar] [CrossRef]
- Mukhopadhyay, S.; Kuhn, R.J.; Rossmann, M.G. A structural perspective of the flavivirus life cycle. Nat. Rev. Microbiol. 2005, 3, 13–22. [Google Scholar] [CrossRef]
- Bhatnagar, P.; Sreekanth, G.P.; Murali-Krishna, K.; Chandele, A.; Sitaraman, R. Dengue virus non-structural protein 5 as a versatile, multi-functional effector in host–pathogen interactions. Front. Cell. Infect. Microbiol. 2021, 11, 574067. [Google Scholar] [CrossRef] [PubMed]
- Thurmond, S.; Wang, B.; Song, J.; Hai, R. Suppression of type I interferon signaling by flavivirus NS5. Viruses 2018, 10, 712. [Google Scholar] [CrossRef] [Green Version]
- Ashour, J.; Laurent-Rolle, M.; Shi, P.Y.; Garcia-Sastre, A. NS5 of dengue virus mediates STAT2 binding and degradation. J. Virol. 2009, 83, 5408–5418. [Google Scholar] [CrossRef] [Green Version]
- Mazzon, M.; Jones, M.; Davidson, A.; Chain, B.; Jacobs, M. Dengue virus NS5 inhibits interferon-alpha signaling by blocking signal transducer and activator of transcription 2 phosphorylation. J. Infect. Dis. 2009, 200, 1261–1270. [Google Scholar] [CrossRef] [Green Version]
- Perry, S.T.; Buck, M.D.; Lada, S.M.; Schindler, C.; Shresta, S. STAT2 mediates innate immunity to Dengue virus in the absence of STAT1 via the type I interferon receptor. PLoS Pathog. 2011, 7, e1001297. [Google Scholar] [CrossRef] [Green Version]
- Castillo Ramirez, J.A.; Urcuqui-Inchima, S. Dengue virus control of type I IFN responses: A history of manipulation and control. J. Interferon Cytokine Res. 2015, 35, 421–430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, A.; Hou, S.; Airo, A.M.; Limonta, D.; Mancinelli, V.; Branton, W.; Power, C.; Hobman, T.C. Zika virus inhibits type-I interferon production and downstream signaling. EMBO Rep. 2016, 17, 1766–1775. [Google Scholar] [CrossRef] [Green Version]
- Grant, A.; Ponia, S.S.; Tripathi, S.; Balasubramaniam, V.; Miorin, L.; Sourisseau, M.; Schwarz, M.C.; Sánchez-Seco, M.P.; Evans, M.J.; Best, S.M.; et al. Zika virus targets human STAT2 to inhibit type I interferon signaling. Cell Host Microbe 2016, 19, 882–890. [Google Scholar] [CrossRef] [Green Version]
- Best, S. The many faces of the flavivirus NS5 protein in antagonism of type I interferon signaling. J. Virol. 2017, 91, e01970-e16. [Google Scholar] [CrossRef] [Green Version]
- Wang, B.; Thurmond, S.; Zhou, K.; Sánchez-Aparicio, M.T.; Fang, J.; Lu, J.; Gao, L.; Ren, W.; Cui, Y.; Veit, E.C.; et al. Structural basis for STAT2 suppression by flavivirus NS5. Nat. Struct. Mol. Biol. 2020, 27, 875–885. [Google Scholar] [CrossRef]
- Muñoz-Jordán, J.L.; Sánchez-Burgos, G.G.; Laurent-Rolle, M.; García-Sastre, A. Inhibition of interferon signaling by dengue virus. Proc. Natl. Acad. Sci. USA 2003, 100, 14333–14338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ashour, J.; Morrison, J.; Laurent-Rolle, M.; Belicha-Villanueva, A.; Plumlee, C.R.; Bernal-Rubio, D.; Williams, K.L.; Harris, E.; Fernandez-Sesma, A.; Schindler, C.; et al. Mouse STAT2 restricts early dengue virus replication. Cell Host Microbe 2010, 8, 410–421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Landau, L.J.B.; Sampaio de Oliveira Fam, B.; Yépez, Y.; Caldas-Garcia, G.B.; Pissinatti, A.; Falótico, T.; Reales, G.; Schüler-Faccini, L.; Sortica, V.A.; Bortolini, M.C. Evolutionary analysis of the anti-viral STAT2 gene of primates and rodents: Signature of different stages of an arms race. Infect. Genet. Evol. 2021, 95, 105030. [Google Scholar] [CrossRef] [PubMed]
- Rogers, M.C.; Miranda-Katz, M.; Zhang, Y.; Oury, T.D.; Uccellini, M.B.; García-Sastre, A.; Williams, J.V. STAT2 limits host species specificity of human metapneumovirus. Viruses 2020, 12, 724. [Google Scholar] [CrossRef]
- Miorin, L.; Laurent-Rolle, M.; Pisanelli, G.; Hendrick Co, P.; Albrecht, R.A.; García-Sastre, A.; Morrison, J. Host-specific NS5 ubiquitination determines yellow fever virus tropism. J. Virol. 2019, 93, e00151-e19. [Google Scholar] [CrossRef] [Green Version]
- Laurent-Rolle, M.; Morrison, J. The role of NS5 protein in determination of host cell range for yellow fever virus. DNA Cell Biol. 2019, 38, 1414–1417. [Google Scholar] [CrossRef]
- Park, C.; Lecomte, M.J.; Schindler, C. Murine Stat2 is uncharacteristically divergent. Nucleic Acids Res. 1999, 27, 4191–4199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morrison, J.; Laurent-Rolle, M.; Maestre, A.M.; Rajsbaum, R.; Pisanelli, G.; Simon, V.; Mulder, L.C.F.; Fernandez-Sesma, A.; García-Sastre, A. Dengue virus co-opts UBR4 to degrade STAT2 and antagonize type I interferon signaling. PLoS Pathog. 2013, 9, e1003265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tasaki, T.; Mulder, L.C.F.; Iwamatsu, A.; Lee, M.J.; Davydov, I.V.; Varshavsky, A.; Muesing, M.; Kwon, Y.T. A family of mammalian E3 ubiquitin ligases that contain the UBR box motif and recognize N-degrons. Mol. Cell. Biol. 2005, 25, 7120–7136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aslam, B.; Ahmad, J.; Ali, A.; Paracha, R.Z.; Tareen, S.H.K.; Khusro, S.; Ahmad, T.; Muhammad, S.A.; Niazi, U.; Azevedo, V. Structural modeling and analysis of dengue-mediated inhibition of interferon signaling pathway. Genet. Mol. Res. 2015, 14, 4215––4237. [Google Scholar] [CrossRef]
- Parisien, J.-P.; Lenoir, J.J.; Alvarado, G.; Horvath, C.M. The human STAT2 coiled-coil domain contains a degron for Zika virus interferon evasion. J. Virol. 2021, JVI0130121. [Google Scholar] [CrossRef]
- Fleming, S.B. Viral inhibition of the IFN-induced JAK/STAT signalling pathway: Development of live attenuated vaccines by mutation of viral-encoded IFN-antagonists. Vaccines 2016, 4, 23. [Google Scholar] [CrossRef] [Green Version]
- Laurent-Rolle, M.; Boer, E.F.; Lubick, K.J.; Wolfinbarger, J.B.; Carmody, A.B.; Rockx, B.; Liu, W.; Ashour, J.; Shupert, W.L.; Holbrook, M.R.; et al. The NS5 protein of the virulent West Nile virus NY99 strain is a potent antagonist of type I interferon-mediated JAK-STAT signaling. J. Virol. 2010, 84, 3503–3515. [Google Scholar] [CrossRef] [Green Version]
- Fanunza, E.; Carletti, F.; Quartu, M.; Grandi, N.; Ermellino, L.; Milia, J.; Corona, A.; Capobianchi, M.R.; Ippolito, G.; Tramontano, E. Zika virus NS2A inhibits interferon signaling by degradation of STAT1 and STAT2. Virulence 2021, 12, 1580–1596. [Google Scholar] [CrossRef]
- Whelan, S.P.; Barr, J.N.; Wertz, G.W. Transcription and replication of nonsegmented negative-strand RNA viruses. Curr. Top. Microbiol. Immunol. 2004, 283, 61–119. [Google Scholar] [CrossRef]
- Douglas, J.; Drummond, A.J.; Kingston, R.L. Evolutionary history of cotranscriptional editing in the paramyxoviral phosphoprotein gene. Virus Evol. 2021, 7, veab028. [Google Scholar] [CrossRef]
- Thomas, S.M.; Lamb, R.A.; Paterson, R.G. Two mRNAs that differ by two nontemplated nucleotides encode the amino coterminal proteins P and V of the paramyxovirus SV5. Cell 1988, 54, 891–902. [Google Scholar] [CrossRef]
- Paterson, R.G.; Lamb, R.A. RNA editing by G-nucleotide insertion in mumps virus P-gene mRNA transcripts. J. Virol. 1990, 64, 4137–4145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vidal, S.; Curran, J.; Kolakofsky, D. Editing of the Sendai virus P/C mRNA by G insertion occurs during mRNA synthesis via a virus-encoded activity. J. Virol. 1990, 64, 239–246. [Google Scholar] [CrossRef] [Green Version]
- Vidal, S.; Curran, J.; Kolakofsky, D. A stuttering model for paramyxovirus P mRNA editing. EMBO J. 1990, 9, 2017–2022. [Google Scholar] [CrossRef]
- Jacques, J.P.; Hausmann, S.; Kolakofsky, D. Paramyxovirus mRNA editing leads to G deletions as well as insertions. EMBO J. 1994, 13, 5496–5503. [Google Scholar] [CrossRef] [PubMed]
- Chenik, M.; Chebli, K.; Blondel, D. Translation initiation at alternate in-frame AUG codons in the rabies virus phosphoprotein mRNA is mediated by a ribosomal leaky scanning mechanism. J. Virol. 1995, 69, 707–712. [Google Scholar] [CrossRef] [Green Version]
- Kulkarni, S.; Volchkova, V.; Basler, C.F.; Palese, P.; Volchkov, V.E.; Shaw, M.L. Nipah virus edits its P gene at high frequency to express the V and W proteins. J. Virol. 2009, 83, 3982–3987. [Google Scholar] [CrossRef] [Green Version]
- Ramachandran, A.; Horvath, C.M. Paramyxovirus disruption of interferon signal transduction: STATus report. J. Interferon Cytokine Res. 2009, 29, 531–537. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, A.; Horvath, C.M. Dissociation of paramyxovirus interferon evasion activities: Universal and virus-specific requirements for conserved V protein amino acids in MDA5 interference. J. Virol. 2010, 84, 11152–11163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kato, A.; Kiyotani, K.; Sakai, Y.; Yoshida, T.; Shioda, T.; Nagai, Y. Importance of the cysteine-rich carboxyl-terminal half of V protein for Sendai virus pathogenesis. J. Virol. 1997, 71, 7266–7272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liston, P.; Briedis, D.J. Measles virus V protein binds zinc. Virology 1994, 198, 399–404. [Google Scholar] [CrossRef] [PubMed]
- Paterson, R.G.; Leser, G.P.; Shaughnessy, M.A.; Lamb, R.A. The paramyxovirus SV5 V protein binds two atoms of zinc and is a structural component of virions. Virology 1995, 208, 121–131. [Google Scholar] [CrossRef] [Green Version]
- Madeira, F.; Park, Y.M.; Lee, J.; Buso, N.; Gur, T.; Madhusoodanan, N.; Basutkar, P.; Tivey, A.R.N.; Potter, S.C.; Finn, R.D.; et al. The EMBL-EBI search and sequence analysis tools APIs in 2019. Nucleic Acids Res. 2019, 47, W636–W641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, T.; Chen, X.; Garbutt, K.C.; Zhou, P.; Zheng, N. Structure of DDB1 in complex with a paramyxovirus V protein: Viral hijack of a propeller cluster in ubiquitin ligase. Cell 2006, 124, 105–117. [Google Scholar] [CrossRef] [Green Version]
- Li, T.; Robert, E.I.; van Breugel, P.C.; Strubin, M.; Zheng, N. A promiscuous alpha-helical motif anchors viral hijackers and substrate receptors to the CUL4-DDB1 ubiquitin ligase machinery. Nat. Struct. Mol. Biol. 2010, 17, 105–111. [Google Scholar] [CrossRef] [Green Version]
- Vanmechelen, B.; Bletsa, M.; Laenen, L.; Lopes, A.R.; Vergote, V.; Beller, L.; Deboutte, W.; Korva, M.; Županc, T.A.; de Bellocq, J.G.; et al. Discovery and genome characterization of three new Jeilongviruses, a lineage of paramyxoviruses characterized by their unique membrane proteins. BMC Genomics 2018, 19, 617. [Google Scholar] [CrossRef] [Green Version]
- Nagano, Y.; Sugiyama, A.; Kimoto, M.; Wakahara, T.; Noguchi, Y.; Jiang, X.; Saijo, S.; Shimizu, N.; Yabuno, N.; Yao, M.; et al. The measles virus V protein binding site to STAT2 overlaps that of IRF9. J. Virol. 2020, 94, e01169-20. [Google Scholar] [CrossRef]
- Lin, G.Y.; Paterson, R.G.; Richardson, C.D.; Lamb, R.A. The V protein of the paramyxovirus SV5 interacts with damage-specific DNA binding protein. Virology 1998, 249, 189–200. [Google Scholar] [CrossRef] [Green Version]
- Audsley, M.D.; Marsh, G.A.; Lieu, K.G.; Tachedjian, M.; Joubert, D.A.; Wang, L.-F.; Jans, D.A.; Moseley, G.W. The immune evasion function of J and Beilong virus V proteins is distinct from that of other paramyxoviruses, consistent with their inclusion in the proposed genus Jeilongvirus. J. Gen. Virol. 2016, 97, 581–592. [Google Scholar] [CrossRef]
- Shiyanov, P.; Nag, A.; Raychaudhuri, P. Cullin 4A associates with the UV-damaged DNA-binding protein DDB. J. Biol. Chem. 1999, 274, 35309–35312. [Google Scholar] [CrossRef] [Green Version]
- Landsberg, C.D.; Megger, D.A.; Hotter, D.; Rückborn, M.U.; Eilbrecht, M.; Rashidi-Alavijeh, J.; Howe, S.; Heinrichs, S.; Sauter, D.; Sitek, B.; et al. A mass spectrometry-based profiling of interactomes of viral DDB1- and cullin ubiquitin ligase-binding proteins reveals NF-κB inhibitory activity of the HIV-2-encoded Vpx. Front. Immunol. 2018, 9, 2978. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andrejeva, J.; Poole, E.; Young, D.F.; Goodbourn, S.; Randall, R.E. The p127 subunit (DDB1) of the UV-DNA damage repair binding protein is essential for the targeted degradation of STAT1 by the V protein of the paramyxovirus simian virus 5. J. Virol. 2002, 76, 11379–11386. [Google Scholar] [CrossRef] [Green Version]
- Parisien, J.P.; Lau, J.F.; Rodriguez, J.J.; Sullivan, B.M.; Moscona, A.; Parks, G.D.; Lamb, R.A.; Horvath, C.M. The V protein of human parainfluenza virus 2 antagonizes type I interferon responses by destabilizing signal transducer and activator of transcription 2. Virology 2001, 283, 230–239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ulane, C.M.; Kentsis, A.; Cruz, C.D.; Parisien, J.-P.; Schneider, K.L.; Horvath, C.M. Composition and assembly of STAT-targeting ubiquitin ligase complexes: Paramyxovirus V protein carboxyl terminus is an oligomerization domain. J. Virol. 2005, 79, 10180–10189. [Google Scholar] [CrossRef] [Green Version]
- Liang, B. Structures of the Mononegavirales polymerases. J. Virol. 2020, 94, e00175-e20. [Google Scholar] [CrossRef] [PubMed]
- Hall, C.B.; Weinberg, G.A.; Iwane, M.K.; Blumkin, A.K.; Edwards, K.M.; Staat, M.A.; Auinger, P.; Griffin, M.R.; Poehling, K.A.; Erdman, D.; et al. The burden of respiratory syncytial virus infection in young children. N. Engl. J. Med. 2009, 360, 588–598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heinze, B.; Frey, S.; Mordstein, M.; Schmitt-Gräff, A.; Ehl, S.; Buchholz, U.J.; Collins, P.L.; Staeheli, P.; Krempl, C.D. Both nonstructural proteins NS1 and NS2 of pneumonia virus of mice are inhibitors of the interferon type I and type III responses in vivo. J. Virol. 2011, 85, 4071–4084. [Google Scholar] [CrossRef] [Green Version]
- Barik, S. Respiratory syncytial virus mechanisms to interfere with type 1 interferons. Curr. Top. Microbiol. Immunol. 2013, 372, 173–191. [Google Scholar] [CrossRef] [PubMed]
- Sedeyn, K.; Schepens, B.; Saelens, X. Respiratory syncytial virus nonstructural proteins 1 and 2: Exceptional disrupters of innate immune responses. PLoS Pathog. 2019, 15, e1007984. [Google Scholar] [CrossRef]
- Kikkert, M. Innate immune evasion by human respiratory RNA viruses. J. Innate. Immun. 2020, 12, 4–20. [Google Scholar] [CrossRef]
- Spann, K.M.; Tran, K.C.; Collins, P.L. Effects of nonstructural proteins NS1 and NS2 of human respiratory syncytial virus on interferon regulatory factor 3, NF-kappaB, and proinflammatory cytokines. J. Virol. 2005, 79, 5353–5362. [Google Scholar] [CrossRef] [Green Version]
- Lo, M.S.; Brazas, R.M.; Holtzman, M.J. Respiratory syncytial virus nonstructural proteins NS1 and NS2 mediate inhibition of Stat2 expression and alpha/beta interferon responsiveness. J. Virol. 2005, 79, 9315–9319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramaswamy, M.; Shi, L.; Varga, S.M.; Barik, S.; Behlke, M.A.; Look, D.C. Respiratory syncytial virus nonstructural protein 2 specifically inhibits type I interferon signal transduction. Virology 2006, 344, 328–339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ling, Z.; Tran, K.C.; Teng, M.N. Human respiratory syncytial virus nonstructural protein NS2 antagonizes the activation of beta interferon transcription by interacting with RIG-I. J. Virol. 2009, 83, 3734–3742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swedan, S.; Musiyenko, A.; Barik, S. Respiratory syncytial virus nonstructural proteins decrease levels of multiple members of the cellular interferon pathways. J. Virol. 2009, 83, 9682–9693. [Google Scholar] [CrossRef] [Green Version]
- Goswami, R.; Majumdar, T.; Dhar, J.; Chattopadhyay, S.; Bandyopadhyay, S.K.; Verbovetskaya, V.; Sen, G.C.; Barik, S. Viral degradasome hijacks mitochondria to suppress innate immunity. Cell Res. 2013, 23, 1025–1042. [Google Scholar] [CrossRef] [Green Version]
- Vasou, A.; Paulus, C.; Narloch, J.; Gage, Z.O.; Rameix-Welti, M.-A.; Eléouët, J.-F.; Nevels, M.; Randall, R.E.; Adamson, C.S. Modular cell-based platform for high throughput identification of compounds that inhibit a viral interferon antagonist of choice. Antiviral Res. 2018, 150, 79–92. [Google Scholar] [CrossRef]
- Swedan, S.; Andrews, J.; Majumdar, T.; Musiyenko, A.; Barik, S. Multiple functional domains and complexes of the two nonstructural proteins of human respiratory syncytial virus contribute to interferon suppression and cellular location. J. Virol. 2011, 85, 10090–100100. [Google Scholar] [CrossRef] [Green Version]
- Dhar, J.; Barik, S. Unique nonstructural proteins of Pneumonia Virus of Mice (PVM) promote degradation of interferon (IFN) pathway components and IFN-stimulated gene proteins. Sci. Rep. 2016, 6, 38139. [Google Scholar] [CrossRef]
- Ribaudo, M.; Barik, S. The nonstructural proteins of Pneumoviruses are remarkably distinct in substrate diversity and specificity. Virol. J. 2017, 14, 215. [Google Scholar] [CrossRef] [Green Version]
- Chatterjee, S.; Luthra, P.; Esaulova, E.; Agapov, E.; Yen, B.C.; Borek, D.M.; Edwards, M.R.; Mittal, A.; Jordan, D.S.; Ramanan, P.; et al. Structural basis for human respiratory syncytial virus NS1-mediated modulation of host responses. Nat. Microbiol. 2017, 2, 17101. [Google Scholar] [CrossRef] [PubMed]
- Pei, J.; Wagner, N.D.; Zou, A.J.; Chatterjee, S.; Borek, D.; Cole, A.R.; Kim, P.J.; Basler, C.F.; Otwinowski, Z.; Gross, M.L.; et al. Structural basis for IFN antagonism by human respiratory syncytial virus nonstructural protein 2. Proc. Natl. Acad. Sci. USA 2021, 118, e2020587118. [Google Scholar] [CrossRef]
- Yang, L.; He, J.; Wang, R.; Zhang, X.; Lin, S.; Ma, Z.; Zhang, Y. Nonstructural protein 11 of porcine reproductive and respiratory syndrome virus induces STAT2 degradation to inhibit interferon signaling. J. Virol. 2019, 93, e01352-e19. [Google Scholar] [CrossRef]
- Zhang, M.; Li, X.; Deng, Z.; Chen, Z.; Liu, Y.; Gao, Y.; Wu, W.; Chen, Z. Structural biology of the Arterivirus nsp11 endoribonucleases. J. Virol. 2017, 91, e01309–e01316. [Google Scholar] [CrossRef] [Green Version]
- Khan, S.; Zimmermann, A.; Basler, M.; Groettrup, M.; Hengel, H. A cytomegalovirus inhibitor of gamma interferon signaling controls immunoproteasome induction. J. Virol. 2004, 78, 1831–1842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Döring, M.; Lessin, I.; Frenz, T.; Spanier, J.; Kessler, A.; Tegtmeyer, P.; Dağ, F.; Thiel, N.; Trilling, M.; Lienenklaus, S.; et al. M27 expressed by cytomegalovirus counteracts effective type I interferon induction of myeloid cells but not of plasmacytoid dendritic cells. J. Virol. 2014, 88, 13638–13650. [Google Scholar] [CrossRef] [Green Version]
- Le-Trilling, V.T.K.; Becker, T.; Nachshon, A.; Stern-Ginossar, N.; Schöler, L.; Voigt, S.; Hengel, H.; Trilling, M. The human cytomegalovirus pUL145 isoforms act as viral DDB1-cullin-associated factors to instruct host protein degradation to impede innate immunity. Cell Rep. 2020, 30, 2248–2260. [Google Scholar] [CrossRef] [Green Version]
- Nightingale, K.; Lin, K.-M.; Ravenhill, B.J.; Davies, C.; Nobre, L.; Fielding, C.A.; Ruckova, E.; Fletcher-Etherington, A.; Soday, L.; Nichols, H.; et al. High-definition analysis of host protein stability during human cytomegalovirus infection reveals antiviral factors and viral evasion mechanisms. Cell Host Microbe. 2018, 24, 447–460.e11. [Google Scholar] [CrossRef] [Green Version]
- Nobre, L.V.; Nightingale, K.; Ravenhill, B.J.; Antrobus, R.; Soday, L.; Nichols, J.; Davies, J.A.; Seirafian, S.; Wang, E.C.; Davison, A.J.; et al. Human cytomegalovirus interactome analysis identifies degradation hubs, domain associations and viral protein functions. eLife 2019, 8, e49894. [Google Scholar] [CrossRef] [PubMed]
- Young, D.F.; Andrejeva, L.; Livingstone, A.; Goodbourn, S.; Lamb, R.A.; Collins, P.L.; Elliott, R.M.; Randall, R.E. Virus replication in engineered human cells that do not respond to interferons. J. Virol. 2003, 77, 2174–2181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le-Trilling, V.T.K.; Wohlgemuth, K.; Rückborn, M.U.; Jagnjic, A.; Maaßen, F.; Timmer, L.; Katschinski, B.; Trilling, M. STAT2-dependent immune responses ensure host survival despite the presence of a potent viral antagonist. J. Virol. 2018, 92, e00296-e18. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.-J.; An, H.-J.; Kim, S.-M.; Yoo, S.-M.; Park, J.; Lee, G.-E.; Kim, W.-Y.; Kim, D.J.; Kang, H.C.; Lee, J.Y.; et al. FBXW7-mediated stability regulation of signal transducer and activator of transcription 2 in melanoma formation. Proc. Natl. Acad. Sci. USA 2020, 117, 584–594. [Google Scholar] [CrossRef] [Green Version]
- Horn-Ghetko, D.; Krist, D.T.; Prabu, J.R.; Baek, K.; Mulder, M.P.C.; Klügel, M.; Scott, D.C.; Ovaa, H.; Kleiger, G.; Schulman, B.A. Ubiquitin ligation to F-box protein targets by SCF–RBR E3–E3 super-assembly. Nature 2021, 590, 671–676. [Google Scholar] [CrossRef]
- Palosaari, H.; Parisien, J.-P.; Rodriguez, J.J.; Ulane, C.M.; Horvath, C.M. STAT protein interference and suppression of cytokine signal transduction by measles virus V protein. J. Virol. 2003, 77, 7635–7644. [Google Scholar] [CrossRef] [Green Version]
- Hannah, J.; Zhou, P. Distinct and overlapping functions of the cullin E3 ligase scaffolding proteins CUL4A and CUL4B. Gene 2015, 573, 33–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marín, I. Diversification of the cullin family. BMC Evol, Biol. 2009, 9, 267. [Google Scholar] [CrossRef] [Green Version]
- Jang, S.-M.; Redon, C.E.; Aladjem, M.I. Switching DCAFs: Beyond substrate receptors. Bioessays 2021, 43, e2100057. [Google Scholar] [CrossRef] [PubMed]
- Le-Trilling, V.T.K.; Trilling, M. Ub to no good: How cytomegaloviruses exploit the ubiquitin proteasome system. Virus Res. 2020, 281, 197938. [Google Scholar] [CrossRef]
- Zhang, M.; Li, J.; Yan, H.; Huang, J.; Wang, F.; Liu, T.; Zeng, L.; Zhou, F. ISGylation in innate antiviral immunity and pathogen defense responses: A review. Front. Cell Dev. Biol. 2021, 9, 788410. [Google Scholar] [CrossRef]
- Liu, G.Q.; Lee, J.-H.; Parker, Z.M.; Acharya, D.; Chiang, J.J.; Gent, M.V.; Riedl, W.; Davis-Gardner, M.E.; Wies, E.; Chiang, C.; et al. ISG15-dependent activation of the sensor MDA5 is antagonized by the SARS-CoV-2 papain-like protease to evade host innate immunity. Nat. Microbiol. 2021, 6, 467–478. [Google Scholar] [CrossRef] [PubMed]
- Shin, D.; Mukherjee, R.; Grewe, D.; Bojkova, D.; Baek, K.; Bhattacharya, A.; Schulz, L.; Widera, M.; Mehdipour, A.R.; Tascher, G.; et al. Papain-like protease regulates SARS-CoV-2 viral spread and innate immunity. Nature 2020, 587, 657–662. [Google Scholar] [CrossRef]
- Yusupov, M.M.; Yusupova, G.Z.; Baucom, A.; Lieberman, K.; Earnest, T.N.; Cate, J.H.; Noller, H.F. Crystal structure of the ribosome at 5.5 A resolution. Science 2001, 292, 883–896. [Google Scholar] [CrossRef] [PubMed]
- Petry, S.; Brodersen, D.E.; Murphy 4th, F.V.; Dunham, C.M.; Selmer, M.; Tarry, M.J.; Kelley, A.C.; Ramakrishnan, V. Crystal structures of the ribosome in complex with release factors RF1 and RF2 bound to a cognate stop codon. Cell 2005, 123, 1255–1266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Selmer, M.; Dunham, C.M.; Murphy, F.V.; Weixlbaumer, A.; Petry, S.; Kelley, A.C.; Weir, J.R.; Ramakrishnan, V. Structure of the 70S ribosome complexed with mRNA and tRNA. Science 2006, 313, 1935–1942. [Google Scholar] [CrossRef] [Green Version]
- Korostelev, A.; Trakhanov, S.; Laurberg, M.; Noller, H.F. Crystal structure of a 70S ribosome-tRNA complex reveals functional interactions and rearrangements. Cell 2006, 126, 1065–1077. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cramer, P.; Bushnell, D.A.; Kornberg, R.D. Structural basis of transcription: RNA polymerase II at 2.8 angstrom resolution. Science 2001, 292, 1863–1876. [Google Scholar] [CrossRef] [Green Version]
- Keiffer, T.R.; Ciancanelli, M.J.; Edwards, M.R.; Basler, C.F. Interactions of the Nipah virus P, V, and W proteins across the STAT family of transcription factors. mSphere 2020, 5, e00449-e19. [Google Scholar] [CrossRef]
- Osiak, A.; Utermöhlen, O.; Niendorf, S.; Horak, I.; Knobeloch, K.-P. ISG15, an interferon-stimulated ubiquitin-like protein, is not essential for STAT1 signaling and responses against vesicular stomatitis and lymphocytic choriomeningitis virus. Mol. Cell. Biol. 2005, 25, 6338–6345. [Google Scholar] [CrossRef] [Green Version]
- Lenschow, D.J.; Giannakopoulos, N.V.; Gunn, L.J.; Johnston, C.; O’Guin, A.K.; Schmidt, R.E.; Levine, B.; Virgin 4th, H.W. Identification of interferon-stimulated gene 15 as an antiviral molecule during Sindbis virus infection in vivo. J. Virol. 2005, 79, 13974–13983. [Google Scholar] [CrossRef] [Green Version]
- Straub, C.P.; Lau, W.H.; Preston, F.M.; Headlam, M.J.; Gorman, J.J.; Collins, P.L.; Spann, K.M. Mutation of the elongin C binding domain of human respiratory syncytial virus non-structural protein 1 (NS1) results in degradation of NS1 and attenuation of the virus. Virol. J. 2011, 8, 252. [Google Scholar] [CrossRef] [Green Version]
- Yu, Y.; Xiao, Z.; Ehrlich, E.S.; Yu, X.; Yu, X.-F. Selective assembly of HIV-1 Vif-Cul5-ElonginB-ElonginC E3 ubiquitin ligase complex through a novel SOCS box and upstream cysteines. Genes Dev. 2004, 18, 2867–2872. [Google Scholar] [CrossRef] [Green Version]
- Mehle, A.; Strack, B.; Ancuta, P.; Zhang, C.; McPike, M.; Gabuzda, D. Vif overcomes the innate antiviral activity of APOBEC3G by promoting its degradation in the ubiquitin-proteasome pathway. J. Biol. Chem. 2004, 279, 7792–7798. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Ye, H.-Q.; Zhang, Q.-Y.; Lu, G.; Zhang, B.; Gong, P. A conformation-based intra-molecular initiation factor identified in the flavivirus RNA-dependent RNA polymerase. PLoS Pathog. 2020, 16, e1008484. [Google Scholar] [CrossRef]
- Schwechheimer, C. NEDD8-its role in the regulation of Cullin-RING ligases. Curr. Opin. Plant. Biol. 2018, 45, 112–119. [Google Scholar] [CrossRef] [PubMed]
- Vijayasimha, K.; Dolan, B.P. The many potential fates of non-canonical protein substrates subject to NEDDylation. Cells 2021, 10, 2660. [Google Scholar] [CrossRef]
- Fouad, S.; Wells, O.S.; Hill, M.A.; D’Angiolella, V. Cullin ring ubiquitin ligases (CRLs) in cancer: Responses to ionizing radiation (IR) treatment. Front. Physiol. 2019, 10, 1144. [Google Scholar] [CrossRef] [PubMed]
- Flores-Martínez, Y.A.; Le-Trilling, V.T.K.; Trilling, M. Nedd8-activating enzyme is a druggable host dependency factor of human and mouse cytomegalovirus. Viruses 2021, 13, 1610. [Google Scholar] [CrossRef] [PubMed]
- Le-Trilling, V.T.K.; Megger, D.A.; Katschinski, B.; Landsberg, C.D.; Rückborn, M.U.; Tao, S.; Krawczyk, A.; Bayer, W.; Drexler, I.; Tenbusch, M.; et al. Broad and potent antiviral activity of the NAE inhibitor MLN4924. Sci. Rep. 2016, 6, 19977. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Virus Family/Name of Virus | Viral Protein(s) Promoting Degradation | Host Factors Needed for STAT2 Degradation |
|---|---|---|
| Flaviviridae (Section 2.3.1) | ||
| Dengue | NS5 | UPS, UBR4 |
| Zika | NS5 | UPS |
| Paramyxoviridae (Section 2.3.2) | ||
| PIV2 | V | UPS (DDB1, STAT1) |
| Mumps | V | |
| Pneumoviridae (Section 2.3.3) | ||
| RSV | NS1, NS2 | UPS |
| MPV | NS1, NS2 | UPS |
| Arteriviridae (Section 2.3.4) | ||
| PRRSV | NS11 | |
| Herpesviridae (Section 2.3.5) | ||
| HSV2 | ICP22 | UPS |
| HCMV | pUL145 | UPS |
| MCMV | M27 | UPS |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barik, S. Mechanisms of Viral Degradation of Cellular Signal Transducer and Activator of Transcription 2. Int. J. Mol. Sci. 2022, 23, 489. https://doi.org/10.3390/ijms23010489
Barik S. Mechanisms of Viral Degradation of Cellular Signal Transducer and Activator of Transcription 2. International Journal of Molecular Sciences. 2022; 23(1):489. https://doi.org/10.3390/ijms23010489
Chicago/Turabian StyleBarik, Sailen. 2022. "Mechanisms of Viral Degradation of Cellular Signal Transducer and Activator of Transcription 2" International Journal of Molecular Sciences 23, no. 1: 489. https://doi.org/10.3390/ijms23010489
APA StyleBarik, S. (2022). Mechanisms of Viral Degradation of Cellular Signal Transducer and Activator of Transcription 2. International Journal of Molecular Sciences, 23(1), 489. https://doi.org/10.3390/ijms23010489