
Wnt/β-Catenin Signalling and Its Cofactor BCL9L Have an Oncogenic Effect in Bladder Cancer Cells



Abstract
:1. Introduction
2. Results
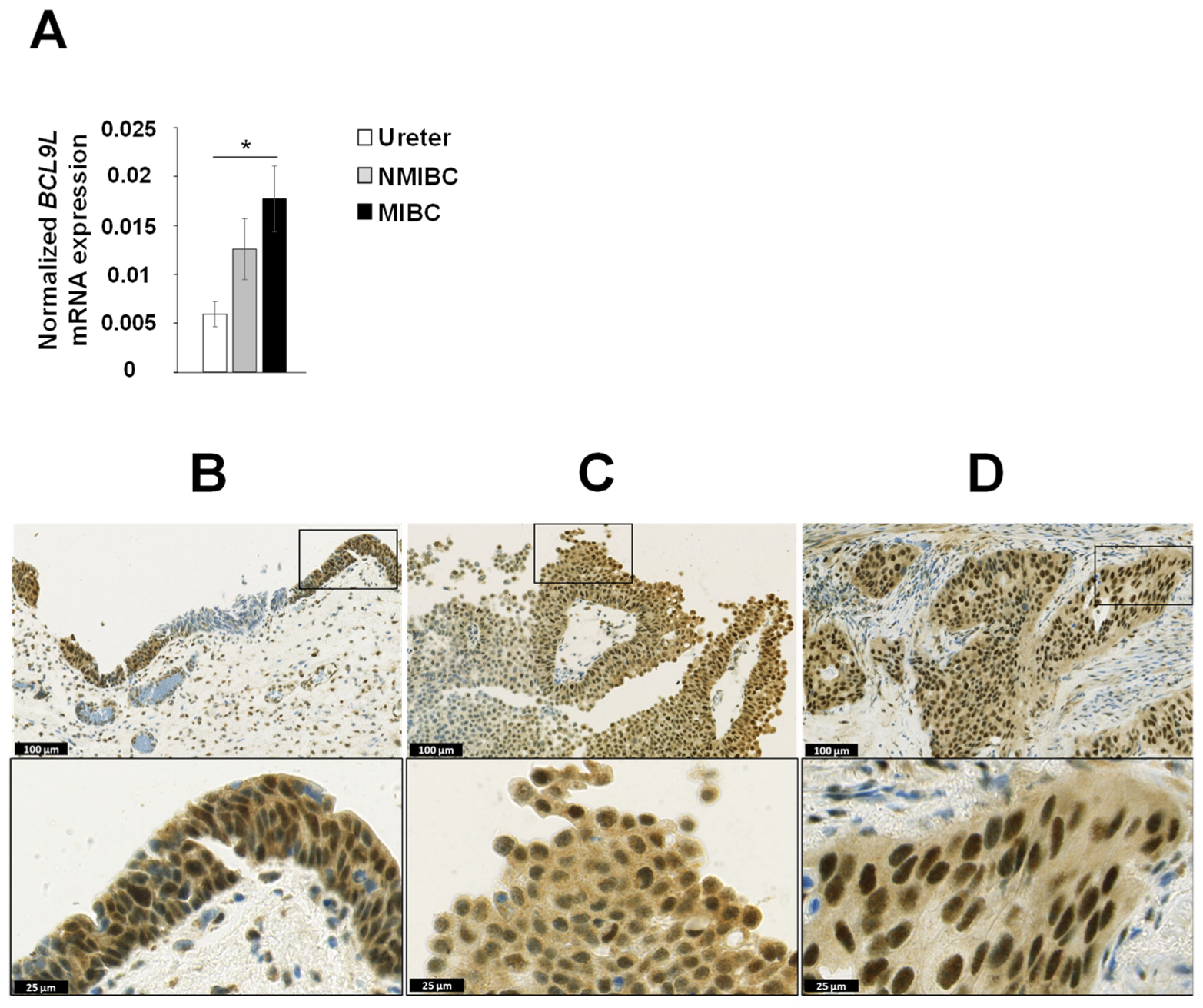
2.1. The Tumour-Associated Gene BCL9L Is Frequent Mutated in Bladder Cancer
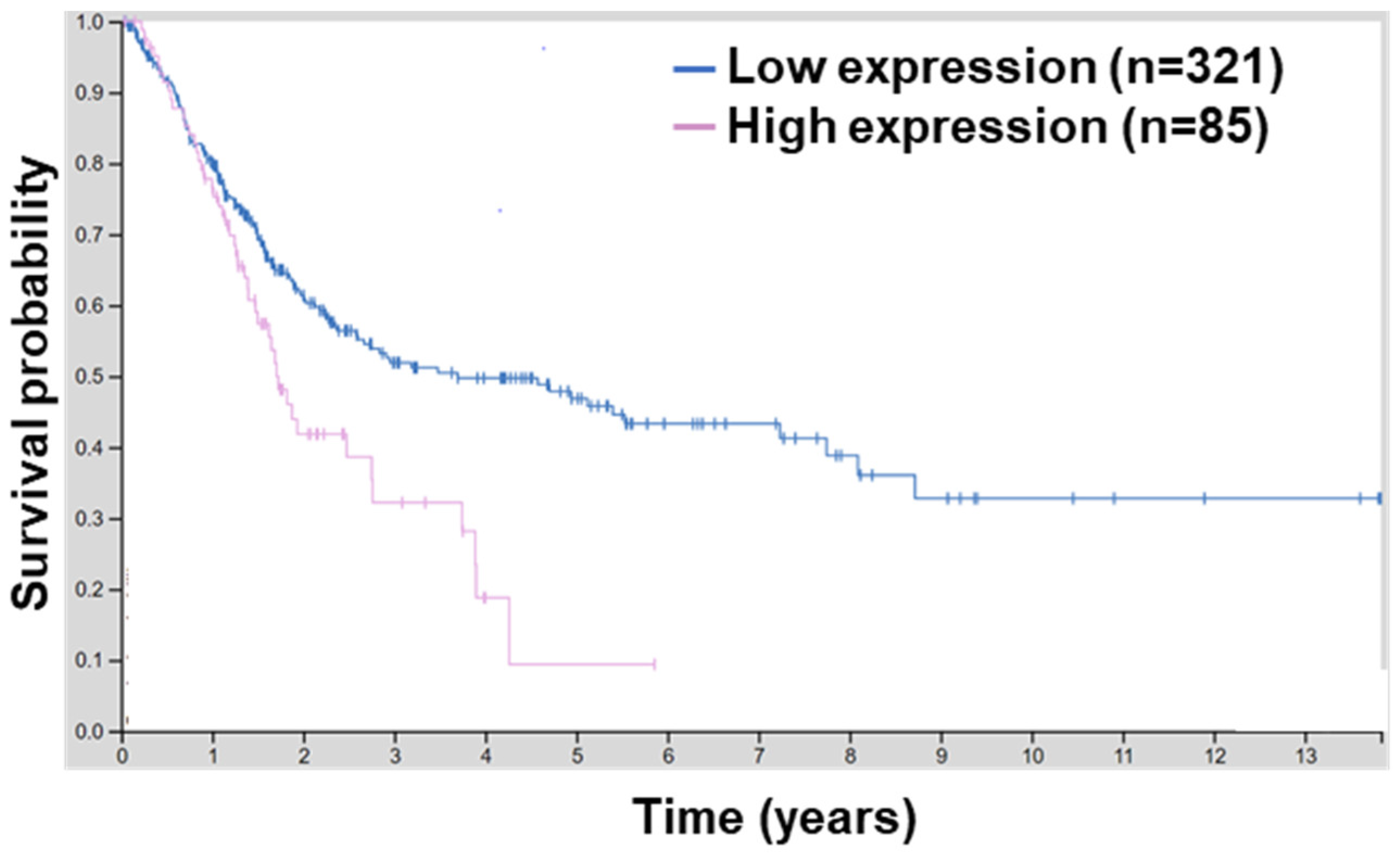
2.2. BCL9L Expression Is Associated with Poor Survival and Bladder Cancer Progression
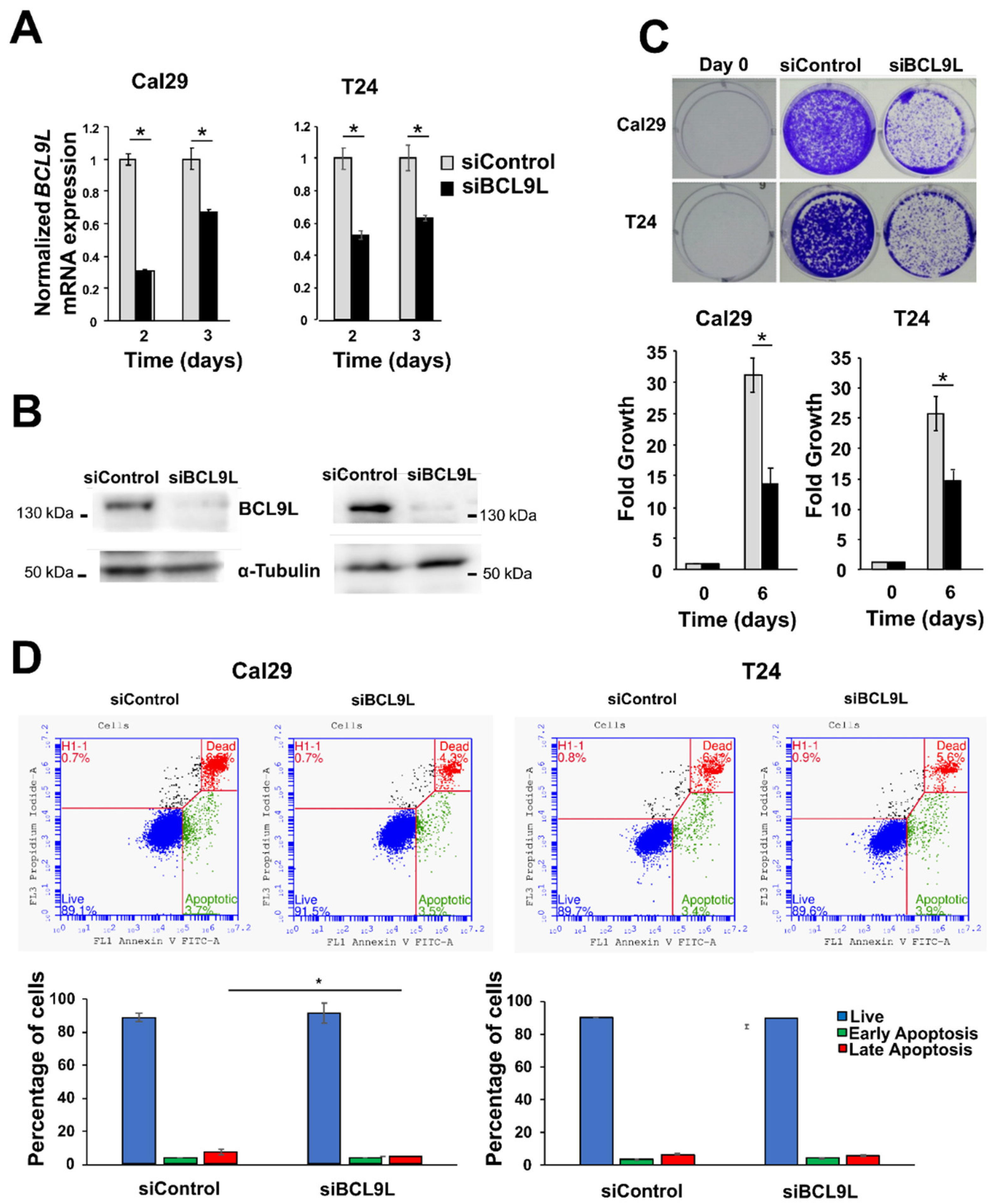
2.3. Knockdown of BCL9L Represses Proliferation Independent of Apoptosis in Bladder Cancer Cells
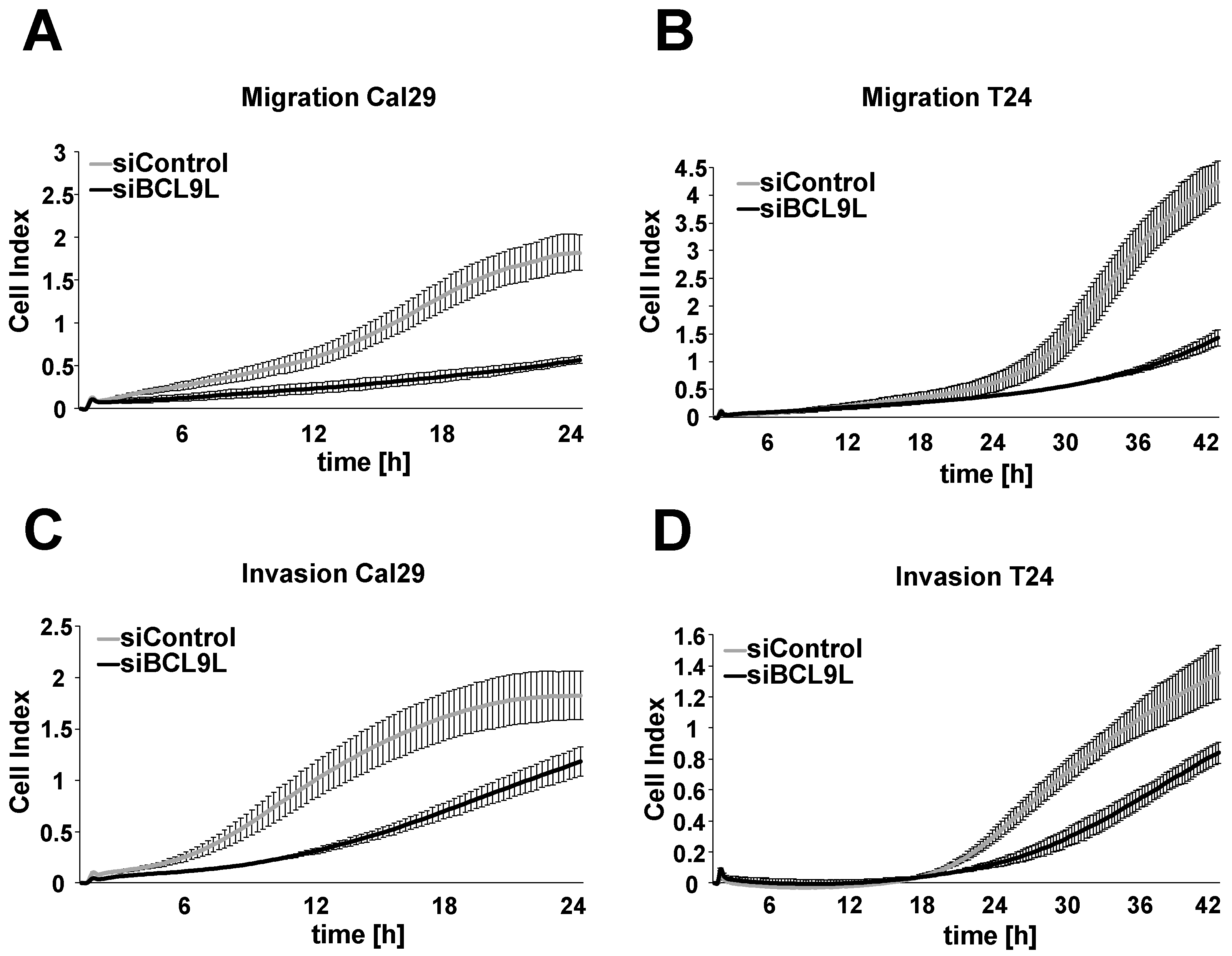
2.4. Knockdown of BCL9L Reduces Migration and Invasion in Bladder Cancer Cells
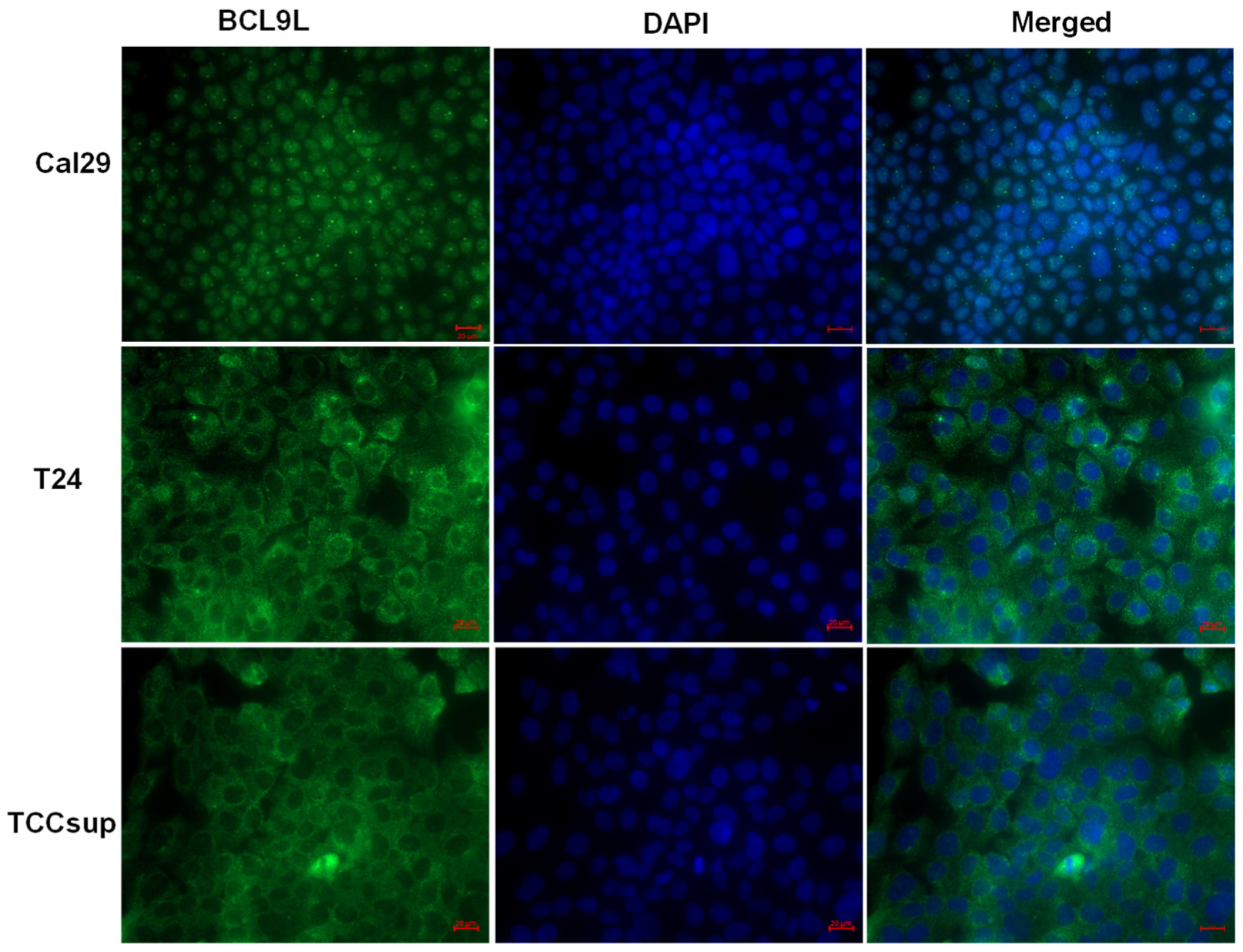
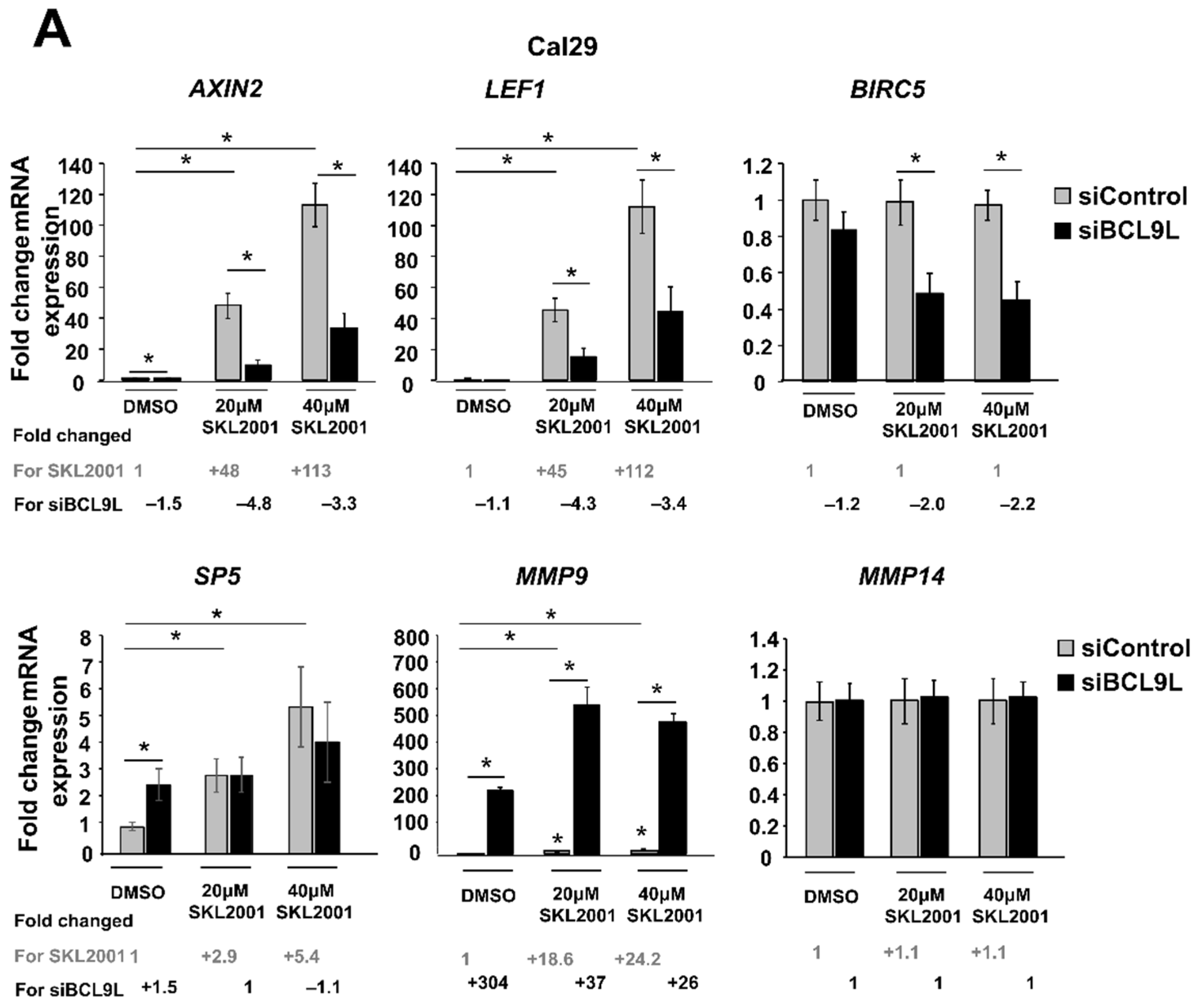
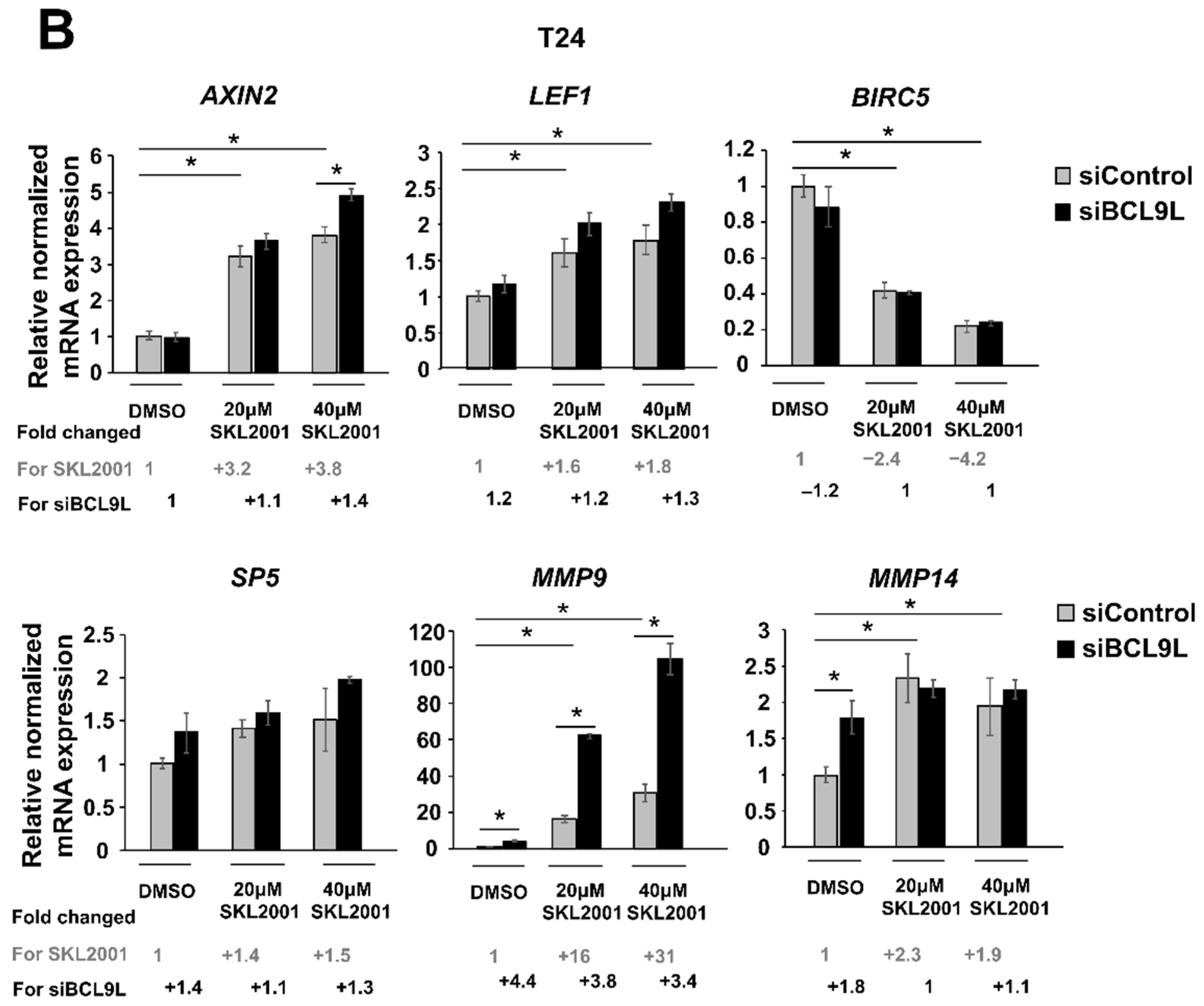
2.5. The Influence of BCL9L on Wnt/β-Catenin Signalling Is Cell Line Specific in Bladder Cancer
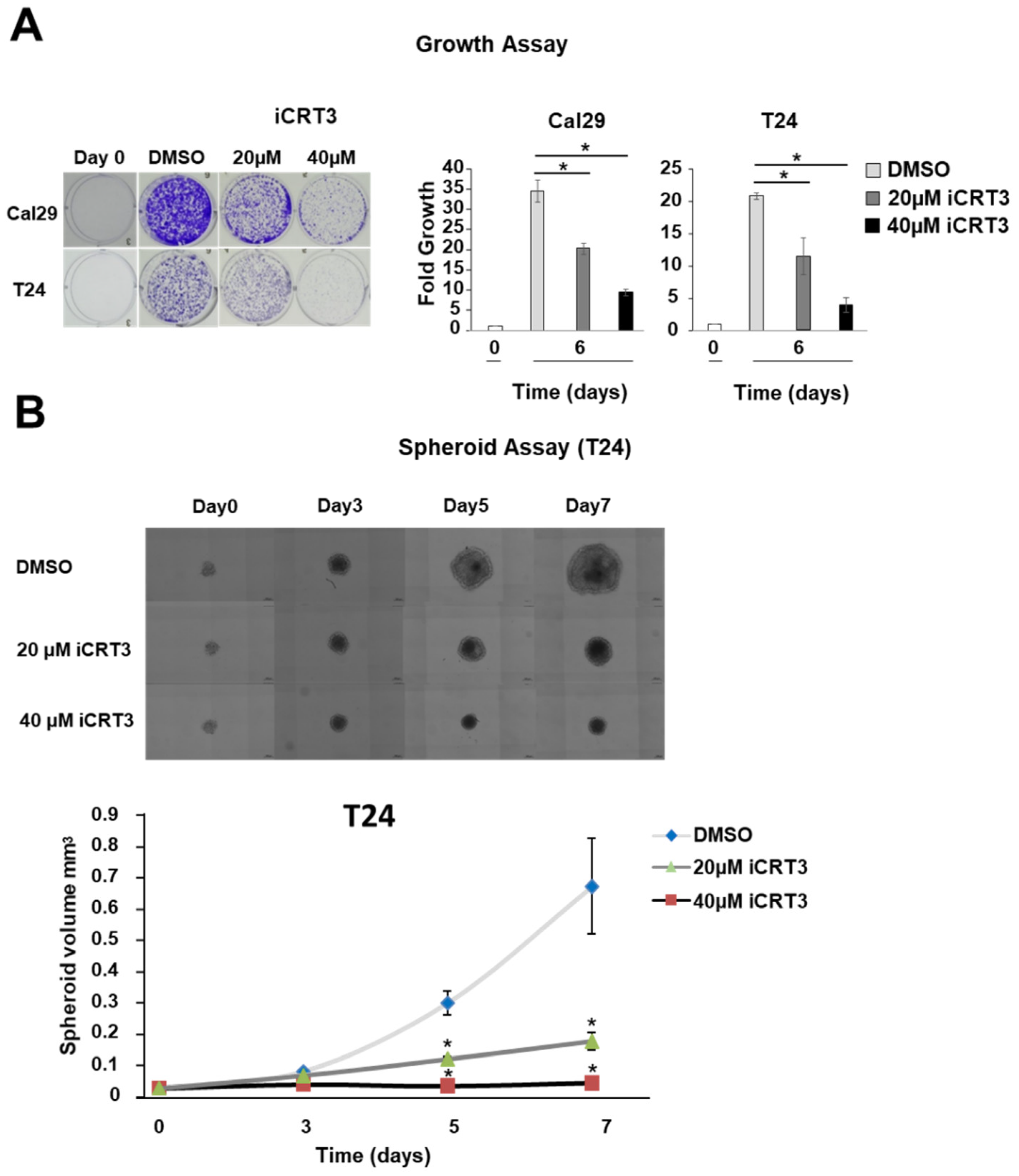
2.6. Inhibition of Wnt/β-Catenin Signalling Reduces Proliferation and Spheroid Growth in BC Cells
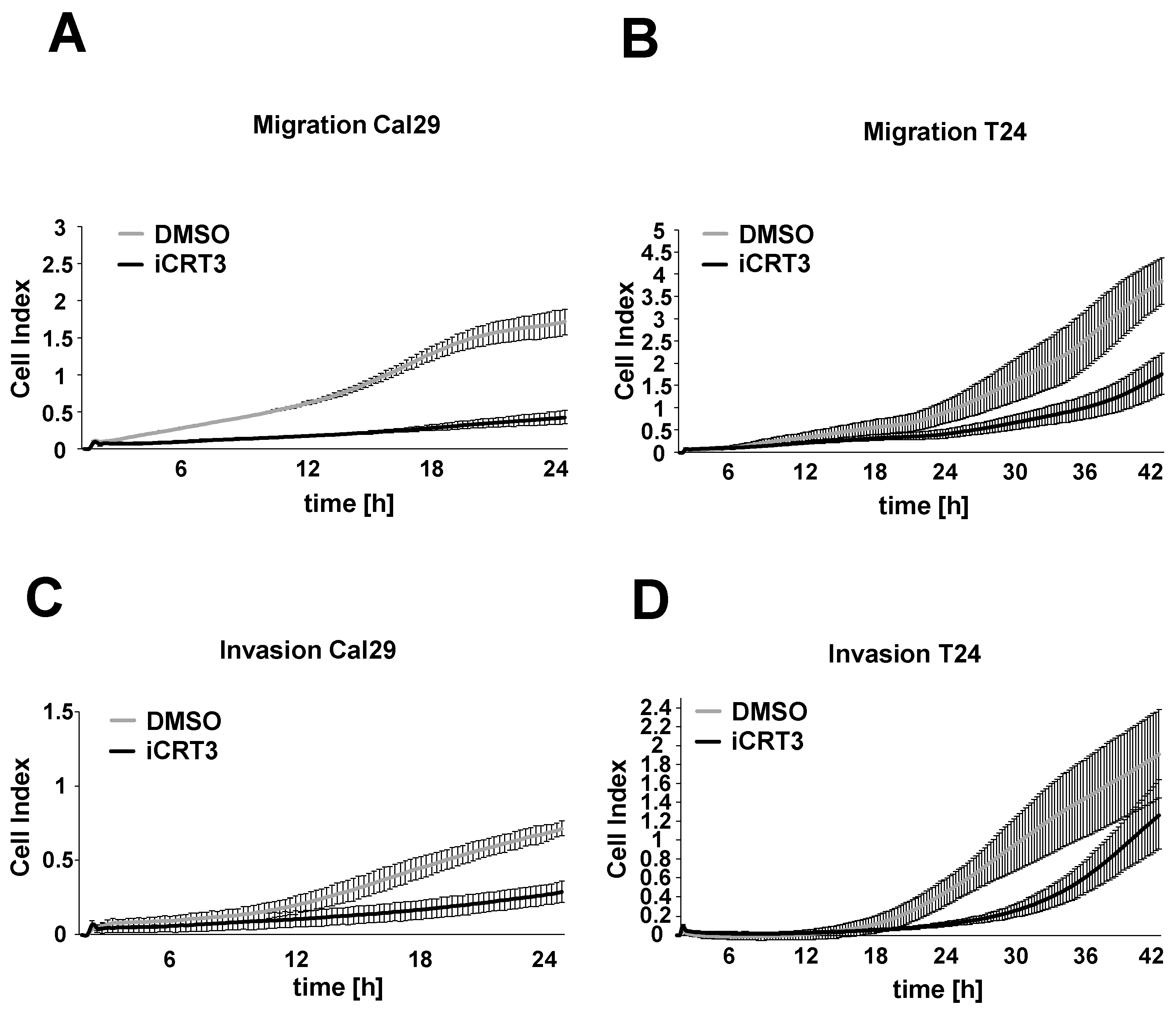
2.7. The Inhibition of Wnt/β-Catenin Signalling Suppresses Migration and Invasion in BC Cells
3. Discussion
4. Materials and Methods
4.1. Patients Material
4.2. Immunohistochemical Analysis
4.3. Cell Culture
4.4. UTR Mutation Analysis by Dual-Luciferase Reporter System Assay
4.5. Total RNA Extraction
4.6. cDNA Synthesis and Reverse Transcription Quantitative PCR (RT-qPCR)
4.7. Protein Analysis by Western Blotting
4.8. Protein Analysis by Immunofluorescence
4.9. Wnt/β-Catenin Signalling Analysis
4.10. Proliferation and Spheroid Formation Assay
4.11. Apoptosis Assay
4.12. Migration and Invasion by xCELLigence Real-Time Cellular Analysis System
4.13. Data and Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Babjuk, M.; Burger, M.; Capoun, O.; Cohen, D.; Compérat, E.M.; Dominguez Escrig, J.L.; Gontero, P.; Liedberg, F.; Masson-Lecomte, A.; Mostafid, A.H.; et al. European Association of Urology Guidelines on Non-muscle-invasive Bladder Cancer (Ta, T1, and Carcinoma in Situ). Eur. Urol. 2022, 81, 75–94. [Google Scholar] [CrossRef] [PubMed]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Freedman, N.D.; Silverman, D.T.; Hollenbeck, A.R.; Schatzkin, A.; Abnet, C.C. Association between smoking and risk of bladder cancer among men and women. Chechens 2011, 306, 87–106. [Google Scholar] [CrossRef] [PubMed]
- Cumberbatch, M.G.; Windsor-Shellard, B.; Catto, J.W. The contemporary landscape of occupational bladder cancer within the United Kingdom: A meta-analysis of risks over the last 80 years. BJU Int. 2017, 119, 100–109. [Google Scholar] [CrossRef] [Green Version]
- Smith, N.D.; Prasad, S.M.; Patel, A.R.; Weiner, A.B.; Pariser, J.J.; Razmaria, A.; Maene, C.; Schuble, T.; Pierce, B.; Steinberg, G.D. Bladder Cancer Mortality in the United States: A Geographic and Temporal Analysis of Socioeconomic and Environmental Factors. J. Urol. 2016, 195, 290–296. [Google Scholar] [CrossRef]
- Sylvester, R.J.; van der Meijden, A.P.; Oosterlinck, W.; Witjes, J.A.; Bouffioux, C.; Denis, L.; Newling, D.W.; Kurth, K. Predicting recurrence and progression in individual patients with stage Ta T1 bladder cancer using EORTC risk tables: A combined analysis of 2596 patients from seven EORTC trials. Eur. Urol. 2006, 49, 466–467. [Google Scholar] [CrossRef]
- Czerniak, B.; Dinney, C.; McConkey, D. Origins of Bladder Cancer. Annu. Rev. Pathol. 2016, 11, 149–174. [Google Scholar] [CrossRef]
- Smith, A.B.; Deal, A.M.; Woods, M.E.; Wallen, E.M.; Pruthi, R.S.; Chen, R.C.; Milowsky, M.I.; Nielsen, M.E. Muscle-invasive bladder cancer: Evaluating treatment and survival in the National Cancer Data Base. BJU Int. 2014, 114, 719–726. [Google Scholar] [CrossRef]
- Witjes, J.A.; Bruins, H.M.; Cathomas, R.; Compérat, E.M.; Cowan, N.C.; Gakis, G.; Hernández, V.; Linares Espinós, E.; Lorch, A.; Neuzillet, Y.; et al. European Association of Urology Guidelines on Muscle-invasive and Metastatic Bladder Cancer: Summary of the 2020 Guidelines. Eur. Urol. 2021, 79, 82–104. [Google Scholar] [CrossRef]
- Schrier, B.P.; Hollander, M.P.; van Rhijn, B.W.; Kiemeney, L.A.; Witjes, J.A. Prognosis of muscle-invasive bladder cancer: Difference between primary and progressive tumours and implications for therapy. Eur. Urol. 2004, 45, 292–296. [Google Scholar] [CrossRef]
- Steinhart, Z.; Angers, S. Wnt signaling in development and tissue homeostasis. Development 2018, 145, dev146589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taciak, B.; Pruszynska, I.; Kiraga, L.; Bialasek, M.; Krol, M. Wnt signaling pathway in development and cancer. J. Physiol. Pharm. 2018, 69, 185–196. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, X. Targeting the Wnt/β-catenin signaling pathway in cancer. J. Hematol. Oncol. 2020, 13, 165. [Google Scholar] [CrossRef] [PubMed]
- Nusse, R.; Clevers, H. Wnt/β-Catenin Signaling, Disease, and Emerging Therapeutic Modalities. Cell 2017, 169, 985–999. [Google Scholar] [CrossRef]
- Ren, J.; Yang, Y.; Peng, T.; Xu, D. Predictive value of β-catenin in bladder cancer: A systematic review and meta-analysis. Biosci. Rep. 2020, 40, BSR20202127. [Google Scholar] [CrossRef]
- Pierzynski, J.A.; Hildebrandt, M.A.; Kamat, A.M.; Lin, J.; Ye, Y.; Dinney, C.P.; Wu, X. Genetic Variants in the Wnt/β-Catenin Signaling Pathway as Indicators of Bladder Cancer Risk. J. Urol. 2015, 194, 1771–1776. [Google Scholar] [CrossRef]
- Garg, M.; Maurya, N. WNT/β-catenin signaling in urothelial carcinoma of bladder. World J. Nephrol. 2019, 8, 83–94. [Google Scholar] [CrossRef]
- Schmid, S.C.; Sathe, A.; Guerth, F.; Seitz, A.K.; Heck, M.M.; Maurer, T.; Schwarzenböck, S.M.; Krause, B.J.; Schulz, W.A.; Stoehr, R.; et al. Wntless promotes bladder cancer growth and acts synergistically as a molecular target in combination with cisplatin. Urol. Oncol. 2017, 35, e541–e544. [Google Scholar] [CrossRef]
- Logan, C.Y.; Nusse, R. The Wnt signaling pathway in development and disease. Annu. Rev. Cell Dev. Biol. 2004, 20, 781–810. [Google Scholar] [CrossRef] [Green Version]
- Stamos, J.L.; Weis, W.I. The β-catenin destruction complex. Cold Spring Harb. Perspect. Biol. 2013, 5, a007898. [Google Scholar] [CrossRef]
- Schaefer, K.N.; Peifer, M. Wnt/Beta-Catenin Signaling Regulation and a Role for Biomolecular Condensates. Dev. Cell 2019, 48, 429–444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chodaparambil, J.V.; Pate, K.T.; Hepler, M.R.; Tsai, B.P.; Muthurajan, U.M.; Luger, K.; Waterman, M.L.; Weis, W.I. Molecular functions of the TLE tetramerization domain in Wnt target gene repression. Embo J. 2014, 33, 719–731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lien, W.H.; Fuchs, E. Wnt some lose some: Transcriptional governance of stem cells by Wnt/β-catenin signaling. Genes Dev. 2014, 28, 1517–1532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Danieau, G.; Morice, S.; Rédini, F.; Verrecchia, F.; Royer, B.B. New Insights about the Wnt/β-Catenin Signaling Pathway in Primary Bone Tumors and Their Microenvironment: A Promising Target to Develop Therapeutic Strategies? Int. J. Mol. Sci. 2019, 20, 3751. [Google Scholar] [CrossRef] [Green Version]
- Katoh, M.; Katoh, M. Identification and characterization of human BCL9L gene and mouse Bcl9l gene In Silico. Int. J. Mol. Med. 2003, 12, 643–649. [Google Scholar] [CrossRef] [PubMed]
- Sampietro, J.; Dahlberg, C.L.; Cho, U.S.; Hinds, T.R.; Kimelman, D.; Xu, W. Crystal structure of a beta-catenin/BCL9/Tcf4 complex. Mol. Cell 2006, 24, 293–300. [Google Scholar] [CrossRef]
- Sustmann, C.; Flach, H.; Ebert, H.; Eastman, Q.; Grosschedl, R. Cell-type-specific function of BCL9 involves a transcriptional activation domain that synergizes with beta-catenin. Mol. Cell Biol. 2008, 28, 3526–3537. [Google Scholar] [CrossRef] [Green Version]
- Adachi, S.; Jigami, T.; Yasui, T.; Nakano, T.; Ohwada, S.; Omori, Y.; Sugano, S.; Ohkawara, B.; Shibuya, H.; Nakamura, T.; et al. Role of a BCL9-related β-catenin-binding protein, B9L, in tumorigenesis induced by aberrant activation of Wnt signaling. Cancer Res. 2004, 64, 8496–8501. [Google Scholar] [CrossRef] [Green Version]
- Cantù, C.; Pagella, P.; Shajiei, T.D.; Zimmerli, D.; Valenta, T.; Hausmann, G.; Basler, K.; Mitsiadis, T.A. A cytoplasmic role of Wnt/b-catenin transcriptional cofactors Bcl9, Bcl9l, and Pygopus in tooth enamel formation. Sci. Signal. 2017, 10, eaah4598. [Google Scholar] [CrossRef] [Green Version]
- Cantù, C.; Zimmerli, D.; Hausmann, G.; Valenta, T.; Moor, A.; Aguet, M.; Basler, K. Pax6-dependent, but β-catenin-independent, function of Bcl9 proteins in mouse lens development. Genes Dev. 2014, 28, 1879–1884. [Google Scholar] [CrossRef] [Green Version]
- Huge, N.; Sandbothe, M.; Schröder, A.K.; Stalke, A.; Eilers, M.; Schäffer, V.; Schlegelberger, B.; Illig, T.; Vajen, B.; Skawran, B. Wnt status-dependent oncogenic role of BCL9 and BCL9L in hepatocellular carcinoma. Hepatol. Int. 2020, 14, 373–384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zatula, N.; Wiese, M.; Bunzendahl, J.; Birchmeier, W.; Perske, C.; Bleckmann, A.; Brembeck, F.H. The BCL9-2 proto-oncogene governs estrogen receptor alpha expression in breast tumorigenesis. Oncotarget 2014, 5, 6770–6787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, K.; Lim, H.Y.; Shi, S.; Lee, J.; Deng, S.; Xie, T.; Zhu, Z.; Wang, Y.; Pocalyko, D.; Yang, W.J.; et al. Genomic landscape of copy number aberrations enables the identification of oncogenic drivers in hepatocellular carcinoma. Hepatology 2013, 58, 706–717. [Google Scholar] [CrossRef] [PubMed]
- Hyeon, J.; Ahn, S.; Lee, J.J.; Song, D.H.; Park, C.K. Prognostic Significance of BCL9 Expression in Hepatocellular Carcinoma. Korean J. Pathol. 2013, 47, 130–136. [Google Scholar] [CrossRef] [PubMed]
- Hoffmans, R.; Basler, K. BCL9-2 binds Arm/β-catenin in a Tyr142-independent manner and requires Pygopus for its function in Wg/Wnt signaling. Mech. Dev. 2007, 124, 59–67. [Google Scholar] [CrossRef]
- Gay, D.M.; Ridgway, R.A.; Müeller, M.; Hodder, M.C.; Hedley, A.; Clark, W.; Leach, J.D.; Jackstadt, R.; Nixon, C.; Huels, D.J.; et al. Loss of BCL9/9l suppresses Wnt driven tumourigenesis in models that recapitulate human cancer. Nat. Commun. 2019, 10, 723. [Google Scholar] [CrossRef] [Green Version]
- Mani, M.; Carrasco, D.E.; Zhang, Y.; Takada, K.; Gatt, M.E.; Dutta-Simmons, J.; Ikeda, H.; Diaz-Griffero, F.; Pena-Cruz, V.; Bertagnolli, M.; et al. BCL9 promotes tumor progression by conferring enhanced proliferative, metastatic, and angiogenic properties to cancer cells. Cancer Res. 2009, 69, 7577–7586. [Google Scholar] [CrossRef] [Green Version]
- Brembeck, F.H.; Wiese, M.; Zatula, N.; Grigoryan, T.; Dai, Y.; Fritzmann, J.; Birchmeier, W. BCL9-2 promotes early stages of intestinal tumor progression. Gastroenterology 2011, 141, 1359–1370.e3. [Google Scholar] [CrossRef]
- de la Roche, M.; Worm, J.; Bienz, M. The function of BCL9 in Wnt/beta-catenin signaling and colorectal cancer cells. BMC Cancer 2008, 8, 199. [Google Scholar] [CrossRef] [Green Version]
- Elsarraj, H.S.; Hong, Y.; Valdez, K.E.; Michaels, W.; Hook, M.; Smith, W.P.; Chien, J.; Herschkowitz, J.I.; Troester, M.A.; Beck, M.; et al. Expression profiling of in vivo ductal carcinoma in situ progression models identified B cell lymphoma-9 as a molecular driver of breast cancer invasion. Breast Cancer Res. 2015, 17, 128. [Google Scholar] [CrossRef] [Green Version]
- Kotolloshi, R.; Hölzer, M.; Gajda, M.; Grimm, M.O.; Steinbach, D. SLC35F2, a Transporter Sporadically Mutated in the Untranslated Region, Promotes Growth, Migration, and Invasion of Bladder Cancer Cells. Cells 2021, 10, 80. [Google Scholar] [CrossRef] [PubMed]
- Robertson, A.G.; Kim, J.; Al-Ahmadie, H.; Bellmunt, J.; Guo, G.; Cherniack, A.D.; Hinoue, T.; Laird, P.W.; Hoadley, K.A.; Akbani, R.; et al. Comprehensive Molecular Characterization of Muscle-Invasive Bladder Cancer. Cell 2017, 171, 540–556.e25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pongsavee, M.; Yamkamon, V.; Dakeng, S.; O-charoenrat, P.; Smith, D.R.; Saunders, G.F.; Patmasiriwat, P. The BRCA1 3′-UTR: 5711+421T/T_5711+1286T/T genotype is a possible breast and ovarian cancer risk factor. Genet Test Mol. Biomark. 2009, 13, 307–317. [Google Scholar] [CrossRef] [PubMed]
- Bisio, A.; Nasti, S.; Jordan, J.J.; Gargiulo, S.; Pastorino, L.; Provenzani, A.; Quattrone, A.; Queirolo, P.; Bianchi-Scarrà, G.; Ghiorzo, P.; et al. Functional analysis of CDKN2A/p16INK4a 5′-UTR variants predisposing to melanoma. Hum. Mol. Genet 2010, 19, 1479–1491. [Google Scholar] [CrossRef] [Green Version]
- Gochhait, S.; Bukhari, S.I.; Bairwa, N.; Vadhera, S.; Darvishi, K.; Raish, M.; Gupta, P.; Husain, S.A.; Bamezai, R.N. Implication of BRCA2 -26G>A 5′ untranslated region polymorphism in susceptibility to sporadic breast cancer and its modulation by p53 codon 72 Arg>Pro polymorphism. Breast Cancer Res. 2007, 9, R71. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Xiao, N.; Guo, W.; Wu, Y.; Cai, Z.; He, Q.; Zhang, L.; Chen, X.; Sun, C.; Wang, J.; et al. The hOGG1 gene 5′-UTR variant c.-53G>C contributes to the risk of gastric cancer but not colorectal cancer in the Chinese population: The functional variation of hOGG1 for gastric cancer risk. J. Cancer Res. Clin. Oncol. 2011, 137, 1477–1485. [Google Scholar] [CrossRef]
- Wang, W.; Sun, J.; Li, F.; Li, R.; Gu, Y.; Liu, C.; Yang, P.; Zhu, M.; Chen, L.; Tian, W.; et al. A frequent somatic mutation in CD274 3′-UTR leads to protein over-expression in gastric cancer by disrupting miR-570 binding. Hum. Mutat. 2012, 33, 480–484. [Google Scholar] [CrossRef]
- Uhlen, M.; Zhang, C.; Lee, S.; Sjöstedt, E.; Fagerberg, L.; Bidkhori, G.; Benfeitas, R.; Arif, M.; Liu, Z.; Edfors, F.; et al. A pathology atlas of the human cancer transcriptome. Science 2017, 357, eaan2507. [Google Scholar] [CrossRef] [Green Version]
- Ishibashi, H.; Suzuki, T.; Suzuki, S.; Moriya, T.; Kaneko, C.; Takizawa, T.; Sunamori, M.; Handa, M.; Kondo, T.; Sasano, H. Sex steroid hormone receptors in human thymoma. J. Clin. Endocrinol. Metab. 2003, 88, 2309–2317. [Google Scholar] [CrossRef]
- Brembeck, F.H.; Schwarz-Romond, T.; Bakkers, J.; Wilhelm, S.; Hammerschmidt, M.; Birchmeier, W. Essential role of BCL9-2 in the switch between β-catenin’s adhesive and transcriptional functions. Genes Dev. 2004, 18, 2225–2230. [Google Scholar] [CrossRef] [Green Version]
- De Sousa, E.M.F.; Medema, J.P. Axing Wnt signals. Cell Res. 2012, 22, 9–11. [Google Scholar] [CrossRef] [PubMed]
- Gonsalves, F.C.; Klein, K.; Carson, B.B.; Katz, S.; Ekas, L.A.; Evans, S.; Nagourney, R.; Cardozo, T.; Brown, A.M.; DasGupta, R. An RNAi-based chemical genetic screen identifies three small-molecule inhibitors of the Wnt/wingless signaling pathway. Proc. Natl. Acad. Sci. USA 2011, 108, 5954–5963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weinstein, J.N.; Akbani, R.; Broom, B.M.; Wang, W.; Verhaak, R.G.W.; McConkey, D.; Lerner, S.; Morgan, M.; Creighton, C.J.; Smith, C.; et al. Comprehensive molecular characterization of urothelial bladder carcinoma. Nature 2014, 507, 315–322. [Google Scholar] [CrossRef] [Green Version]
- Lamy, P.; Nordentoft, I.; Birkenkamp-Demtröder, K.; Houlberg Thomsen, M.B.; Villesen, P.; Vang, S.; Hedegaard, J.; Borre, M.; Jensen, J.B.; Høyer, S.; et al. Paired exome analysis reveals clonal evolution and potential therapeutic targets in urothelial carcinoma. Cancer Res. 2016, 76, 5894–5906. [Google Scholar] [CrossRef] [Green Version]
- Paiss, T.; Wöhr, G.; Hautmann, R.E.; Mattfeldt, T.; Müller, M.; Haeussler, J.; Vogel, W. Some tumors of the bladder are polyclonal in origin. J. Urol. 2002, 167, 718–723. [Google Scholar] [CrossRef]
- Sannino, G.; Armbruster, N.; Bodenhöfer, M.; Haerle, U.; Behrens, D.; Buchholz, M.; Rothbauer, U.; Sipos, B.; Schmees, C. Role of BCL9L in transforming growth factor-β (TGF-β)-induced epithelial-to-mesenchymal-transition (EMT) and metastasis of pancreatic cancer. Oncotarget 2016, 7, 73725–73738. [Google Scholar] [CrossRef] [Green Version]
- Moor, A.E.; Anderle, P.; Cantù, C.; Rodriguez, P.; Wiedemann, N.; Baruthio, F.; Deka, J.; André, S.; Valenta, T.; Moor, M.B.; et al. BCL9/9L-β-catenin Signaling is Associated with Poor Outcome in Colorectal Cancer. EBioMedicine 2015, 2, 1932–1943. [Google Scholar] [CrossRef] [Green Version]
- Toya, H.; Oyama, T.; Ohwada, S.; Togo, N.; Sakamoto, I.; Horiguchi, J.; Koibuchi, Y.; Adachi, S.; Jigami, T.; Nakajima, T.; et al. Immunohistochemical expression of the beta-catenin-interacting protein B9L is associated with histological high nuclear grade and immunohistochemical ErbB2/HER-2 expression in breast cancers. Cancer Sci. 2007, 98, 484–490. [Google Scholar] [CrossRef]
- Sun, R.; Liu, Z.; Han, L.; Yang, Y.; Wu, F.; Jiang, Q.; Zhang, H.; Ma, R.; Miao, J.; He, K.; et al. MiR-22 and miR-214 targeting BCL9L inhibit proliferation, metastasis, and epithelial-mesenchymal transition by down-regulating Wnt signaling in colon cancer. FASEB J. 2019, 33, 5411–5424. [Google Scholar] [CrossRef]
- Cadigan, K.M.; Nusse, R. Wnt signaling: A common theme in animal development. Genes Dev. 1997, 11, 3286–3305. [Google Scholar] [CrossRef] [Green Version]
- Gammons, M.; Bienz, M. Multiprotein complexes governing Wnt signal transduction. Curr Opin Cell Biol. 2018, 51, 42–49. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Feng, M.; Xiao, T.; Guo, B.; Liu, D.; Liu, C.; Pei, J.; Liu, Q.; Xiao, Y.; Rosin-Arbesfeld, R.; et al. BCL9/BCL9L promotes tumorigenicity through immune-dependent and independent mechanisms in triple negative breast cancer. Oncogene 2021, 40, 2982–2997. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, I.; Morton, J.P.; Singh, L.B.; Radulescu, S.M.; Ridgway, R.A.; Patel, S.; Woodgett, J.; Winton, D.J.; Taketo, M.M.; Wu, X.R.; et al. β-Catenin activation synergizes with PTEN loss to cause bladder cancer formation. Oncogene 2011, 30, 178–189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmad, I.; Patel, R.; Liu, Y.; Singh, L.B.; Taketo, M.M.; Wu, X.R.; Leung, H.Y.; Sansom, O.J. Ras mutation cooperates with β-catenin activation to drive bladder tumourigenesis. Cell Death Dis. 2011, 2, e124. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Miao, S.; Han, X.; Li, C.; Zhang, M.; Cui, K.; Xiong, T.; Chen, Z.; Wang, C.; Xu, H. MicroRNA-3619-5p suppresses bladder carcinoma progression by directly targeting β-catenin and CDK2 and activating p21. Cell Death Dis. 2018, 9, 960. [Google Scholar] [CrossRef]
- Buchert, M.; Athineos, D.; Abud, H.E.; Burke, Z.D.; Faux, M.C.; Samuel, M.S.; Jarnicki, A.G.; Winbanks, C.E.; Newton, I.P.; Meniel, V.S.; et al. Genetic dissection of differential signaling threshold requirements for the Wnt/beta-catenin pathway in vivo. PLoS Genet 2010, 6, e1000816. [Google Scholar] [CrossRef] [Green Version]
- Lv, J.; Liu, Y.; Cheng, F.; Li, J.; Zhou, Y.; Zhang, T.; Zhou, N.; Li, C.; Wang, Z.; Ma, L.; et al. Cell softness regulates tumorigenicity and stemness of cancer cells. Embo J. 2021, 40, e106123. [Google Scholar] [CrossRef]
- Yu, Z.; Pestell, T.G.; Lisanti, M.P.; Pestell, R.G. Cancer stem cells. Int. J. Biochem. Cell Biol. 2012, 44, 2144–2151. [Google Scholar] [CrossRef] [Green Version]
- Pan, Y.; Ma, S.; Cao, K.; Zhou, S.; Zhao, A.; Li, M.; Qian, F.; Zhu, C. Therapeutic approaches targeting cancer stem cells. J. Cancer Res. 2018, 14, 1469–1475. [Google Scholar] [CrossRef]
- Kripnerova, M.; Parmar, H.S.; Pesta, M.; Kohoutova, M.; Kuncova, J.; Drbal, K.; Rajtmajerova, M.; Hatina, J. Urothelial Cancer Stem Cell Heterogeneity. Adv. Exp Med. Biol. 2019, 1139, 127–151. [Google Scholar] [CrossRef]
- Balla, M.M.; Ningthoujam, R.S.; Kumar, M.; Bandekar, J.R.; Pandey, B.N. Cellular and spectroscopic characterization of cancer stem cell-like cells derived from A549 lung carcinoma. J. Cancer Res. 2016, 12, 1144–1152. [Google Scholar] [CrossRef]
- Hatina, J.; Parmar, H.S.; Kripnerova, M.; Hepburn, A.; Heer, R. Urothelial Carcinoma Stem Cells: Current Concepts, Controversies, and Methods. Methods Mol. Biol. 2018, 1655, 121–136. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, I. The role of WNT signalling in urothelial cell carcinoma. Ann. R. Coll. Surg. Engl. 2015, 97, 481–486. [Google Scholar] [CrossRef] [Green Version]
- Dimov, I.; Visnjic, M.; Stefanovic, V. Urothelial cancer stem cells. Sci. World J. 2010, 10, 1400–1415. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Lin, K.; Yang, Z.; Han, N.; Quan, X.; Guo, X.; Li, C. Bladder cancer stem cells: Clonal origin and therapeutic perspectives. Oncotarget 2017, 8, 66668–66679. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Xu, H.; Shen, J.; Yang, Y.; Wu, S.; Xiao, J.; Xu, Y.; Liu, X.Y.; Chu, L. RGD-modifided oncolytic adenovirus exhibited potent cytotoxic effect on CAR-negative bladder cancer-initiating cells. Cell Death Dis. 2015, 6, e1760. [Google Scholar] [CrossRef] [Green Version]
- Wezel, F.; Lustig, J.; Azoitei, A.; Liu, J.; Meessen, S.; Najjar, G.; Zehe, V.; Faustmann, P.; Zengerling, F.; John, A.; et al. Grainyhead-Like 3 Influences Migration and Invasion of Urothelial Carcinoma Cells. Int. J. Mol. Sci. 2021, 22, 2959. [Google Scholar] [CrossRef]
- Rasmussen, R. Quantification on the LightCycler. In Rapid Cycle Real-Time PCR: Methods and Applications; Meuer, S., Wittwer, C., Nakagawara, K.-I., Eds.; Springer: Berlin/Heidelberg, Germany, 2001; pp. 21–34. [Google Scholar] [CrossRef]
- Pfaffl, M.W. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001, 29, e45. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient | Primary NMIBC | MIBC | ||
|---|---|---|---|---|
| No. | Age | Sex | TNM-T/Grading | TNM-T/Grading |
| 1 | 67 | m | pTa HG | pT4a HG |
| 2 | 79 | f | pT1 HG | pT2b HG |
| 3 | 69 | m | pTa HG | pT4a HG |
| 4 | 78 | f | pT1 HG | pT4a HG |
| 5 | 65 | m | pTa HG | pT2a HG |
| 6 | 71 | m | pT1 HG | pT2a HG |
| 7 | 74 | m | pTa HG | pT3a HG |
| 8 | 73 | m | pTa LG | pT2 HG |
| 9 | 63 | m | pTa HG | pT2a HG |
| 10 | 76 | m | pTa HG | pT3b HG |
| 11 | 76 | m | pTa HG | pT4a HG |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kotolloshi, R.; Gajda, M.; Grimm, M.-O.; Steinbach, D. Wnt/β-Catenin Signalling and Its Cofactor BCL9L Have an Oncogenic Effect in Bladder Cancer Cells. Int. J. Mol. Sci. 2022, 23, 5319. https://doi.org/10.3390/ijms23105319
Kotolloshi R, Gajda M, Grimm M-O, Steinbach D. Wnt/β-Catenin Signalling and Its Cofactor BCL9L Have an Oncogenic Effect in Bladder Cancer Cells. International Journal of Molecular Sciences. 2022; 23(10):5319. https://doi.org/10.3390/ijms23105319
Chicago/Turabian StyleKotolloshi, Roland, Mieczyslaw Gajda, Marc-Oliver Grimm, and Daniel Steinbach. 2022. "Wnt/β-Catenin Signalling and Its Cofactor BCL9L Have an Oncogenic Effect in Bladder Cancer Cells" International Journal of Molecular Sciences 23, no. 10: 5319. https://doi.org/10.3390/ijms23105319
APA StyleKotolloshi, R., Gajda, M., Grimm, M. -O., & Steinbach, D. (2022). Wnt/β-Catenin Signalling and Its Cofactor BCL9L Have an Oncogenic Effect in Bladder Cancer Cells. International Journal of Molecular Sciences, 23(10), 5319. https://doi.org/10.3390/ijms23105319