
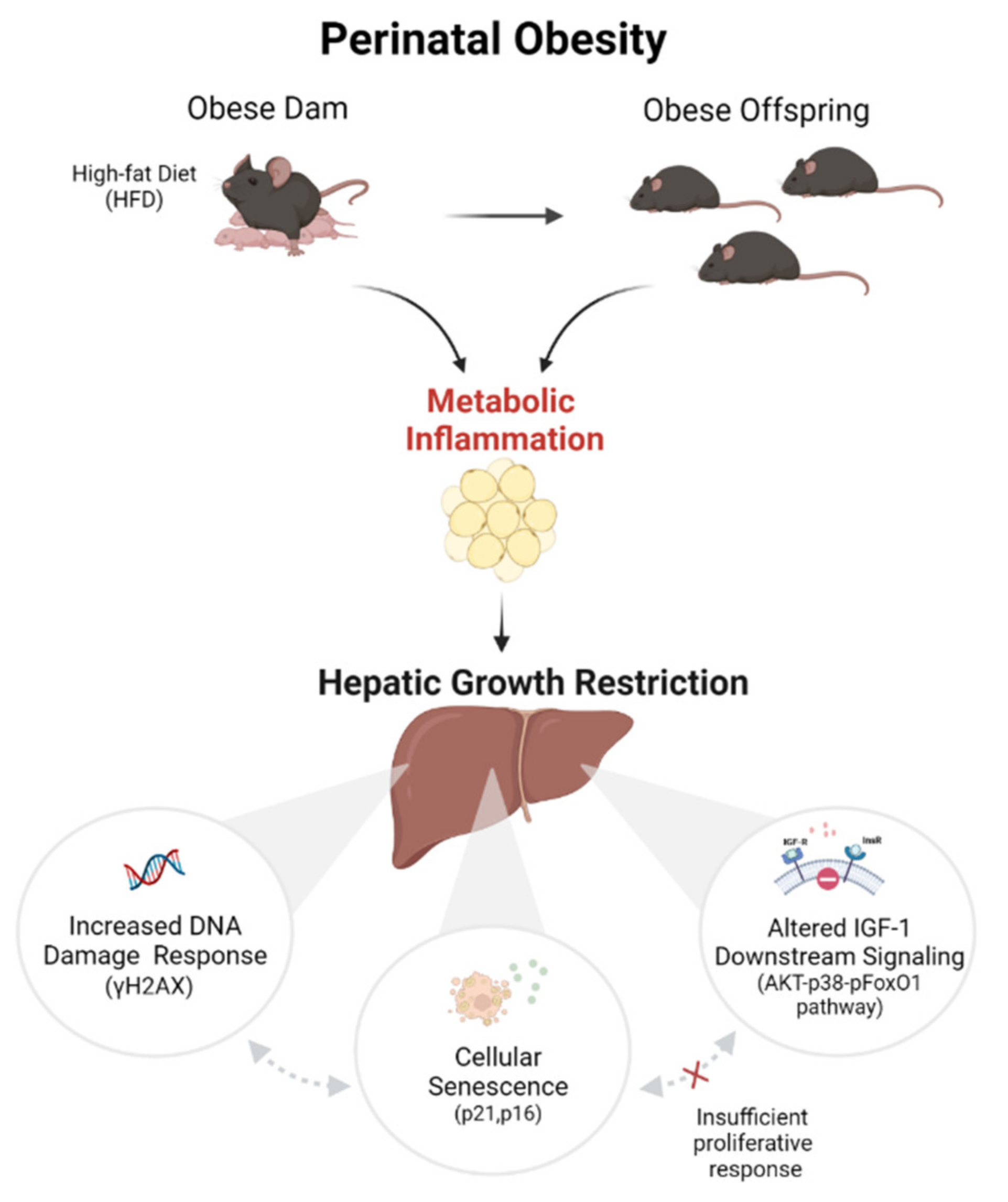
Perinatal Obesity Induces Hepatic Growth Restriction with Increased DNA Damage Response, Senescence, and Dysregulated Igf-1-Akt-Foxo1 Signaling in Male Offspring of Obese Mice

 , , , , , , , ,
, , , , , , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
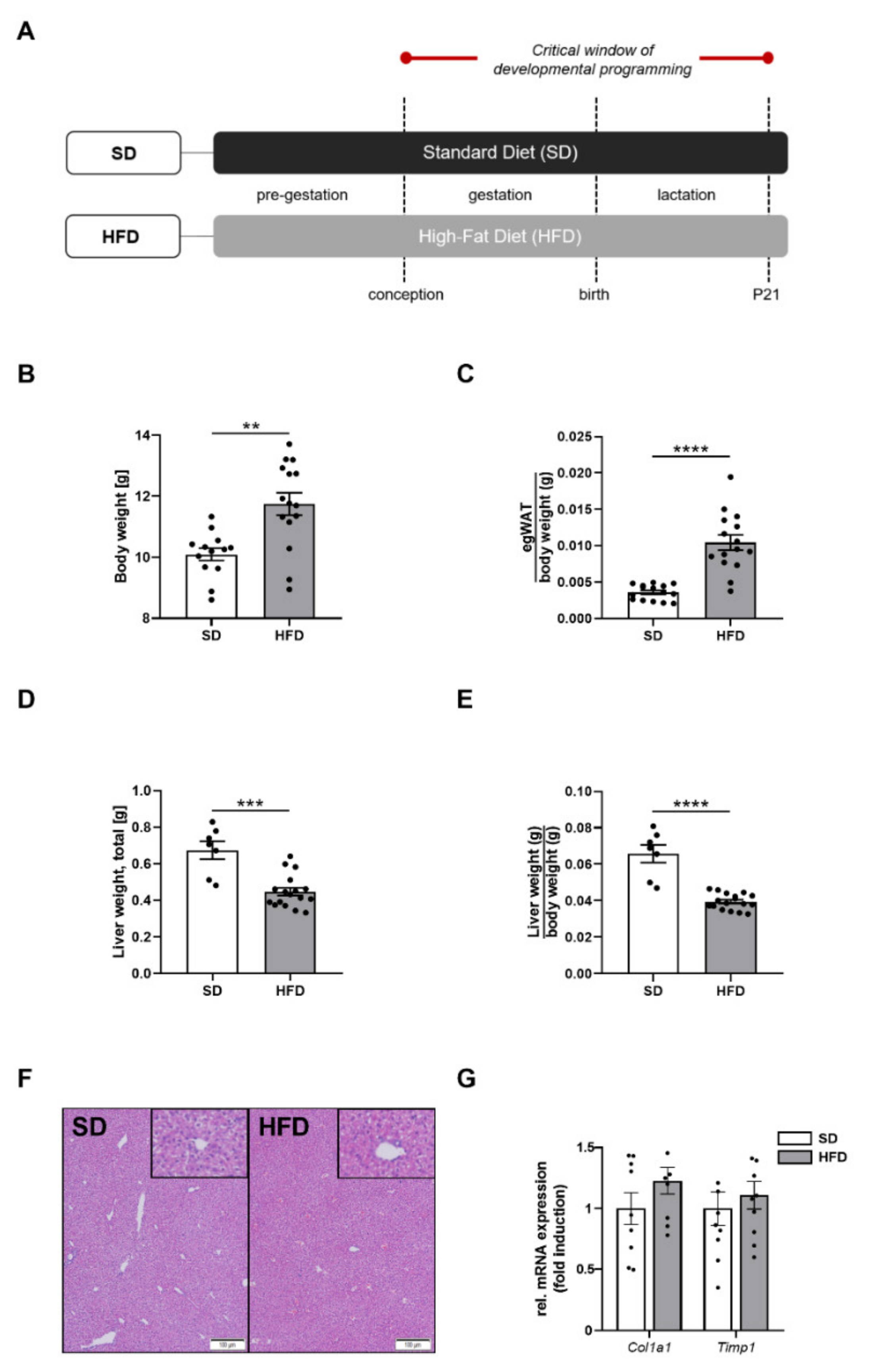
2.1. Perinatal Obesity Induces an Obese Body Composition and Hepatic Growth Restriction in the Male Offspring
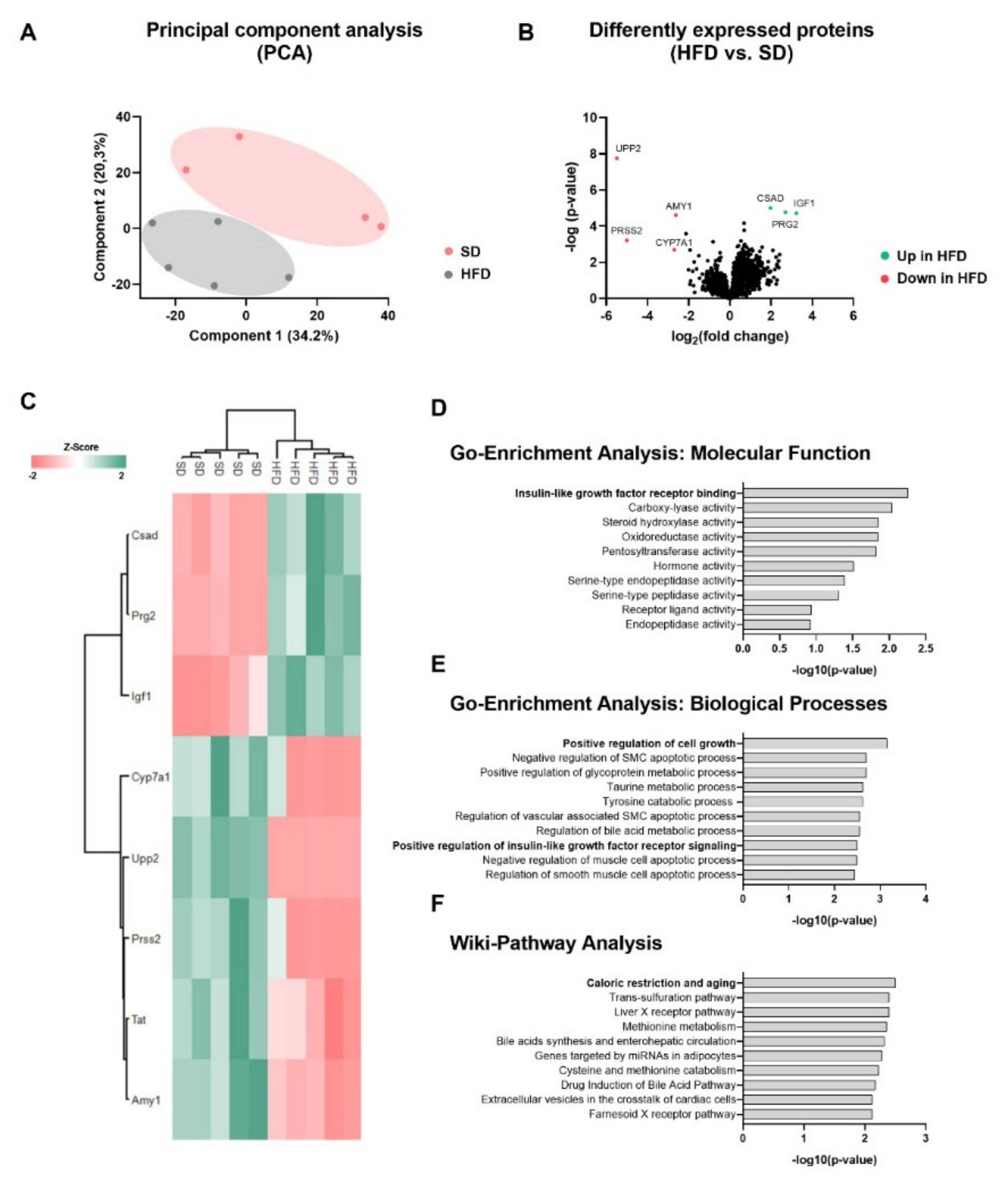
2.2. Perinatal Obesity Induces a Specific Liver Proteome Signature and Changes Hepatic Molecular Processes in the Offspring
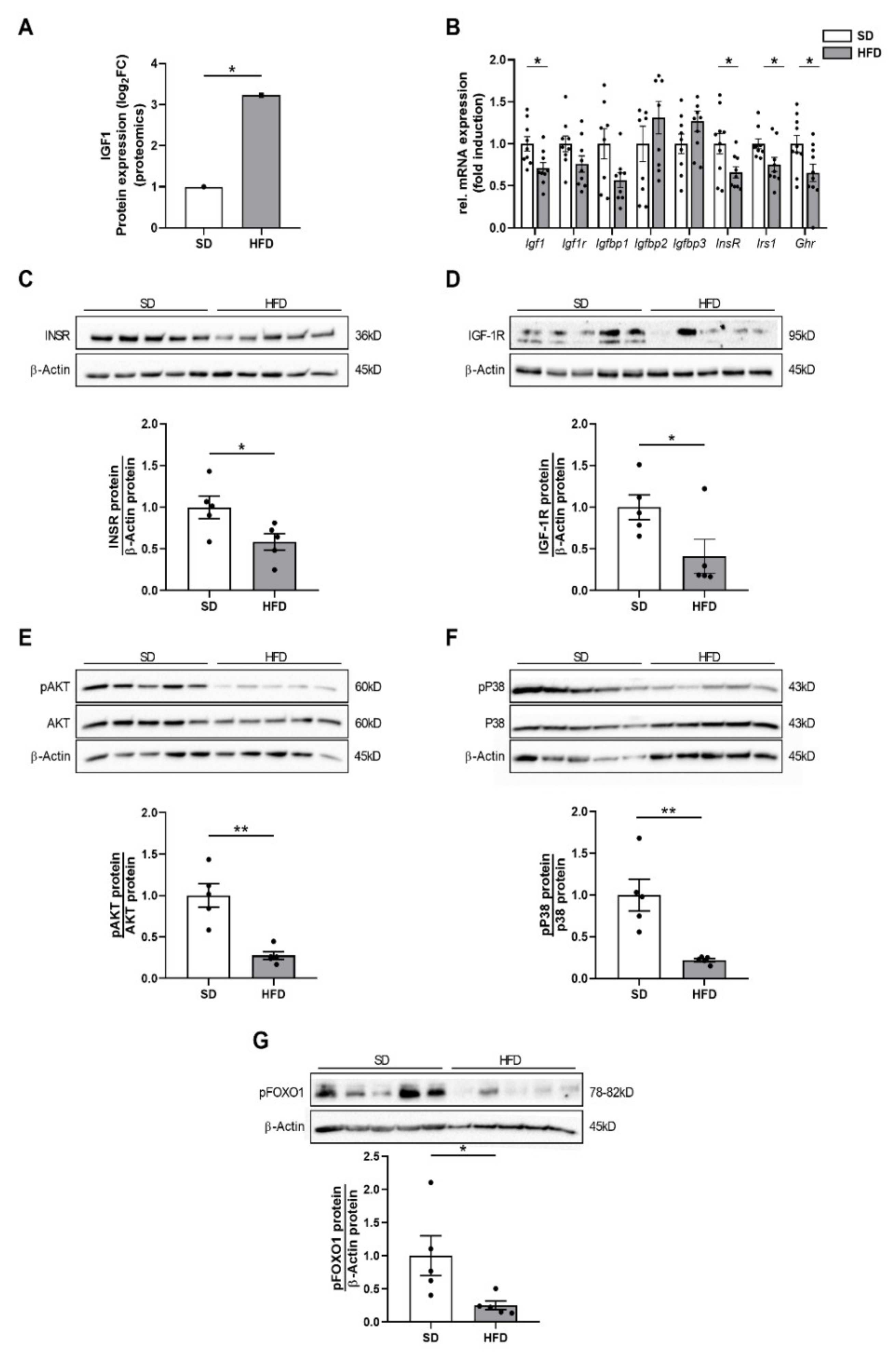
2.3. Perinatal Obesity Mediates Altered Insulin and Insulin-Like Growth Factor 1 Signaling in the Livers of the Male Offspring
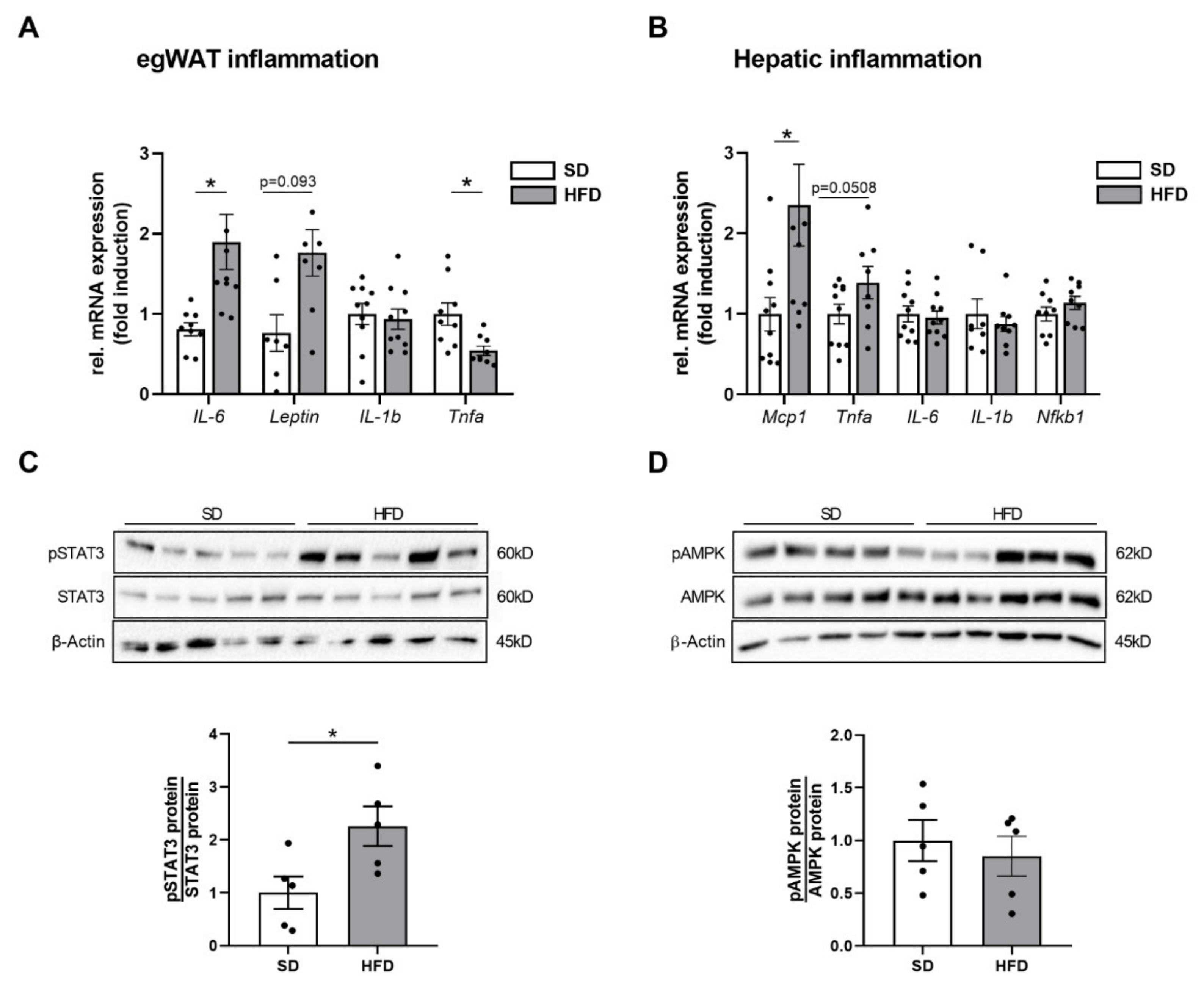
2.4. Perinatal Obesity Induces Expression of Inflammatory Markers in Egwat and Activation of Hepatic STAT3 Signaling in the Offspring
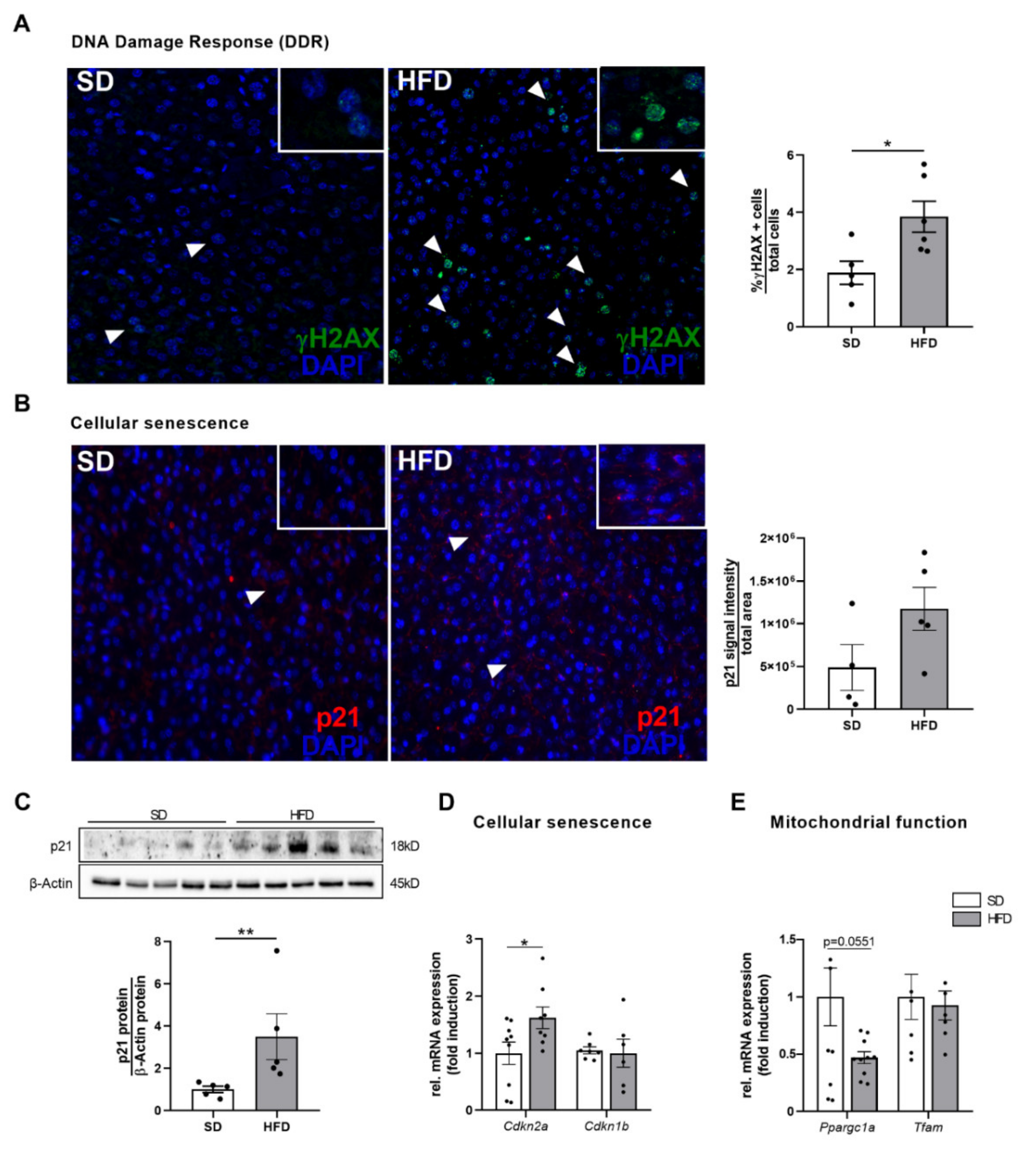
2.5. Hepatic Growth Restriction after Perinatal Obesity Is Associated with DNA Damage Response and Expression of Cellular Senescence Markers
2.6. Hepatic Growth Restriction in HFD Offspring of Obese Dams Is Related to Markers of Increased Cell Proliferation
3. Discussion
- (1)
- DNA damage response in the liver after perinatal obesity
- (2)
- Cellular senescence in the liver after perinatal obesity
- (3)
- Dysregulated insulin/IGF-1-AKT-pFoxO1 signaling in the liver after perinatal obesity
- (4)
- Activation of an inflammatory egWAT- liver axis after perinatal obesity might trigger aging processes
Limitations of the Study
4. Materials and Methods
4.1. Animal Procedures
Animal Studies
4.2. Tissue Preparation
4.3. Histological Analysis of the Liver Tissue
4.4. RNA Extraction and Real-Time Reverse Transcription Polymerase Chain Reaction (RT-PCR)
4.5. Protein Isolation and Immunoblotting
4.6. Proteomic Analysis
4.7. Immunofluorescent Staining
4.8. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Abarca-Gómez, L.; Abdeen, Z.; Hamid, Z.; Abu-Rmeileh, N.M.; Acosta-Cazares, B.; Acuin, C.; Adams, R.J.; Aekplakorn, W.; Afsana, K.; Aguilar-Salinas, C.A.; et al. Worldwide trends in body-mass index, underweight, overweight, and obesity from 1975 to 2016: A pooled analysis of 2416 population-based measurement studies in 128·9 million children, adolescents, and adults. Lancet 2017, 390, 2627–2642. [Google Scholar] [CrossRef] [Green Version]
- Blüher, M. Obesity: Global epidemiology and pathogenesis. Nat. Rev. Endocrinol. 2019, 15, 288–298. [Google Scholar] [CrossRef] [PubMed]
- Poston, L.; Caleyachetty, R.; Cnattingius, S.; Corvalán, C.; Uauy, R.; Herring, S.; Gillman, M.W. Preconceptional and maternal obesity: Epidemiology and health consequences. Lancet Diabetes Endocrinol. 2016, 4, 1025–1036. [Google Scholar] [CrossRef]
- Heslehurst, N.; Rankin, J.; Wilkinson, J.R.; Summerbell, C.D. A nationally representative study of maternal obesity in England, UK: Trends in incidence and demographic inequalities in 619 323 births 1989. Int. J. Obes. 2010, 34, 420–428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Catalano, P.M.; Shankar, K. Obesity and pregnancy: Mechanisms of short term and long term adverse consequences for mother and child. BMJ 2017, 356, 1–16. [Google Scholar] [CrossRef]
- Butler, E.A.; Cohen, E.; Berger, H.; Ray, J.G. Change in Pre-Pregnancy Body Mass Index in Relation to COVID-19 Pandemic. J. Obs. Gynaecol. Can. 2021, 21, 750–757. [Google Scholar] [CrossRef]
- Godfrey, K.; Reynolds, R.; Prescott, S.; Nyirenda, M.; Jaddoe, V.W.; Eriksson, J.G.; Broekman, B.F. Influence of maternal obesity on the long-term health of offspring. Lancet Diabetes Endocrinol. 2017, 5, 53–64. [Google Scholar] [CrossRef] [Green Version]
- Shrestha, N.; Ezechukwu, H.C.; Holland, O.J.; Hryciw, D.H. Developmental programming of peripheral diseases in offspring exposed to maternal obesity during pregnancy. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2020, 319, R507–R516. [Google Scholar] [CrossRef]
- Schoonejans, J.M.; Ozanne, S.E. Developmental programming by maternal obesity: Lessons from animal models. Diabet. Med. 2021, 38, e14694. [Google Scholar] [CrossRef]
- Barker, D.J.P. The origins of the developmental origins theory. J. Intern. Med. 2007, 261, 412–417. [Google Scholar] [CrossRef]
- Kuiper-Makris, C.; Selle, J.; Nüsken, E.; Dötsch, J.; Alejandre Alcazar, M.A. Perinatal Nutritional and Metabolic Pathways: Early Origins of Chronic Lung Diseases. Front. Med. 2021, 8, 667315. [Google Scholar] [CrossRef]
- Lumbers, E.R.; Kandasamy, Y.; Delforce, S.J.; Boyce, A.C.; Gibson, K.J.; Pringle, K.G. Programming of Renal Development and Chronic Disease in Adult Life. Front. Physiol. 2020, 11, 757. [Google Scholar] [CrossRef]
- Wesolowski, S.; El Kasmi, K.; Jonscher, K.; Friedman, J. Developmental origins of NAFLD: A womb with a clue. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 81–96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scheja, L.; Heeren, J. The endocrine function of adipose tissues in health and cardiometabolic disease. Nat. Rev. Endocrinol. 2019, 15, 507–524. [Google Scholar] [CrossRef]
- Mau, T.; Yung, R. Adipose tissue inflammation in aging. Exp. Gerontol. 2018, 105, 27–31. [Google Scholar] [CrossRef]
- Kasper, P.; Breuer, S.; Hoffmann, T.; Vohlen, C.; Janoschek, R.; Schmitz, L.; Appel, S.; Fink, G.; Hünseler, C.; Quaas, A.; et al. Maternal Exercise Mediates Hepatic Metabolic Programming via Activation of AMPK-PGC1α Axis in the Offspring of Obese Mothers. Cells 2021, 10, 1247. [Google Scholar] [CrossRef] [PubMed]
- Yakar, S.; Adamo, M.L. Insulin-like growth factor 1 physiology: Lessons from mouse models. Endocrinol. Metab. Clin. North. Am. 2012, 41, 231–247. [Google Scholar] [CrossRef] [Green Version]
- Adamek, A.; Kasprzak, A. Insulin-Like Growth Factor (IGF) System in Liver Diseases. Int. J. Mol. Sci. 2018, 19, 1308. [Google Scholar] [CrossRef] [Green Version]
- Kasper, P.; Vohlen, C.; Dinger, K.; Mohr, J.; Hucklenbruch-Rother, E.; Janoschek, R.; Köth, J.; Matthes, J.; Appel, S.; Dötsch, J.; et al. Renal metabolic programming is linked to the dynamic regulation of a Leptin-Klf15 axis and Akt/AMPKα signaling in male offspring of obese dams. Endocrinology 2017, 158, 3399–3415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aguirre, G.A.; De Ita, J.R.; de la Garza, R.G.; Castilla-Cortazar, I. Insulin-like growth factor-1 deficiency and metabolic syndrome. J. Transl. Med. 2016, 14, 3. [Google Scholar] [CrossRef] [Green Version]
- Schmidt-Arras, D.; Rose-John, S. IL-6 pathway in the liver: From physiopathology to therapy. J. Hepatol. 2016, 64, 1403–1415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varela-Rey, M.; Beraza, N.; Lu, S.C.; Mato, J.M.; Martínez-Chantar, M.L. Role of AMP-activated protein kinase in the control of hepatocyte priming and proliferation during liver regeneration. Exp. Biol. Med. 2011, 236, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Kim, I.H.; Kisseleva, T.; Brenner, D.A. Aging and liver disease. Curr. Opin. Gastroenterol. 2015, 31, 184–191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, A.; Singh, K.; Almasan, A. Histone H2AX phosphorylation: A marker for DNA damage. Methods Mol. Biol. 2012, 920, 613–626. [Google Scholar]
- Wang, C.; Jurk, D.; Maddick, M.; Nelson, G.; Martin-Ruiz, C.; von Zglinicki, T. DNA damage response and cellular senescence in tissues of aging mice. Aging Cell 2009, 8, 311–323. [Google Scholar] [CrossRef]
- Pestell, R.G. New roles of cyclin D1. Am. J. Pathol. 2013, 183, 3–9. [Google Scholar] [CrossRef] [Green Version]
- Sun, B.; Karin, M. NF-kappaB signaling, liver disease and hepatoprotective agents. Oncogene 2008, 27, 6228–6244. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Liu, X.; Wang, H.; Liu, S.; Hu, N.; Li, X. Akt Regulated Phosphorylation of GSK-3β/Cyclin D1, p21 and p27 Contributes to Cell Proliferation Through Cell Cycle Progression From G1 to S/G2M Phase in Low-Dose Arsenite Exposed HaCat Cells. Front. Pharmacol. 2019, 10, 1176. [Google Scholar] [CrossRef] [Green Version]
- Böhm, F.; Köhler, U.A.; Speicher, T.; Werner, S. Regulation of liver regeneration by growth factors and cytokines. EMBO Mol. Med. 2010, 2, 294–305. [Google Scholar] [CrossRef]
- Moh, A.; Iwamoto, Y.; Chai, G.-X.; Zhang, S.S.M.; Kano, A.; Yang, D.D.; Zhang, W.; Wang, J.; Jacoby, J.J.; Gao, B.; et al. Role of STAT3 in liver regeneration: Survival, DNA synthesis, inflammatory reaction and liver mass recovery. Lab. Investig. 2007, 87, 1018–1028. [Google Scholar] [CrossRef] [Green Version]
- Dinger, K.; Koningsbruggen-Rietschel, S.V.; Dötsch, J.; Alejandre Alcazar, M.A. Identification of Critical Windows of Metabolic Programming of Metabolism and Lung Function in Male Offspring of Obese Dams. Clin. Transl. Sci. 2020, 13, 1065–1070. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.-M.; Zhang, X.-Y.; Lu, X.-Y.; Yu, S.R.; Wang, X.; Zou, Y.; Zuo, Z.Y.; Liu, D.Y.; Zhou, B.G. Erzhi Pill(®) Protected Experimental Liver Injury Against Apoptosis via the PI3K/Akt/Raptor/Rictor Pathway. Front. Pharmacol. 2018, 9, 283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mortensen, O.H.; Olsen, H.L.; Frandsen, L.; Nielsen, P.E.; Nielsen, F.C.; Grunnet, N.; Quistorff, B. Gestational protein restriction in mice has pronounced effects on gene expression in newborn offspring’s liver and skeletal muscle; protective effect of taurine. Pediatr. Res. 2010, 67, 47–53. [Google Scholar] [CrossRef] [Green Version]
- Howell, K.R.; Powell, T.L. Effects of maternal obesity on placental function and fetal development. Reproduction 2017, 153, R97–R108. [Google Scholar] [CrossRef] [PubMed]
- Lewandowska, M. Maternal Obesity and Risk of Low Birth Weight, Fetal Growth Restriction, and Macrosomia: Multiple Analyses. Nutrients 2021, 13, 1213. [Google Scholar] [CrossRef] [PubMed]
- Guzzardi, M.A.; Liistro, T.; Gargani, L.; Ait Ali, L.; D’Angelo, G.; Rocchiccioli, S.; La Rosa, F.; Kemeny, A.; Sanguinetti, E.; Ucciferri, N.; et al. Maternal Obesity and Cardiac Development in the Offspring: Study in Human Neonates and Minipigs. JACC Cardiovasc. Imaging 2018, 11, 1750–1755. [Google Scholar] [CrossRef]
- Moritz, K.M.; Wintour, E.M.; Black, M.J.; Bertram, J.F.; Caruana, G. Factors influencing mammalian kidney development: Implications for health in adult life. Adv. Anat. Embryol. Cell Biol. 2008, 196, 1–78. [Google Scholar]
- Lessard-Beaudoin, M.; Laroche, M.; Demers, M.-J.; Grenier, G.; Graham, R.K. Characterization of age-associated changes in peripheral organ and brain region weights in C57BL/6 mice. Exp. Gerontol. 2015, 63, 27–34. [Google Scholar] [CrossRef]
- Dollé, M.E.T.; Kuiper, R.V.; Roodbergen, M.; Robinson, J.; de Vlugt, S.; Wijnhoven, S.W.; Beems, R.B.; de la Fonteyne, L.; de With, P.; van der Pluijm, I.; et al. Broad segmental progeroid changes in short-lived Ercc1(-/Δ7) mice. Pathobiol. Aging Age Relat. Dis. 2011, 1, 7219. [Google Scholar] [CrossRef] [Green Version]
- White, R.R.; Milholland, B.; de Bruin, A.; Curran, S.; Laberge, R.M.; Van Steeg, H.; Campisi, J.; Maslov, A.Y.; Vijg, J. Controlled induction of DNA double-strand breaks in the mouse liver induces features of tissue ageing. Nat. Commun. 2015, 6, 6790. [Google Scholar] [CrossRef] [Green Version]
- Włodarczyk, M.; Nowicka, G. Obesity, DNA Damage, and Development of Obesity-Related Diseases. Int. J. Mol. Sci. 2019, 20, 1146. [Google Scholar] [CrossRef] [PubMed]
- Bankoglu, E.E.; Tschopp, O.; Schmitt, J.; Burkard, P.; Jahn, D.; Geier, A.; Stopper, H. Role of PTEN in Oxidative Stress and DNA Damage in the Liver of Whole-Body Pten Haplodeficient Mice. PLoS ONE 2016, 11, e0166956. [Google Scholar] [CrossRef] [PubMed]
- Di Micco, R.; Krizhanovsky, V.; Baker, D.; d’Adda di Fagagna, F. Cellular senescence in ageing: From mechanisms to therapeutic opportunities. Nat. Rev. Mol. Cell Biol. 2021, 22, 75–95. [Google Scholar] [CrossRef]
- Ferreira-Gonzalez, S.; Rodrigo-Torres, D.; Gadd, V.L.; Forbes, S.J. Cellular Senescence in Liver Disease and Regeneration. Semin. Liver Dis. 2021, 41, 50–66. [Google Scholar] [CrossRef] [PubMed]
- Engelmann, C.; Tacke, F. The Potential Role of Cellular Senescence in Non-Alcoholic Fatty Liver Disease. Int. J. Mol. Sci. 2022, 23, 652. [Google Scholar] [CrossRef]
- Ogrodnik, M.; Miwa, S.; Tchkonia, T.; Tiniakos, D.; Wilson, C.L.; Lahat, A.; Day, C.P.; Burt, A.; Palmer, A.; Anstee, Q.M.; et al. Cellular senescence drives age-dependent hepatic steatosis. Nat. Commun. 2017, 8, 15691. [Google Scholar] [CrossRef]
- Sell, C. Minireview: The Complexities of IGF/Insulin Signaling in Aging: Why Flies and Worms Are Not Humans. Mol. Endocrinol. 2015, 29, 1107–1113. [Google Scholar] [CrossRef] [Green Version]
- Beyer, T.A.; Xu, W.; Teupser, D.; Auf dem Keller, U.; Bugnon, P.; Hildt, E.; Thiery, J.; Kan, Y.W.; Werner, S. Impaired liver regeneration in Nrf2 knockout mice: Role of ROS-mediated insulin/IGF-1 resistance. EMBO J. 2008, 27, 212–223. [Google Scholar] [CrossRef]
- Desbois-Mouthon, C.; Wendum, D.; Cadoret, A.; Rey, C.; Leneuve, P.; Blaise, A.; Housset, C.; Tronche, F.; Le Bouc, Y.; Holzenberger, M. Hepatocyte proliferation during liver regeneration is impaired in mice with liver-specific IGF-1R knockout. FASEB J. 2006, 20, 773–775. [Google Scholar] [CrossRef] [Green Version]
- Morales-Garza, L.A.; Puche, J.E.; Aguirre, G.A.; Muñoz, Ú.; García-Magariño, M.; De la Garza, R.G.; Castilla-Cortazar, I. Experimental approach to IGF-1 therapy in CCl(4)-induced acute liver damage in healthy controls and mice with partial IGF-1 deficiency. J. Transl. Med. 2017, 15, 96. [Google Scholar] [CrossRef] [Green Version]
- Tosh, D.N.; Fu, Q.; Callaway, C.W.; McKnight, R.A.; McMillen, I.C.; Ross, M.G.; Lane, R.H.; Desai, M. Epigenetics of programmed obesity: Alteration in IUGR rat hepatic IGF1 mRNA expression and histone structure in rapid vs. delayed postnatal catch-up growth. Am. J. Physiol. Gastrointest. Liver Physiol. 2010, 299, G1023–G1029. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Connor, J.C.; McCusker, R.H.; Strle, K.; Johnson, R.W.; Dantzer, R.; Kelley, K.W. Regulation of IGF-I function by proinflammatory cytokines: At the interface of immunology and endocrinology. Cell Immunol. 2008, 252, 91–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Mansoori, L.; Al-Jaber, H.; Prince, M.S.; Elrayess, M.A. Role of Inflammatory Cytokines, Growth Factors and Adipokines in Adipogenesis and Insulin Resistance. Inflammation 2022, 45, 31–44. [Google Scholar] [CrossRef] [PubMed]
- Dinger, K.; Kasper, P.; Hucklenbruch-Rother, E.; Vohlen, C.; Jobst, E.; Janoschek, R.; Bae-Gartz, I.; van Koningsbruggen-Rietschel, S.; Plank, C.; Dötsch, J.; et al. Early-onset obesity dysregulates pulmonary adipocytokine/insulin signaling and induces asthma-like disease in mice. Sci. Rep. 2016, 6, 24168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Litzenburger, T.; Huber, E.-K.; Dinger, K.; Wilke, R.; Vohlen, C.; Selle, J.; Kadah, M.; Persigehl, T.; Heneweer, C.; Dötsch, J.; et al. Maternal high-fat diet induces long-term obesity with sex-dependent metabolic programming of adipocyte differentiation, hypertrophy and dysfunction in the offspring. Clin. Sci. 2020, 134, 921–939. [Google Scholar] [CrossRef]
- Bae-Gartz, I.; Kasper, P.; Großmann, N.; Breuer, S.; Janoschek, R.; Kretschmer, T.; Appel, S.; Schmitz, L.; Vohlen, C.; Quaas, A.; et al. Maternal exercise conveys protection against NAFLD in the offspring via hepatic metabolic programming. Sci. Rep. 2020, 10, 15424. [Google Scholar] [CrossRef]
- Bruce, K.D.; Cagampang, F.R.; Argenton, M.; Zhang, J.; Ethirajan, P.L.; Burdge, G.C.; Bateman, A.C.; Clough, G.F.; Poston, L.; Hanson, M.A.; et al. Maternal high-fat feeding primes steatohepatitis in adult mice offspring, involving mitochondrial dysfunction and altered lipogenesis gene expression. Hepatology 2009, 50, 1796–1808. [Google Scholar] [CrossRef]
- Breuer, S.; Kasper, P.; Vohlen, C.; Janoschek, R.; Hoffmann, T.; Appel, S.; Müller-Limberger, E.; Mesaros, A.; Rose-John, S.; Garbers, C.; et al. Brain-Restricted Inhibition of IL-6 Trans-Signaling Mildly Affects Metabolic Consequences of Maternal Obesity in Male Offspring. Nutrients 2021, 13, 3735. [Google Scholar] [CrossRef]
- Demichev, V.; Messner, C.B.; Vernardis, S.I.; Lilley, K.S.; Ralser, M. DIA-NN: Neural networks and interference correction enable deep proteome coverage in high throughput. Nat. Methods 2020, 17, 41–44. [Google Scholar] [CrossRef]
- Hirani, D.; Alvira, C.M.; Danopoulos, S.; Milla, C.; Donato, M.; Tian, L.; Mohr, J.; Dinger, K.; Vohlen, C.; Selle, J.; et al. Macrophage-derived IL-6 trans-signaling as a novel target in the pathogenesis of bronchopulmonary dysplasia. Eur. Respir. J. 2021, 59, 2002248. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kasper, P.; Selle, J.; Vohlen, C.; Wilke, R.; Kuiper-Makris, C.; Klymenko, O.; Bae-Gartz, I.; Schömig, C.; Quaas, A.; Schumacher, B.; et al. Perinatal Obesity Induces Hepatic Growth Restriction with Increased DNA Damage Response, Senescence, and Dysregulated Igf-1-Akt-Foxo1 Signaling in Male Offspring of Obese Mice. Int. J. Mol. Sci. 2022, 23, 5609. https://doi.org/10.3390/ijms23105609
Kasper P, Selle J, Vohlen C, Wilke R, Kuiper-Makris C, Klymenko O, Bae-Gartz I, Schömig C, Quaas A, Schumacher B, et al. Perinatal Obesity Induces Hepatic Growth Restriction with Increased DNA Damage Response, Senescence, and Dysregulated Igf-1-Akt-Foxo1 Signaling in Male Offspring of Obese Mice. International Journal of Molecular Sciences. 2022; 23(10):5609. https://doi.org/10.3390/ijms23105609
Chicago/Turabian StyleKasper, Philipp, Jaco Selle, Christina Vohlen, Rebecca Wilke, Celien Kuiper-Makris, Oleksiy Klymenko, Inga Bae-Gartz, Charlotte Schömig, Alexander Quaas, Björn Schumacher, and et al. 2022. "Perinatal Obesity Induces Hepatic Growth Restriction with Increased DNA Damage Response, Senescence, and Dysregulated Igf-1-Akt-Foxo1 Signaling in Male Offspring of Obese Mice" International Journal of Molecular Sciences 23, no. 10: 5609. https://doi.org/10.3390/ijms23105609
APA StyleKasper, P., Selle, J., Vohlen, C., Wilke, R., Kuiper-Makris, C., Klymenko, O., Bae-Gartz, I., Schömig, C., Quaas, A., Schumacher, B., Demir, M., Bürger, M., Lang, S., Martin, A., Steffen, H. -M., Goeser, T., Dötsch, J., & Alcazar, M. A. A. (2022). Perinatal Obesity Induces Hepatic Growth Restriction with Increased DNA Damage Response, Senescence, and Dysregulated Igf-1-Akt-Foxo1 Signaling in Male Offspring of Obese Mice. International Journal of Molecular Sciences, 23(10), 5609. https://doi.org/10.3390/ijms23105609