
Impaired LEF1 Activation Accelerates iPSC-Derived Keratinocytes Differentiation in Hutchinson-Gilford Progeria Syndrome

 ,
,
Abstract
:1. Introduction
2. Results
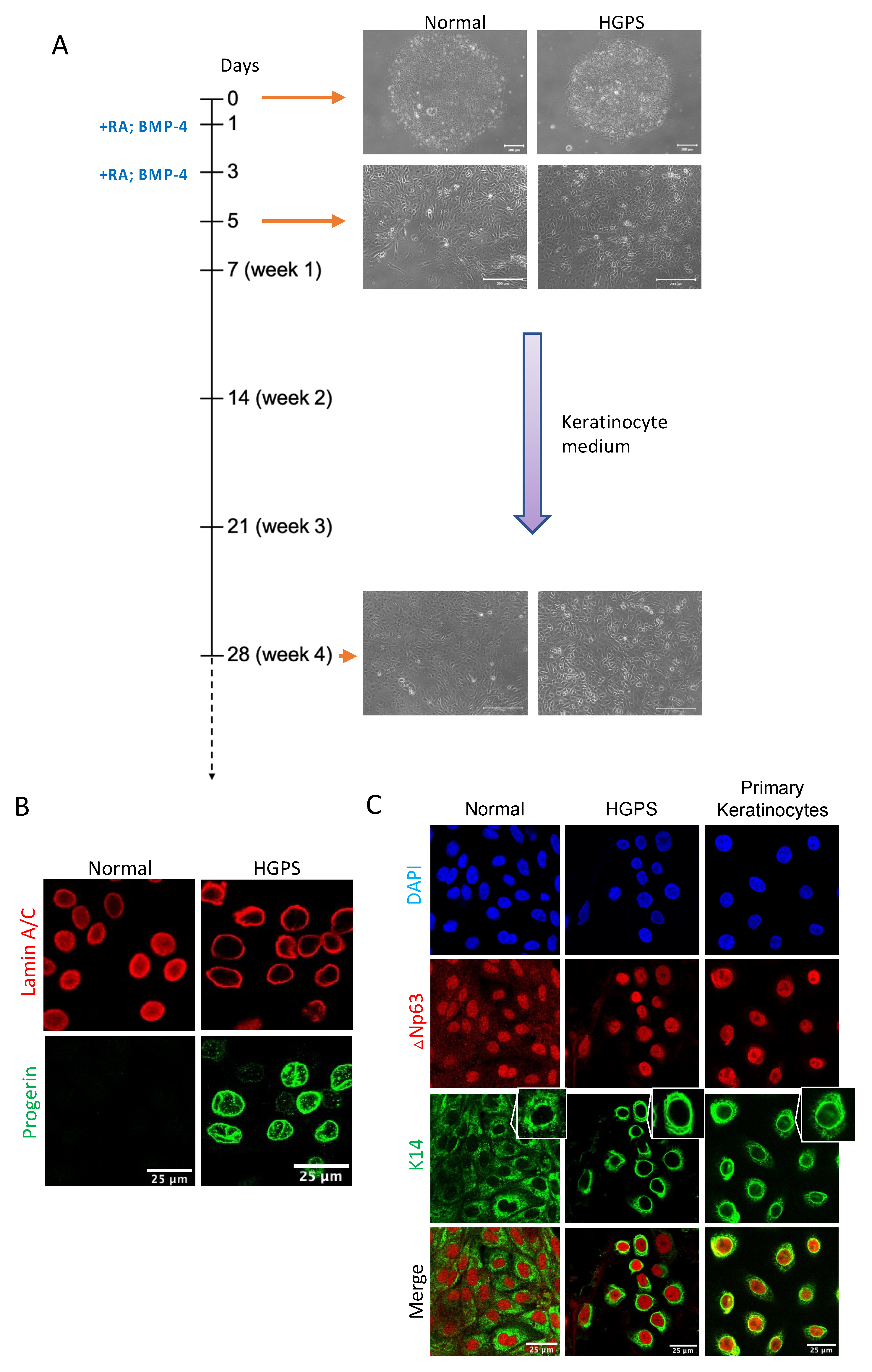
2.1. Differentiating Normal and HGPS-Derived iPSCs into Keratinocytes
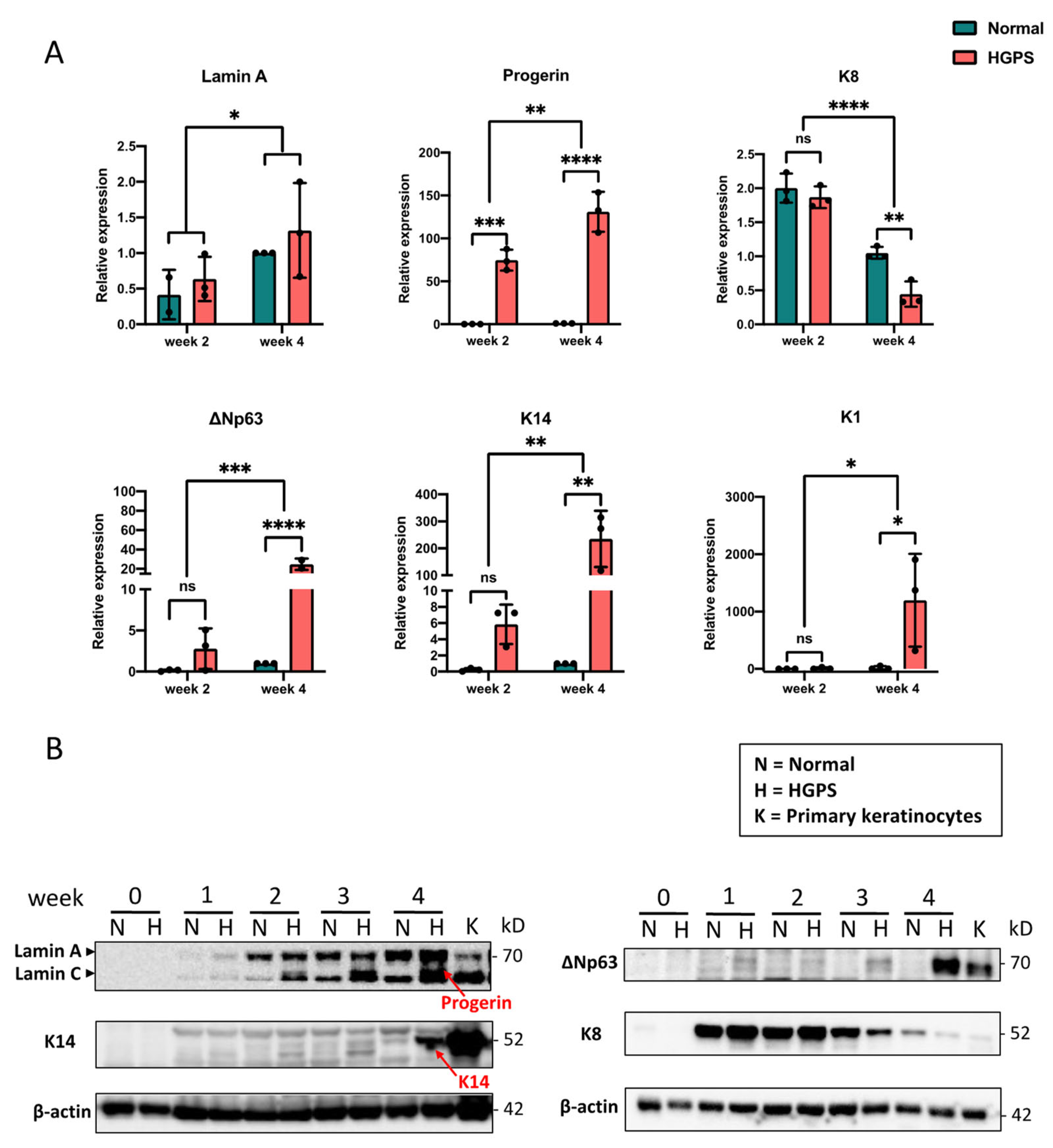
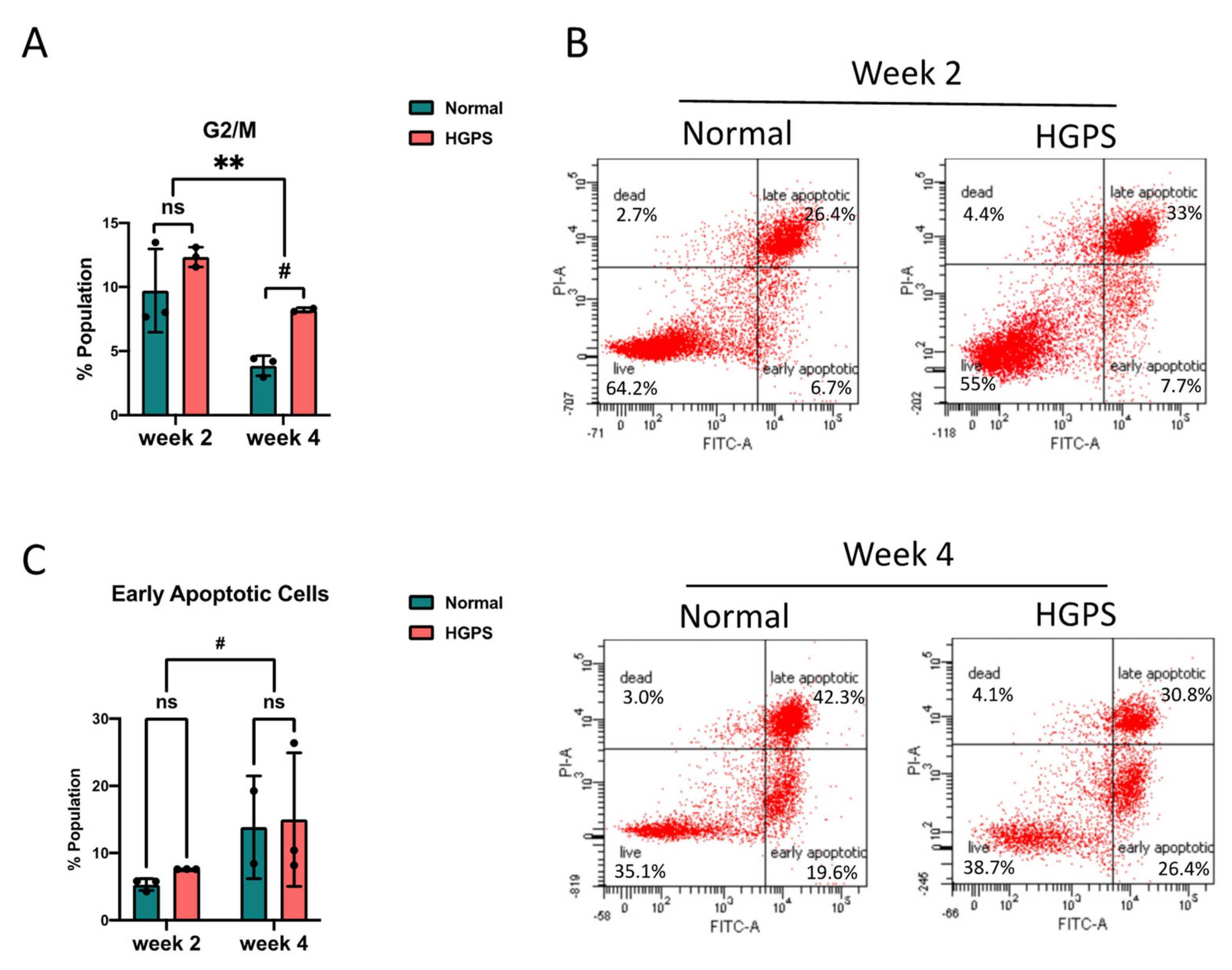
2.2. Dysregulated Cell Cycle in HGPS iPSCs-Keratinocytes Differentiation
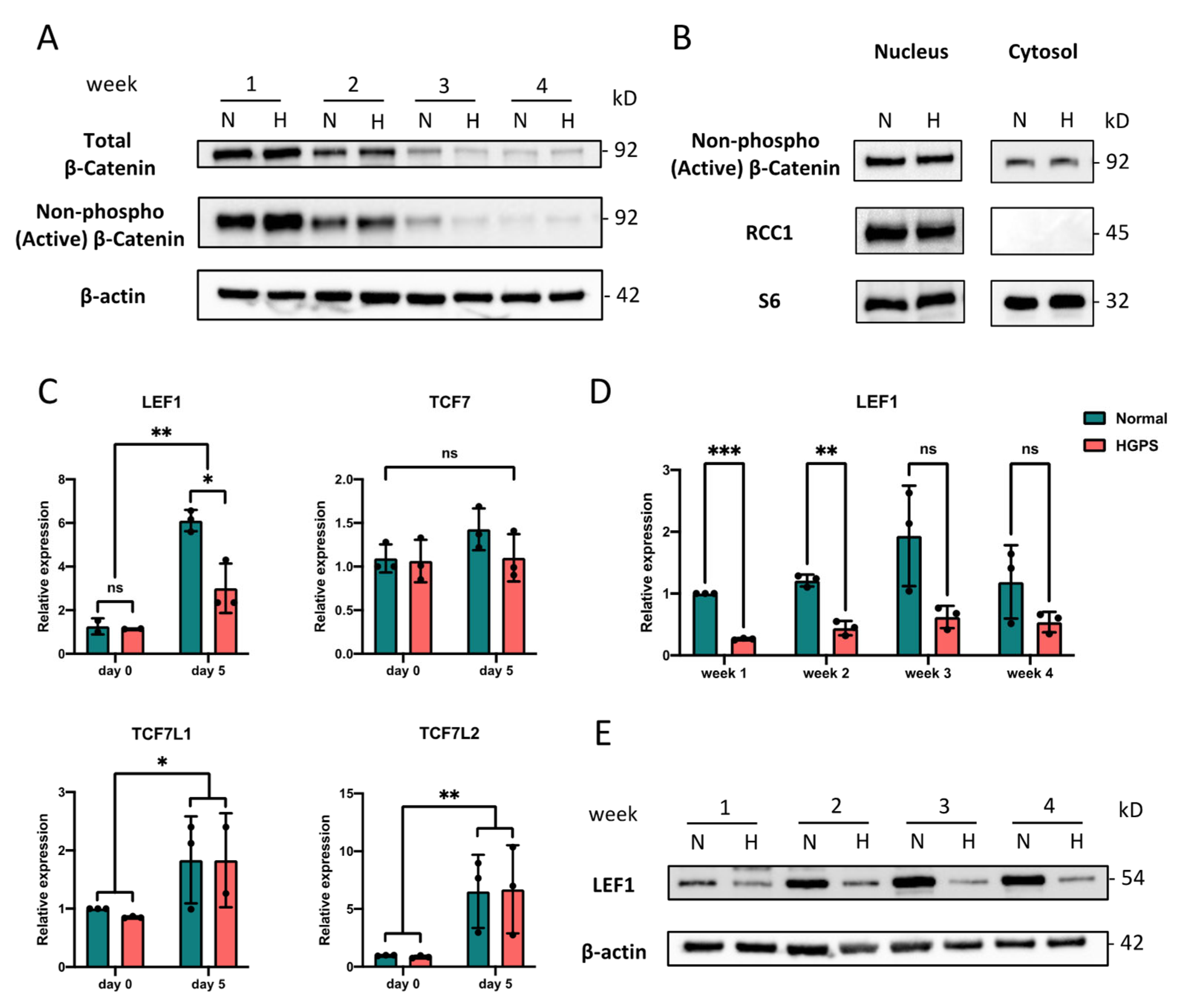
2.3. LEF1 Is Down-Regulated in Early HGPS iPSCs-Keratinocytes Differentiation
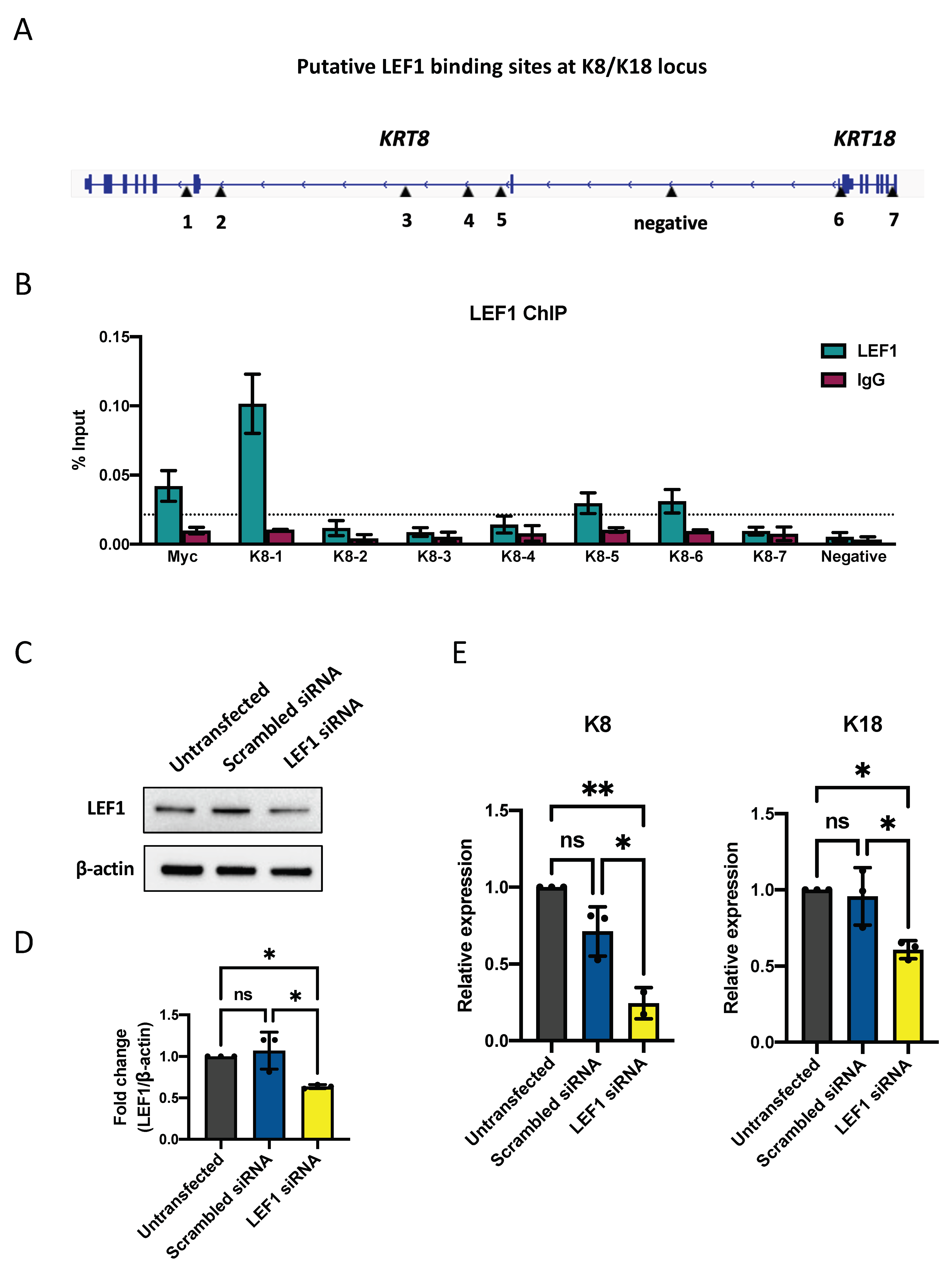
2.4. LEF1 Regulates Keratinocytes Differentiation through K8/K18
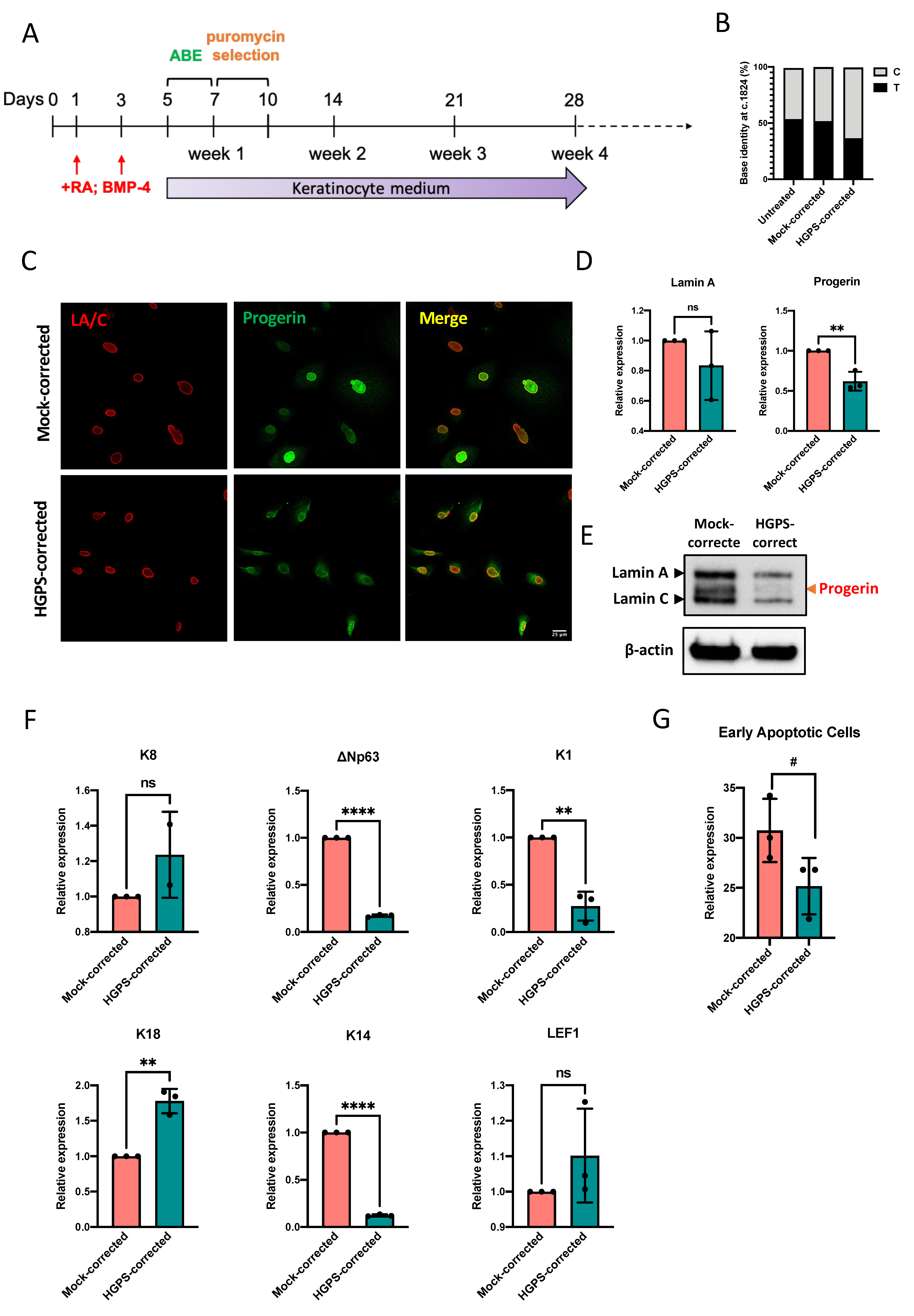
2.5. Correcting the HGPS Mutation in iPSCs-Keratinocytes Differentiation with ABE
3. Discussion
3.1. Stem Cell Depletion May Contribute to the Accelerated Keratinocyte Commitment in HGPS iPSCs Differentiation
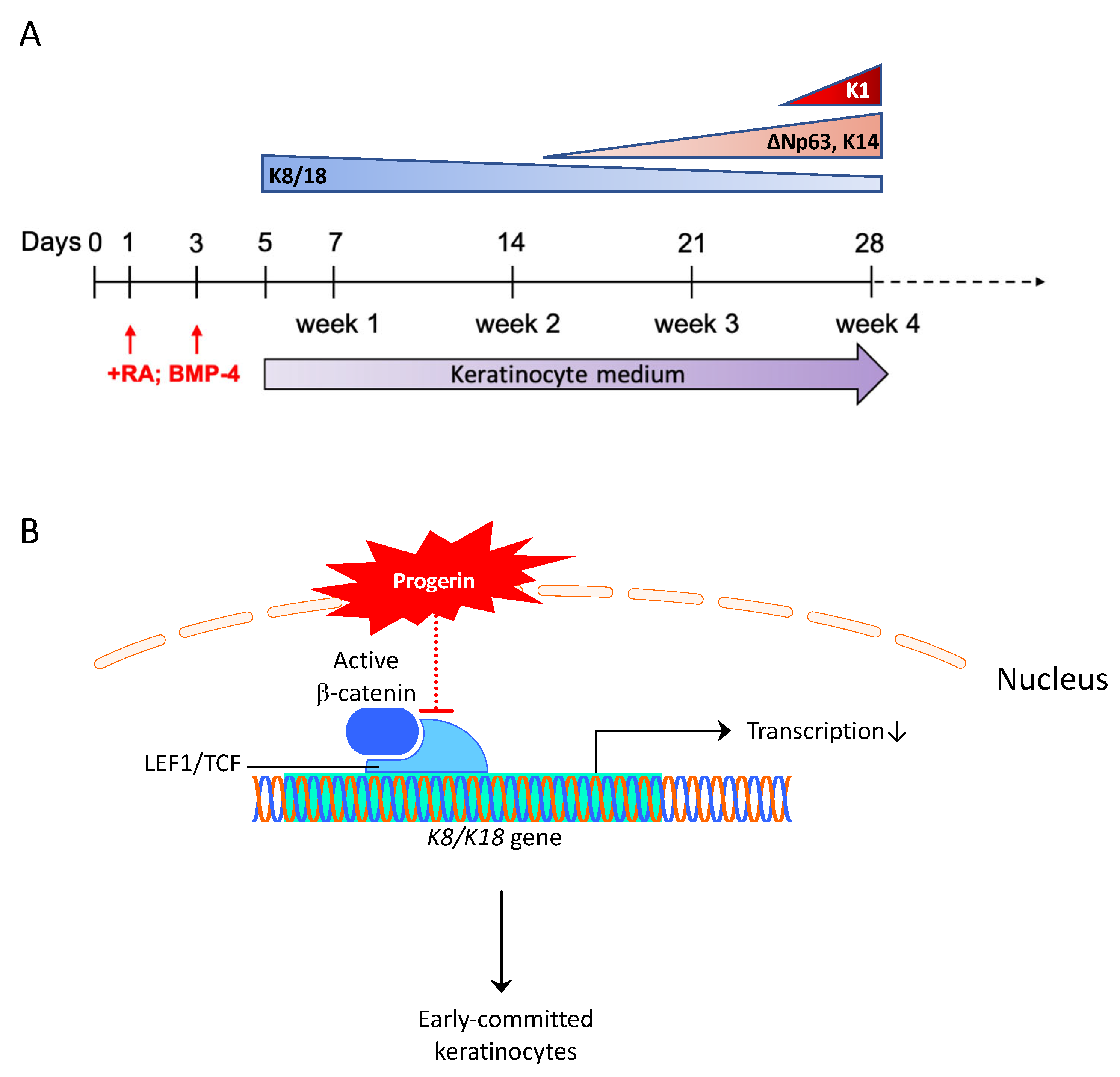
3.2. LEF1 Is an Early Regulator in Epidermal Development, and Its Down-Regulation Accelerates the Keratinocyte Lineage Commitment in HGPS
3.3. ABE in HGPS iPSCs-Keratinocytes Differentiation
4. Materials and Methods
4.1. Cell Culture and Keratinocytes Differentiation from iPSCs
4.2. Adenine Base Editor Lentiviral Vectors Production
4.3. Immunocytochemistry
4.4. Western Blot
4.5. RNA Isolation and Real-Time Quantitative PCR
4.6. Cell Cycle Analysis
4.7. Apoptosis Assay
4.8. Chromatin Immunoprecipitation
4.9. Silencing Experiments
4.10. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Eriksson, M.; Brown, W.T.; Gordon, L.B.; Glynn, M.W.; Singer, J.; Scott, L.; Erdos, M.R.; Robbins, C.M.; Moses, T.Y.; Berglund, P.; et al. Recurrent de Novo Point Mutations in Lamin A Cause Hutchinson-Gilford Progeria Syndrome. Nature 2003, 423, 293–298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Merideth, M.A.; Gordon, L.B.; Clauss, S.; Sachdev, V.; Smith, A.C.M.; Perry, M.B.; Brewer, C.C.; Zalewski, C.; Kim, H.J.; Solomon, B.; et al. Phenotype and Course of Hutchinson-Gilford Progeria Syndrome. N. Engl. J. Med. 2008, 358, 592–604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gordon, L.B.; Brown, W.T.; Collins, F.S. Hutchinson-Gilford Progeria Syndrome. GeneReviews 2019, 66, 753–756. [Google Scholar]
- De Sandre-Giovannoli, A.; Bernard, R.; Cau, P.; Navarro, C.; Amiel, J.; Boccaccio, I.; Lyonnet, S.; Stewart, C.L.; Munnich, A.; Le Merrer, M.; et al. Lamin A Truncation in Hutchinson-Gilford Progeria. Science 2003, 300, 2055. [Google Scholar] [CrossRef]
- Shumaker, D.K.; Dechat, T.; Kohlmaier, A.; Adam, S.A.; Bozovsky, M.R.; Erdos, M.R.; Eriksson, M.; Goldman, A.E.; Khuon, S.; Collins, F.S.; et al. Mutant Nuclear Lamin A Leads to Progressive Alterations of Epigenetic Control in Premature Aging. Proc. Natl. Acad. Sci. USA 2006, 103, 8703–8708. [Google Scholar] [CrossRef] [Green Version]
- Goldman, R.D.; Shumaker, D.K.; Erdos, M.R.; Eriksson, M.; Goldman, A.E.; Gordon, L.B.; Gruenbaum, Y.; Khuon, S.; Mendez, M.; Varga, R.; et al. Accumulation of Mutant Lamin A Causes Progressive Changes in Nuclear Architecture in Hutchinson–Gilford Progeria Syndrome. Proc. Natl. Acad. Sci. USA 2004, 101, 8963–8968. [Google Scholar] [CrossRef] [Green Version]
- McCord, R.P.; Nazario-Toole, A.; Zhang, H.; Chines, P.S.; Zhan, Y.; Erdos, M.R.; Collins, F.S.; Dekker, J.; Cao, K. Correlated Alterations in Genome Organization, Histone Methylation, and DNA-Lamin A/C Interactions in Hutchinson-Gilford Progeria Syndrome. Genome Res. 2013, 23, 260–269. [Google Scholar] [CrossRef] [Green Version]
- Millington, P.F. Skin; Cambridge University Press: Cambridge, UK, 1983; pp. 49–50. ISBN 978-0-521-10681-8. [Google Scholar]
- Fuchs, E.; Raghavan, S. Getting under the Skin of Epidermal Morphogenesis. Nat. Rev. Genet. 2002, 3, 199–209. [Google Scholar] [CrossRef]
- Sagelius, H.; Rosengardten, Y.; Hanif, M.; Erdos, M.R.; Rozell, B.; Collins, F.S.; Eriksson, M. Targeted Transgenic Expression of the Mutation Causing Hutchinson-Gilford Progeria Syndrome Leads to Proliferative and Degenerative Epidermal Disease. J. Cell Sci. 2008, 121, 969–978. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Panteleyev, A.A.; Owens, D.M.; Djabali, K.; Stewart, C.L.; Worman, H.J. Epidermal Expression of the Truncated Prelamin A Causing Hutchinson-Gilford Progeria Syndrome: Effects on Keratinocytes, Hair and Skin. Hum. Mol. Genet. 2008, 17, 2357–2369. [Google Scholar] [CrossRef] [Green Version]
- Rosengardten, Y.; Mckenna, T.; Grochová, D.; Eriksson, M. Stem Cell Depletion in Hutchinson-Gilford Progeria Syndrome. Aging Cell 2011, 10, 1011–1020. [Google Scholar] [CrossRef] [PubMed]
- Sola-Carvajal, A.; Revêchon, G.; Helgadottir, H.; Whisenant, D.; Hagblom, R.; Döhla, J.; Katajisto, P.; Brodin, D.; Fagerström-Billai, F.; Viceconte, N.; et al. Accumulation of Progerin Affects the Symmetry of Cell Division and Is Associated with Impaired Wnt Signaling and the Mislocalization of Nuclear Envelope Proteins. J. Investig. Dermatol. 2019, 139, 2272–2280.e12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mckenna, T.; Rosengardten, Y.; Viceconte, N.; Baek, J.H.; Grochová, D.; Eriksson, M. Embryonic Expression of the Common Progeroid Lamin A Splice Mutation Arrests Postnatal Skin Development. Aging Cell 2014, 13, 292–302. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.Y.; Lai, J.K.; Xiong, Z.M.; Ren, M.; Moorer, M.C.; Stains, J.P.; Cao, K. Diminished Canonical β-Catenin Signaling During Osteoblast Differentiation Contributes to Osteopenia in Progeria. J. Bone Miner. Res. 2018, 33, 2059–2070. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hernandez, L.; Roux, K.J.; Wong, E.S.M.; Mounkes, L.C.; Mutalif, R.; Navasankari, R.; Rai, B.; Cool, S.; Jeong, J.W.; Wang, H.; et al. Functional Coupling between the Extracellular Matrix and Nuclear Lamina by Wnt Signaling in Progeria. Dev. Cell 2010, 19, 413–425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koblan, L.W.; Erdos, M.R.; Wilson, C.; Cabral, W.A.; Levy, J.M.; Xiong, Z.M.; Tavarez, U.L.; Davison, L.M.; Gete, Y.G.; Mao, X.; et al. In Vivo Base Editing Rescues Hutchinson–Gilford Progeria Syndrome in Mice. Nature 2021, 589, 608–614. [Google Scholar] [CrossRef]
- Gaudelli, N.M.; Komor, A.C.; Rees, H.A.; Packer, M.S.; Badran, A.H.; Bryson, D.I.; Liu, D.R. Programmable Base Editing of T to G C in Genomic DNA without DNA Cleavage. Nature 2017, 551, 464–471. [Google Scholar] [CrossRef]
- Gete, Y.G.; Koblan, L.W.; Mao, X.; Trappio, M.; Mahadik, B.; Fisher, J.P.; Liu, D.R.; Cao, K. Mechanisms of Angiogenic Incompetence in Hutchinson–Gilford Progeria via Downregulation of Endothelial NOS. Aging Cell 2021, 20, e13388. [Google Scholar] [CrossRef]
- Xiong, Z.-M.; LaDana, C.; Wu, D.; Cao, K. An Inhibitory Role of Progerin in the Gene Induction Network of Adipocyte Differentiation from IPS Cells. Aging 2013, 5, 288–303. [Google Scholar] [CrossRef] [Green Version]
- Atchison, L.; Zhang, H.; Cao, K.; Truskey, G.A. A Tissue Engineered Blood Vessel Model of Hutchinson-Gilford Progeria Syndrome Using Human IPSC-Derived Smooth Muscle Cells. Sci. Rep. 2017, 7, 1–12. [Google Scholar] [CrossRef]
- Atchison, L.; Abutaleb, N.O.; Snyder-Mounts, E.; Gete, Y.; Ladha, A.; Ribar, T.; Cao, K.; Truskey, G.A. IPSC-Derived Endothelial Cells Affect Vascular Function in a Tissue-Engineered Blood Vessel Model of Hutchinson-Gilford Progeria Syndrome. Stem Cell Rep. 2020, 14, 325–337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Xiong, Z.-M.; Cao, K. Mechanisms Controlling the Smooth Muscle Cell Death in Progeria via Down-Regulation of Poly(ADP-Ribose) Polymerase 1. Proc. Natl. Acad. Sci. USA 2014, 111, E2261–E2270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kogut, I.; Roop, D.R.; Bilousova, G. Differentiation of Human Induced Pluripotent Stem Cells into a Keratinocyte Lineage. Methods Mol. Biol. 2014, 1195, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Itoh, M.; Kiuru, M.; Cairo, M.S.; Christiano, A.M. Generation of Keratinocytes from Normal and Recessive Dystrophic Epidermolysis Bullosa-Induced Pluripotent Stem Cells. Proc. Natl. Acad. Sci. USA 2011, 108, 8797–8802. [Google Scholar] [CrossRef] [Green Version]
- Jackson, B.W.; Grund, C.; Winter, S.; Franke, W.W.; Illmensee, K. Formation of Cytoskeletal Elements during Mouse Embryogenesis. II. Epithelial Differentiation and Intermediate-Sized Filaments in Early Postimplantation Embryos. Differentiation 1981, 20, 203–216. [Google Scholar] [CrossRef] [PubMed]
- Romano, R.A.; Smalley, K.; Magraw, C.; Serna, V.A.; Kurita, T.; Raghavan, S.; Sinha, S. ΔNp63 Knockout Mice Reveal Its Indispensable Role as a Master Regulator of Epithelial Development and Differentiation. Development 2012, 139, 772–782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagarajan, P.; Romano, R.A.; Sinha, S. Transcriptional Control of the Differentiation Program of Interfollicular Epidermal Keratinocytes. Crit. Rev. Eukaryot. Gene Expr. 2008, 18, 57–79. [Google Scholar] [CrossRef]
- Fuchs, E. Scratching the Surface of Skin Development. Nature 2007, 445, 834–842. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L. Keratins in Skin Epidermal Development and Diseases. In Keratin; IntechOpen: London UK, 2018. [Google Scholar] [CrossRef] [Green Version]
- Fuchs, E.; Green, H. Changes in Keratin Gene Expression during Terminal Differentiation of the Keratinocyte. Cell 1980, 19, 1033–1042. [Google Scholar] [CrossRef]
- Dechat, T.; Shimi, T.; Adam, S.A.; Rusinol, A.E.; Andres, D.A.; Spielmann, H.P.; Sinensky, M.S.; Goldman, R.D. Alterations in Mitosis and Cell Cycle Progression Caused by a Mutant Lamin A Known to Accelerate Human Aging. Proc. Natl. Acad. Sci. USA 2007, 104, 4955–4960. [Google Scholar] [CrossRef] [Green Version]
- Cao, K.; Capell, B.C.; Erdos, M.R.; Djabali, K.; Collins, F.S. A Lamin A Protein Isoform Overexpressed in Hutchinson-Gilford Progeria Syndrome Interferes with Mitosis in Progeria and Normal Cells. Proc. Natl. Acad. Sci. USA 2007, 104, 4949–4954. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, R.P.; Kuhn, S.; Yin, D.; Hotz, J.; Maher, F.A.; Robey, R.W.; Gottesman, M.M.; Horibata, S. Cross-Resistance of Cisplatin Selected Cells to Anti-Microtubule Agents: Role of General Survival Mechanisms. Transl. Oncol. 2021, 14, 100917. [Google Scholar] [CrossRef] [PubMed]
- Lim, X.; Nusse, R. Wnt Signaling in Skin Development, Homeostasis, and Disease. Cold Spring Harb. Perspect. Biol. 2013, 5, a00802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Halaschek-Wiener, J.; Brooks-Wilson, A. Progeria of Stem Cells: Stem Cell Exhaustion in Hutchinson-Gilford Progeria Syndrome. J. Gerontol. Ser. A 2007, 62, 3–8. [Google Scholar] [CrossRef] [Green Version]
- Gonzalo, S.; Kreienkamp, R. DNA Repair Defects and Genome Instability in Hutchinson-Gilford Progeria Syndrome. Curr. Opin. Cell Biol. 2015, 34, 75–83. [Google Scholar] [CrossRef] [Green Version]
- Roberts, N.; Jahangiri, M.; Xu, Q. Progenitor Cells in Vascular Disease. J. Cell. Mol. Med. 2005, 9, 583–591. [Google Scholar] [CrossRef] [Green Version]
- Caplice, N.M.; Doyle, B. Vascular Progenitor Cells: Origin and Mechanisms of Mobilization, Differentiation, Integration, and Vasculogenesis. Stem Cells Dev. 2005, 14, 122–139. [Google Scholar] [CrossRef]
- Armenteros, T.; Andreu, Z.; Hortigüela, R.; Lie, D.C.; Mira, H. BMP and WNT Signalling Cooperate through LEF1 in the Neuronal Specification of Adult Hippocampal Neural Stem and Progenitor Cells. Sci. Rep. 2018, 8, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Cao, K.; Graziotto, J.J.; Blair, C.D.; Mazzulli, J.R.; Erdos, M.R.; Krainc, D.; Collins, F.S. Rapamycin Reverses Cellular Phenotypes and Enhances Mutant Protein Clearance in Hutchinson-Gilford Progeria Syndrome Cells. Sci. Transl. Med. 2011, 3, 89ra58. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Sequence | |
|---|---|---|
| ACTB | Forward | 5′-CTGGAACGGTGAAGGTGACA-3′ |
| Reverse | 5′-AAGGGACTTCCTGTAACAATGCA-3′ | |
| LMNA | Forward | 5′-GCAACAAGTCCAATGAGGACCA-3′ |
| Reverse | 5′-CATGATGCTGCAGTTCTGGGGGCTCTGGAT-3′ | |
| Progerin | Forward | 5′-GCAACAAGTCCAATGAGGACCA-3′ |
| Reverse | 5′-CATGATGCTGCAGTTCTGGGGGCTCTGGAC-3′ | |
| K8 | Forward | 5′-CAGAAGTCCTACAAGGTGTCCA-3′ |
| Reverse | 5′-CTCTGGTTGACCGTAACTGCG-3′ | |
| K18 | Forward | 5’-GGCATCCAGAACGAGAAGGAG-3’ |
| Reverse | 5’-ATTGTCCACAGTATTTGCGAAGA-3’ | |
| ∆Np63 | Forward | 5’-ACCTGGAAAACAATGCCCAGA-3’ |
| Reverse | 5’-GGCAATCTGTCCCTCGTTGA-3’ | |
| K14 | Forward | 5’-TGAGCCGCATTCTGAACGAG-3’ |
| Reverse | 5’-GATGACTGCGATCCAGAGGA-3’ | |
| K1 | Forward | 5′-GGTGCTTATATGACCAAGGTGG-3′ |
| Reverse | 5′-ATGCTGTCCAGGTCGAGACT-3′ | |
| LEF1 | Forward | 5′-CGATGACGGAAAGCATCCAG-3′ |
| Reverse | 5′-CCACCCGGAGACAAGGGATA-3′ | |
| TCF7 | Forward | 5′-AACATTTCAACAGCCCACATCC-3′ |
| Reverse | 5′-GGAGTAGAAGCCAGAGAGGTC-3′ | |
| TCF7L1 | Forward | 5′-CCACAGTCAAGGACACGAGG-3′ |
| Reverse | 5′-TCGATCTCTGGGGAGAGGTG-3′ | |
| TCF7L2 | Forward | 5′-CCTCACGCCTCTTATCACGTA-3′ |
| Reverse | 5′-AGGCGATAGTGGGTAATACGG-3′ |
| LEF1 Binding Site | Sequence | |
|---|---|---|
| MYC | Forward | 5′-CCCAAAAAAAGGCACGGAA-3′ |
| Reverse | 5′-TATTGGAAATGCGGTCATGC-3′ | |
| K8-1 | Forward | 5′-TCCCAGGAGCCAGTCAGCGT-3′ |
| Reverse | 5′-GCCCATTCAGCCCGTGTCCC-3′ | |
| K8-2 | Forward | 5′-GCTCCCAACGGGCCAGAGGA-3′ |
| Reverse | 5′-GCCTGGGGTGCCTTGCTCAG-3′ | |
| K8-3 | Forward | 5′-ACCTTCAGACCACCCTCCCCC-3′ |
| Reverse | 5′-ACCCTGGGAAGAGGCAGCAGA-3′ | |
| K8-4 | Forward | 5′-CGTCCTGTGAACTGACCCTCCC-3′ |
| Reverse | 5′-TGTTTTCCTGCTACTGCACA-3′ | |
| K8-5 | Forward | 5′-CCCCTCCGTCATCCTGGCCT-3′ |
| Reverse | 5′-CCTGGGGCTGGCTGGTAGGA-3′ | |
| K8-6 | Forward | 5′-CTGCGCCCGGTCAAGACTCC-3′ |
| Reverse | 5′-CCGGGCATGGACACGGACAG-3′ | |
| K8-7 | Forward | 5′-CACAGACCCGGGCAGAGGGA-3′ |
| Reverse | 5′-TCTTCCAGCAGGCGGCGGTA-3′ | |
| Negative | Forward | 5′-GCCACTTGGGAGGCTGAGGC-3′ |
| Reverse | 5′-TTGAGCCACTGCACCCTGCC-3′ |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mao, X.; Xiong, Z.-M.; Xue, H.; Brown, M.A.; Gete, Y.G.; Yu, R.; Sun, L.; Cao, K. Impaired LEF1 Activation Accelerates iPSC-Derived Keratinocytes Differentiation in Hutchinson-Gilford Progeria Syndrome. Int. J. Mol. Sci. 2022, 23, 5499. https://doi.org/10.3390/ijms23105499
Mao X, Xiong Z-M, Xue H, Brown MA, Gete YG, Yu R, Sun L, Cao K. Impaired LEF1 Activation Accelerates iPSC-Derived Keratinocytes Differentiation in Hutchinson-Gilford Progeria Syndrome. International Journal of Molecular Sciences. 2022; 23(10):5499. https://doi.org/10.3390/ijms23105499
Chicago/Turabian StyleMao, Xiaojing, Zheng-Mei Xiong, Huijing Xue, Markus A. Brown, Yantenew G. Gete, Reynold Yu, Linlin Sun, and Kan Cao. 2022. "Impaired LEF1 Activation Accelerates iPSC-Derived Keratinocytes Differentiation in Hutchinson-Gilford Progeria Syndrome" International Journal of Molecular Sciences 23, no. 10: 5499. https://doi.org/10.3390/ijms23105499
APA StyleMao, X., Xiong, Z. -M., Xue, H., Brown, M. A., Gete, Y. G., Yu, R., Sun, L., & Cao, K. (2022). Impaired LEF1 Activation Accelerates iPSC-Derived Keratinocytes Differentiation in Hutchinson-Gilford Progeria Syndrome. International Journal of Molecular Sciences, 23(10), 5499. https://doi.org/10.3390/ijms23105499