
Vitamin K-Dependent Protein Activation: Normal Gamma-Glutamyl Carboxylation and Disruption in Disease


{kind=link}
{kind=link}
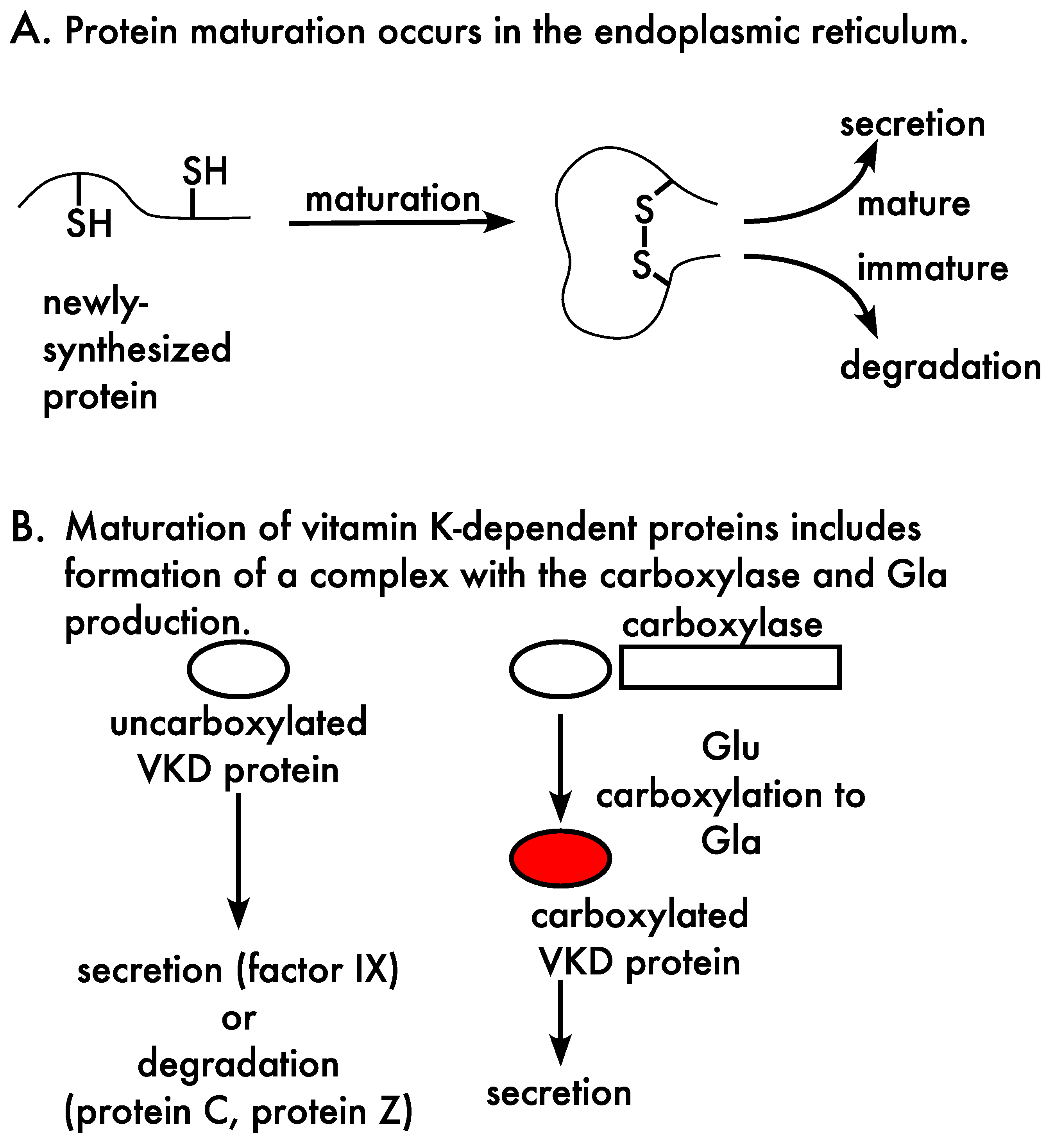{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Vitamin K-Dependent Proteins Are Essential to Human Health
3. Multiple Proteins Cooperate in Activating VKD Proteins during Secretion
3.1. Carboxylation, Secretion and Quality Control
3.2. The Carboxylation Reaction
3.3. The Gamma-Glutamyl Carboxylase
3.4. Vitamin K Oxidoreductases (VKORs)

4. Multiple Gla Residues Are Required for VKD Protein Function
5. Disruption of Vitamin K-Dependent Protein Carboxylation in Disease
6. Approaches for Analyzing VKD Protein Carboxylation
Funding
Conflicts of Interest
References
- Berkner, K.L. The vitamin K–dependent carboxylase. Annu. Rev. Nutr. 2005, 25, 127–149. [Google Scholar] [CrossRef] [PubMed]
- Berkner, K.L. Vitamin K-dependent carboxylation. Vitam. Horm. 2008, 78, 131–156. [Google Scholar] [PubMed]
- Berkner, K.L.; Runge, K.W. The physiology of vitamin K nutriture and vitamin K-dependent protein function in atherosclerosis. J. Thromb. Haemost. 2004, 2, 2118–2132. [Google Scholar] [CrossRef] [PubMed]
- Chatrou, M.L.; Winckers, K.; Hackeng, T.M.; Reutelingsperger, C.P.; Schurgers, L.J. Vascular calcification: The price to pay for anticoagulation therapy with vitamin K-antagonists. Blood Rev. 2012, 26, 155–166. [Google Scholar] [CrossRef] [PubMed]
- Holden, R.M.; Sanfilippo, A.S.; Hopman, W.M.; Zimmerman, D.; Garland, J.S.; Morton, A.R. Warfarin and aortic valve calcification in hemodialysis patients. J. Nephrol. 2007, 20, 417–422. [Google Scholar]
- Lerner, R.G.; Aronow, W.S.; Sekhri, A.; Palaniswamy, C.; Ahn, C.; Singh, T.; Sandhu, R.; McClung, J.A. Warfarin use and the risk of valvular calcification. J. Thromb. Haemost. 2009, 7, 2023–2027. [Google Scholar] [CrossRef]
- Palaniswamy, C.; Sekhri, A.; Aronow, W.S.; Kalra, A.; Peterson, S.J. Association of warfarin use with valvular and vascular calcification: A review. Clin. Cardiol. 2011, 34, 74–81. [Google Scholar] [CrossRef]
- Villines, T.C.; O’Malley, P.G.; Feuerstein, I.M.; Thomas, S.; Taylor, A.J. Does prolonged warfarin exposure potentiate coronary calcification in humans? Results of the warfarin and coronary calcification study. Calcif. Tissue Int. 2009, 85, 494–500. [Google Scholar] [CrossRef]
- Li, Q.; Grange, D.K.; Armstrong, N.L.; Whelan, A.J.; Hurley, M.Y.; Rishavy, M.A.; Hallgren, K.W.; Berkner, K.L.; Schurgers, L.J.; Jiang, Q.; et al. Mutations in the GGCX and ABCC6 genes in a family with pseudoxanthoma elasticum-like phenotypes. J. Investig. Dermatol. 2009, 129, 553–563. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Jiang, Q.; Uitto, J. Ectopic mineralization disorders of the extracellular matrix of connective tissue: Molecular genetics and pathomechanisms of aberrant calcification. Matrix Biol. 2014, 33, 23–28. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Schurgers, L.J.; Smith, A.C.; Tsokos, M.; Uitto, J.; Cowen, E.W. Co-existent pseudoxanthoma elasticum and vitamin K-dependent coagulation factor deficiency: Compound heterozygosity for mutations in the GGCX gene. Am. J. Pathol. 2009, 174, 534–540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Q.; van de Wetering, K.; Uitto, J. Pseudoxanthoma Elasticum as a Paradigm of Heritable Ectopic Mineralization Disorders: Pathomechanisms and Treatment Development. Am. J. Pathol. 2019, 189, 216–225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vanakker, O.M.; Martin, L.; Gheduzzi, D.; Leroy, B.P.; Loeys, B.L.; Guerci, V.I.; Matthys, D.; Terry, S.F.; Coucke, P.J.; Pasquali-Ronchetti, I.; et al. Pseudoxanthoma elasticum-like phenotype with cutis laxa and multiple coagulation factor deficiency represents a separate genetic entity. J. Investig. Dermatol. 2007, 127, 581–587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, B.; Ginsburg, D. Familial multiple coagulation factor deficiencies: New biologic insight from rare genetic bleeding disorders. J. Thromb. Haemost. 2004, 2, 1564–1572. [Google Scholar] [CrossRef]
- Rishavy, M.A.; Hallgren, K.W.; Runge, K.W.; Berkner, K.L. Compound heterozygosity of the gamma-glutamyl carboxylase mutants V255M and S300F causes pseudoxanthoma elasticum-like disease through impaired processivity. bioRxiv. 2019, 668574. [Google Scholar] [CrossRef]
- Stenina, O.; Pudota, B.N.; McNally, B.A.; Hommema, E.L.; Berkner, K.L. Tethered processivity of the vitamin K-dependent carboxylase: Factor IX is efficiently modified in a mechanism which distinguishes Gla’s from Glu’s and which accounts for comprehensive carboxylation in vivo. Biochemistry 2001, 40, 10301–10309. [Google Scholar] [CrossRef]
- Furie, B.; Furie, B.C. Molecular basis of vitamin K-dependent gamma-carboxylation. Blood 1990, 75, 1753–1762. [Google Scholar] [CrossRef] [Green Version]
- Coughlin, S.R. Thrombin signalling and protease-activated receptors. Nature 2000, 407, 258–264. [Google Scholar] [CrossRef]
- Law, L.A.; Graham, D.K.; Di Paola, J.; Branchford, B.R. GAS6/TAM Pathway Signaling in Hemostasis and Thrombosis. Front. Med. 2018, 5, 137. [Google Scholar] [CrossRef] [Green Version]
- van der Meer, J.H.; van der Poll, T.; van ‘t Veer, C. TAM receptors, Gas6, and protein S: Roles in inflammation and hemostasis. Blood 2014, 123, 2460–2469. [Google Scholar] [CrossRef]
- Aghourian, M.N.; Lemarie, C.A.; Bertin, F.R.; Blostein, M.D. Prostaglandin E synthase is upregulated by Gas6 during cancer-induced venous thrombosis. Blood 2016, 127, 769–777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dahlback, B. Vitamin K–Dependent Protein S: Beyond the Protein C Pathway. Semin. Thromb. Hemost. 2018, 44, 176–184. [Google Scholar] [CrossRef] [PubMed]
- Hafizi, S.; Dahlback, B. Gas6 and protein S. Vitamin K-dependent ligands for the Axl receptor tyrosine kinase subfamily. FEBS J. 2006, 273, 5231–5244. [Google Scholar] [CrossRef] [PubMed]
- Happonen, K.E.; Tran, S.; Mörgelin, M.; Prince, R.; Calzavarini, S.; Angelillo-Scherrer, A.; Dahlbäck, B. The Gas6-Axl Protein Interaction Mediates Endothelial Uptake of Platelet Microparticles. J. Biol. Chem. 2016, 291, 10586–10601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mosnier, L.O.; Zlokovic, B.V.; Griffin, J.H. The cytoprotective protein C pathway. Blood 2007, 109, 3161–3172. [Google Scholar] [CrossRef]
- Robins, R.S.; Lemarie, C.A.; Laurance, S.; Aghourian, M.N.; Wu, J.; Blostein, M.D. Vascular Gas6 contributes to thrombogenesis and promotes tissue factor up-regulation after vessel injury in mice. Blood 2013, 121, 692–699. [Google Scholar] [CrossRef] [Green Version]
- Luo, G.; Ducy, P.; McKee, M.D.; Pinero, G.J.; Loyer, E.; Behringer, R.R.; Karsenty, G. Spontaneous calcification of arteries and cartilage in mice lacking matrix GLA protein. Nature 1997, 386, 78–81. [Google Scholar] [CrossRef]
- Price, P.A.; Faus, S.A.; Williamson, M.K. Warfarin causes rapid calcification of the elastic lamellae in rat arteries and heart valves. Arterioscler. Thromb. Vasc. Biol. 1998, 18, 1400–1407. [Google Scholar] [CrossRef] [Green Version]
- Schurgers, L.J.; Joosen, I.A.; Laufer, E.M.; Chatrou, M.L.L.; Herfs, M.; Winkens, M.H.M.; Westenfeld, R.; Veulemans, V.; Krueger, T.; Shanahan, C.; et al. Vitamin K-antagonists accelerate atherosclerotic calcification and induce a vulnerable plaque phenotype. PLoS ONE 2012, 7, e43229. [Google Scholar] [CrossRef] [Green Version]
- Munroe, P.B.; Olgunturk, R.O.; Fryns, J.P.; Van Maldergem, L.; Ziereisen, F.; Yuksel, B.; Gardiner, R.M.; Chung, E. Mutations in the gene encoding the human matrix Gla protein cause Keutel syndrome. Nat. Genet. 1999, 21, 142–144. [Google Scholar] [CrossRef]
- Eitzinger, N.; Surmann-Schmitt, C.; Bösl, M.; Schett, G.; Engelke, K.; Hess, A.; von der Mark, K.; Stock, M. Ucma is not necessary for normal development of the mouse skeleton. Bone 2012, 50, 670–680. [Google Scholar] [CrossRef] [PubMed]
- Willems, B.A.; Furmanik, M.; Caron, M.M.J.; Chatrou, M.L.L.; Kusters, D.H.M.; Welting, T.J.M.; Stock, M.; Rafael, M.S.; Viegas, C.S.B.; Simes, D.C.; et al. Ucma/GRP inhibits phosphate-induced vascular smooth muscle cell calcification via SMAD-dependent BMP signalling. Sci. Rep. 2018, 8, 4961. [Google Scholar] [CrossRef] [PubMed]
- Viegas, C.S.; Rafael, M.S.; Enriquez, J.L.; Teixeira, A.; Vitorino, R.; Luís, I.M.; Costa, R.M.; Santos, S.; Cavaco, S.; Neves, J.; et al. Gla-rich protein acts as a calcification inhibitor in the human cardiovascular system. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 399–408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Viegas, C.S.B.; Herfs, M.; Rafael, M.S.; Enriquez, J.L.; Teixeira, A.; Luís, I.M.; van ‘t Hoofd, C.M.R.; João, A.; Maria, V.L.; Cavaco, S.; et al. Gla-rich protein is a potential new vitamin K target in cancer: Evidences for a direct GRP-mineral interaction. BioMed. Res. Int. 2014, 2014, 340216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Viegas, C.S.B.; Simes, D.C. A dual role for GRP in cardiovascular disease. Aging 2019, 11, 1323–1324. [Google Scholar] [CrossRef]
- Kulman, J.D.; Harris, J.E.; Haldeman, B.A.; Davie, E.W. Primary structure and tissue distribution of two novel proline-rich gamma-carboxyglutamic acid proteins. Proc. Natl. Acad. Sci. USA 1997, 94, 9058–9062. [Google Scholar] [CrossRef] [Green Version]
- Kulman, J.D.; Harris, J.E.; Xie, L.; Davie, E.W. Identification of two novel transmembrane gamma -carboxyglutamic acid proteins expressed broadly in fetal and adult tissues. Proc. Natl. Acad. Sci. USA 2001, 98, 1370–1375. [Google Scholar] [CrossRef]
- McClure, D.B.; Walls, J.D.; Grinnell, B.W. Post-translational processing events in the secretion pathway of human protein C, a complex vitamin K-dependent antithrombotic factor. J. Biol. Chem. 1992, 267, 19710–19717. [Google Scholar] [CrossRef]
- Alberts, B.; Johnson, A.; Lewis, J.; Morgan, D.; Raff, M.; Roberts, K.; Walter, P. Molecular Biology of the Cell, 6th ed.; W. W. Norton & Company: New York, NY, USA, 2014; 1464p. [Google Scholar]
- Jackson, L.P.; Lewis, M.; Kent, H.M.; Edeling, M.A.; Evans, P.R.; Duden, R.; Owen, D.J. Molecular basis for recognition of dilysine trafficking motifs by COPI. Dev. Cell 2012, 23, 1255–1262. [Google Scholar] [CrossRef] [Green Version]
- Souri, M.; Iwata, H.; Zhang, W.G.; Ichinose, A. Unique secretion mode of human protein Z: Its Gla domain is responsible for inefficient, vitamin K–dependent and warfarin-sensitive secretion. Blood 2009, 113, 3857–3864. [Google Scholar] [CrossRef] [Green Version]
- Tokunaga, F.; Wakabayashi, S.; Koide, T. Warfarin causes the degradation of protein C precursor in the endoplasmic reticulum. Biochemistry 1995, 34, 1163–1170. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Bancroft, J.D.; Suttie, J.W. Structural features of the kringle domain determine the intracellular degradation of under-g-carboxylated prothrombin: Studies of chimeric rat/human prothrombin. Proc. Natl. Acad. Sci. USA 1997, 94, 13654–13660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hallgren, K.W.; Hommema, E.L.; McNally, B.A.; Berkner, K.L. Carboxylase overexpression impairs factor IX secretion: Implications for the release of vitamin K-dependent proteins. Biochemistry 2002, 41, 15045–15055. [Google Scholar] [CrossRef] [PubMed]
- Rishavy, M.A.; Berkner, K.L. Vitamin K oxygenation, glutamate carboxylation, and processivity: Defining the three critical facets of catalysis by the vitamin K–dependent carboxylase. Adv. Nutr. 2012, 3, 135–148. [Google Scholar] [CrossRef] [Green Version]
- Caspers, M.; Czogalla, K.J.; Liphardt, K.; Müller, J.; Westhofen, P.; Watzka, M.; Oldenburg, J. Two enzymes catalyze vitamin K 2,3-epoxide reductase activity in mouse: VKORC1 is highly expressed in exocrine tissues while VKORC1L1 is highly expressed in brain. Thromb. Res. 2015, 135, 977–983. [Google Scholar] [CrossRef] [Green Version]
- Spohn, G.; Kleinridders, A.; Wunderlich, F.T.; Watzka, M.; Zaucke, F.; Blum-Bach, K.; Geisen, C.; Seifried, E.; Müller, C.; Paulsson, M.; et al. VKORC1 deficiency in mice causes early postnatal lethality due to severe bleeding. Thromb. Haemost. 2009, 101, 1044–1050. [Google Scholar] [CrossRef] [Green Version]
- Zhu, A.; Sun, H.; Raymond, R.M., Jr.; Furie, B.C.; Furie, B.; Bronstein, M.; Kaufman, R.J.; Westrick, R.; Ginsburg, D. Fatal hemorrhage in mice lacking γ-glutamyl carboxylase. Blood 2007, 109, 5270–5275. [Google Scholar] [CrossRef] [Green Version]
- Lacombe, J.; Rishavy, M.A.; Berkner, K.L.; Ferron, M. VKOR paralog VKORC1L1 supports vitamin K–dependent protein carboxylation in vivo. JCI Insight 2018, 3, e96501. [Google Scholar] [CrossRef] [Green Version]
- Schurgers, L.J.; Spronk, H.M.; Soute, B.A.; Schiffers, P.M.; Demey, J.G.; Vermeer, C. Regression of warfarin-induced medial elastocalcinosis by high intake of vitamin K in rats. Blood 2007, 109, 2823–2831. [Google Scholar] [CrossRef]
- Shea, M.K.; Berkner, K.L.; Ferland, G.; Fu, X.; Holden, R.M.; Booth, S.L. Perspective: Evidence before Enthusiasm—A Critical Review of the Potential Cardiovascular Benefits of Vitamin K. Adv. Nutr. 2021, 12, 632–646. [Google Scholar] [CrossRef]
- Buitenhuis, H.C.; Soute, B.A.; Vermeer, C. Comparison of the vitamins K1, K2 and K3 as cofactors for the hepatic vitamin K-dependent carboxylase. Biochim. Biophys. Acta (BBA)-Gen. Subj. 1990, 1034, 170–175. [Google Scholar] [CrossRef]
- Carlisle, T.L.; Suttie, J.W. Vitamin K dependent carboxylase: Subcellular location of the carboxylase and enzymes involved in vitamin K metabolism in rat liver. Biochemistry 1980, 19, 1161–1167. [Google Scholar] [CrossRef] [PubMed]
- Tie, J.; Wu, S.M.; Jin, D.; Nicchitta, C.V.; Stafford, D.W. A topological study of the human gamma-glutamyl carboxylase. Blood 2000, 96, 973–978. [Google Scholar] [CrossRef] [PubMed]
- Rishavy, M.A.; Hallgren, K.W.; Yakubenko, A.V.; Shtofman, R.L.; Runge, K.W.; Berkner, K.L. Brønsted analysis reveals Lys218 as the carboxylase active site base that deprotonates vitamin K hydroquinone to initiate vitamin K-dependent protein carboxylation. Biochemistry 2006, 45, 13239–13248. [Google Scholar] [CrossRef]
- Begley, G.S.; Furie, B.C.; Czerwiec, E.; Taylor, K.L.; Furie, G.L.; Bronstein, L.; Stenflo, J.; Furie, B. A conserved motif within the vitamin K-dependent carboxylase gene is widely distributed across animal phyla. J. Biol. Chem. 2000, 275, 36245–36249. [Google Scholar] [CrossRef] [Green Version]
- Lin, P.J.; Jin, D.Y.; Tie, J.K.; Presnell, S.R.; Straight, D.L.; Stafford, D.W. The putative vitamin K-dependent gamma-glutamyl carboxylase internal propeptide appears to be the propeptide binding site. J. Biol. Chem. 2002, 277, 28584–28591. [Google Scholar] [CrossRef] [Green Version]
- Mutucumarana, V.P.; Acher, F.; Straight, D.L.; Jin, D.Y.; Stafford, D.W. A conserved region of human vitamin K-dependent carboxylase between residues 393 and 404 is important for its interaction with the glutamate substrate. J. Biol. Chem. 2003, 278, 46488–46493. [Google Scholar] [CrossRef] [Green Version]
- Soute, B.A.M.; Jin, D.-Y.; Spronk, H.M.H.; Mutucumarana, V.P.; Lin, P.-J.; Hackeng, T.M.; Stafford, D.W.; Vermeer, C. Characteristics of recombinant W501S mutated human gamma-glutamyl carboxylase. J. Thromb. Haemost. 2004, 2, 597–604. [Google Scholar] [CrossRef]
- Wu, S.M.; Mutucumarana, V.P.; Geromanos, S.; Stafford, D.W. The propeptide binding site of the bovine g-glutamyl carboxylase. J. Biol. Chem. 1997, 272, 11718–11722. [Google Scholar] [CrossRef] [Green Version]
- Berkner, K.L.; Pudota, B.N. Vitamin K-dependent carboxylation of the carboxylase. Proc. Natl. Acad. Sci. USA 1998, 95, 466–471. [Google Scholar] [CrossRef] [Green Version]
- Hallgren, K.W.; Zhang, D.; Kinter, M.; Willard, B.; Berkner, K.L. Methylation of γ-carboxylated Glu (Gla) allows detection by liquid chromatography-mass spectrometry and the identification of Gla residues in the γ-glutamyl carboxylase. J. Proteome Res. 2013, 12, 2365–2374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pudota, B.N.; Hommema, E.L.; Hallgren, K.W.; McNally, B.A.; Lee, S.; Berkner, K.L. Identification of sequences within the g-carboxylase that represent a novel contact site with vitamin K-dependent proteins and that are required for activity. J. Biol. Chem. 2001, 276, 46878–46886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rishavy, M.A.; Berkner, K.L. Insight into the coupling mechanism of the vitamin K-dependent carboxylase: Mutation of histidine 160 disrupts glutamic acid carbanion formation and efficient coupling of vitamin K epoxidation to glutamic acid carboxylation. Biochemistry 2008, 47, 9836–9846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McMillan, C.W.; Roberts, H.R. Congenital combined deficiency of coagulation factors II, VII, IX and X. Report of a case. N. Engl. J. Med. 1966, 274, 1313–1315. [Google Scholar] [CrossRef]
- Spronk, H.M.; Farah, R.A.; Buchanan, G.R.; Vermeer, C.; Soute, B.A. Novel mutation in the gamma-glutamyl carboxylase gene resulting in congenital combined deficiency of all vitamin K-dependent blood coagulation factors. Blood 2000, 96, 3650–3652. [Google Scholar] [CrossRef]
- Jin, D.Y.; Tie, J.K.; Stafford, D.W. The conversion of vitamin K epoxide to vitamin K quinone and vitamin K quinone to vitamin K hydroquinone uses the same active site cysteines. Biochemistry 2007, 46, 7279–7283. [Google Scholar] [CrossRef] [PubMed]
- Rost, S.; Fregin, A.; Hunerberg, M.; Bevans, C.G.; Muller, C.R.; Oldenburg, J. Site-directed mutagenesis of coumarin-type anticoagulant-sensitive VKORC1: Evidence that highly conserved amino acids define structural requirements for enzymatic activity and inhibition by warfarin. Thromb. Haemost. 2005, 94, 780–786. [Google Scholar]
- Rost, S.; Fregin, A.; Ivaskevicius, V.; Conzelmann, E.; Hörtnagel, K.; Pelz, H.-J.; Lappegard, K.T.; Seifried, E.; Scharrer, I.; Tuddenham, E.G.D.; et al. Mutations in VKORC1 cause warfarin resistance and multiple coagulation factor deficiency type 2. Nature 2004, 427, 537–541. [Google Scholar] [CrossRef]
- Wajih, N.; Sane, D.C.; Hutson, S.M.; Wallin, R. Engineering of a recombinant vitamin K-dependent gamma-carboxylation system with enhanced gamma-carboxyglutamic acid forming capacity: Evidence for a functional CXXC redox center in the system. J. Biol. Chem. 2005, 280, 10540–10547. [Google Scholar] [CrossRef] [Green Version]
- Schulman, S.; Wang, B.; Li, W.; Rapoport, T.A. Vitamin K epoxide reductase prefers ER membrane-anchored thioredoxin-like redox partners. Proc. Natl. Acad. Sci. USA 2010, 107, 15027–15032. [Google Scholar] [CrossRef] [Green Version]
- Fasco, M.J.; Principe, L.M. Vitamin K1 hydroquinone formation catalyzed by DT-diaphorase. Biochem. Biophys. Res. Commun. 1982, 104, 187–192. [Google Scholar] [CrossRef]
- Hildebrandt, E.F.; Suttie, J.W. Mechanism of coumarin action: Sensitivity of vitamin K metabolizing enzymes of normal and warfarin-resistant rat liver. Biochemistry 1982, 21, 2406–2411. [Google Scholar] [CrossRef] [PubMed]
- Wallin, R. Vitamin K antagonism of coumarin anticoagulation. A dehydrogenase pathway in rat liver is responsible for the antagonistic effect. Biochem. J. 1986, 236, 685–693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rishavy, M.A.; Hallgren, K.W.; Wilson, L.A.; Usubalieva, A.; Runge, K.W.; Berkner, K.L. The vitamin K oxidoreductase is a multimer that efficiently reduces vitamin K epoxide to hydroquinone to allow vitamin K-dependent protein carboxylation. J. Biol. Chem. 2013, 288, 31556–31566. [Google Scholar] [CrossRef] [Green Version]
- Rishavy, M.A.; Hallgren, K.W.; Wilson, L.; Singh, S.; Runge, K.W.; Berkner, K.L. Warfarin alters vitamin K metabolism: A surprising mechanism of VKORC1 uncoupling necessitates an additional reductase. Blood 2018, 131, 2826–2835. [Google Scholar] [CrossRef]
- Ingram, B.O.; Turbyfill, J.L.; Bledsoe, P.J.; Jaiswal, A.K.; Stafford, D.W. Assessment of the contribution of NAD(P)H-dependent quinone oxidoreductase 1 (NQO1) to the reduction of vitamin K in wild-type and NQO1-deficient mice. Biochem. J. 2013, 456, 47–54. [Google Scholar] [CrossRef] [Green Version]
- Hammed, A.; Matagrin, B.; Spohn, G.; Prouillac, C.; Benoit, E.; Lattard, V. VKORC1L1, an enzyme rescuing the vitamin K 2,3-epoxide reductase activity in some extrahepatic tissues during anticoagulation therapy. J. Biol. Chem. 2013, 288, 28733–28742. [Google Scholar] [CrossRef] [Green Version]
- Lacombe, J.; Ferron, M. VKORC1L1, An Enzyme Mediating the Effect of Vitamin K in Liver and Extrahepatic Tissues. Nutrients 2018, 10, 970. [Google Scholar] [CrossRef] [Green Version]
- Czogalla, K.J.; Biswas, A.; Höning, K.; Hornung, V.; Liphardt, K.; Watzka, M.; Oldenburg, J. Warfarin and vitamin K compete for binding to Phe55 in human VKOR. Nat. Struct. Mol. Biol. 2017, 24, 77–85. [Google Scholar] [CrossRef]
- Li, S.; Liu, S.; Liu, X.R.; Zhang, M.M.; Li, W. Competitive tight-binding inhibition of VKORC1 underlies warfarin dosage variation and antidotal efficacy. Blood Adv. 2020, 4, 2202–2212. [Google Scholar] [CrossRef]
- Fasco, M.J.; Principe, L.M. R- and S-Warfarin inhibition of vitamin K and vitamin K 2,3-epoxide reductase activities in the rat. J. Biol. Chem. 1982, 257, 4894–4901. [Google Scholar] [CrossRef]
- Matagrin, B.; Hodroge, A.; Montagut-Romans, A.; Andru, J.; Fourel, I.; Besse, S.; Benoit, E.; Lattard, V. New insights into the catalytic mechanism of vitamin K epoxide reductase (VKORC1)—The catalytic properties of the major mutations of rVKORC1 explain the biological cost associated to mutations. FEBS Open Bio 2013, 3, 144–150. [Google Scholar] [CrossRef] [PubMed]
- Tulinsky, A. The structures of domains of blood proteins. Thromb. Haemost. 1991, 66, 16–31. [Google Scholar] [CrossRef] [PubMed]
- Dowd, T.L.; Rosen, J.F.; Li, L.; Gundberg, C.M. The three-dimensional structure of bovine calcium ion-bound osteocalcin using 1H NMR spectroscopy. Biochemistry 2003, 42, 7769–7779. [Google Scholar] [CrossRef] [Green Version]
- Zebboudj, A.F.; Imura, M.; Boström, K. Matrix GLA protein, a regulatory protein for bone morphogenetic protein-2. J. Biol. Chem. 2002, 277, 4388–4394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Young, J.; Liao, Y.; Xiao, Y.; Jalkanen, J.; Lajoie, G.; Karttunen, M.; Goldberg, H.A.; Hunter, G.K. Matrix Gla protein inhibits ectopic calcification by a direct interaction with hydroxyapatite crystals. J. Am. Chem. Soc. 2011, 133, 18406–18412. [Google Scholar] [CrossRef]
- Breyer, W.A.; Matthews, B.W. A structural basis for processivity. Protein Sci. 2001, 10, 1699–1711. [Google Scholar] [CrossRef]
- Presnell, S.R.; Tripathy, A.; Lentz, B.R.; Jin, D.Y.; Stafford, D.W. A novel fluorescence assay to study propeptide interaction with gamma- glutamyl carboxylase. Biochemistry 2001, 40, 11723–11733. [Google Scholar] [CrossRef]
- Malhotra, O.P. Purification and characterization of dicoumarol-induced prothrombins. II. Barium oxalate atypical (5-Gla) variant. Thromb. Res. 1979, 15, 439–448. [Google Scholar] [CrossRef]
- Malhotra, O.P. Purification and characterization of dicoumarol-induced prothrombins. III. Alumina pH 4.6 atypical (2-Gla) variant. Thromb. Res. 1979, 15, 449–463. [Google Scholar] [CrossRef]
- Stanley, T.B.; Jin, D.Y.; Lin, P.J.; Stafford, D.W. The propeptides of the vitamin K-dependent proteins possess different affinities for the vitamin K-dependent carboxylase. J. Biol. Chem. 1999, 274, 16940–16944. [Google Scholar] [CrossRef] [Green Version]
- Hao, Z.; Jin, D.Y.; Stafford, D.W.; Tie, J.K. Vitamin K-dependent carboxylation of coagulation factors: Insights from a cell-based functional study. Haematologica 2020, 105, 2164–2173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riphagen, I.J.; Keyzer, C.A.; Drummen, N.E.A.; De Borst, M.H.; Beulens, J.W.J.; Gansevoort, R.T.; Geleijnse, J.M.; Muskiet, F.A.J.; Navis, G.; Visser, S.T.; et al. Prevalence and Effects of Functional Vitamin K Insufficiency: The PREVEND Study. Nutrients 2017, 9, 1334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shea, M.K.; Booth, S.L. Concepts and Controversies in Evaluating Vitamin K Status in Population-Based Studies. Nutrients 2016, 8, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shea, M.K.; Kritchevsky, S.B.; Hsu, F.-C.; Nevitt, M.; Booth, S.L.; Kwoh, C.K.; McAlindon, T.E.; Vermeer, C.; Drummen, N.; Harris, T.B.; et al. The association between vitamin K status and knee osteoarthritis features in older adults: The Health, Aging and Body Composition Study. Osteoarthr. Cartil. 2015, 23, 370–378. [Google Scholar] [CrossRef] [Green Version]
- Nigwekar, S.U.; Bloch, D.; Nazarian, R.M.; Vermeer, C.; Booth, S.L.; Xu, D.; Thadhani, R.I.; Malhotra, R. Vitamin K–Dependent Carboxylation of Matrix Gla Protein Influences the Risk of Calciphylaxis. J. Am. Soc. Nephrol. 2017, 28, 1717–1722. [Google Scholar] [CrossRef]
- Saifan, C.; Saad, M.; El-Charabaty, E.; El-Sayegh, S. Warfarin-induced calciphylaxis: A case report and review of literature. Int. J. Gen. Med. 2013, 6, 665–669. [Google Scholar] [CrossRef] [Green Version]
- CCastiglione, V.; Pottel, H.; Lieske, J.C.; Lukas, P.; Cavalier, E.; Delanaye, P.; Rule, A.D. Evaluation of inactive Matrix-Gla-Protein (MGP) as a biomarker for incident and recurrent kidney stones. J. Nephrol. 2020, 33, 101–107. [Google Scholar] [CrossRef]
- Cranenburg, E.C.M.; Koos, R.; Schurgers, L.J.; Magdeleyns, E.J.; Schoonbrood, T.H.M.; Landewé, R.B.; Brandenburg, V.M.; Bekers, O.; Vermeer, C. Characterisation and potential diagnostic value of circulating matrix Gla protein (MGP) species. Thromb. Haemost. 2010, 104, 811–822. [Google Scholar] [CrossRef]
- Griffin, T.P.; Islam, N.; Wall, D.; Ferguson, J.; Griffin, D.G.; Griffin, M.D.; O’Shea, P.M. Plasma dephosphorylated-uncarboxylated Matrix Gla-Protein (dp-ucMGP): Reference intervals in Caucasian adults and diabetic kidney disease biomarker potential. Sci. Rep. 2019, 9, 18452. [Google Scholar] [CrossRef] [Green Version]
- Jeannin, A.-C.; Salem, J.-E.; Massy, Z.; Aubert, C.E.; Vemeer, C.; Amouyal, C.; Phan, F.; Halbron, M.; Funck-Brentano, C.; Hartemann, A.; et al. Inactive matrix gla protein plasma levels are associated with peripheral neuropathy in Type 2 diabetes. PLoS ONE 2020, 15, e0229145. [Google Scholar] [CrossRef]
- Li, C.; Li, J.; He, F.; Li, K.; Li, X.; Zhang, Y. Matrix Gla protein regulates adipogenesis and is serum marker of visceral adiposity. Adipocyte 2020, 9, 68–76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, D.H.; Fu, H.D.; Liu, W.L.; Wei, Y.S.; Jiang, D.M. Decreased local and systematic matrix Gla protein (MGP) expression and its link to radiographic progression in ankylosing spondylitis patients. Clin. Lab. 2015, 61, 243–249. [Google Scholar] [CrossRef] [PubMed]
- Machado-Fragua, M.D.; Hoogendijk, E.O.; Struijk, E.A.; Rodriguez-Artalejo, F.; Lopez-Garcia, E.; Beulens, J.W.; Van Ballegooijen, A.J. High dephospho-uncarboxylated matrix Gla protein concentrations, a plasma biomarker of vitamin K, in relation to frailty: The Longitudinal Aging Study Amsterdam. Eur. J. Nutr. 2019, 59, 1243–1251. [Google Scholar] [CrossRef]
- Oikonomaki, T.; Papasotiriou, M.; Ntrinias, T.; Kalogeropoulou, C.; Zabakis, P.; Kalavrizioti, D.; Papadakis, I.; Goumenos, D.S.; Papachristou, E. The effect of vitamin K2 supplementation on vascular calcification in haemodialysis patients: A 1-year follow-up randomized trial. Int. Urol. Nephrol. 2019, 51, 2037–2044. [Google Scholar] [CrossRef]
- Christiadi, D.; Singer, R.F. Calciphylaxis in a dialysis patient successfully treated with high-dose vitamin K supplementation. Clin. Kidney J. 2018, 11, 528–529. [Google Scholar] [CrossRef] [Green Version]
- Jacobs-Kosmin, D.; Dehoratius, R.J. Calciphylaxis: An important imitator of cutaneous vasculitis. Arthritis Rheum. 2007, 57, 533–537. [Google Scholar] [CrossRef]
- Miyata, K.N.; Nast, C.C.; Dai, T.; Dukkipati, R.; LaPage, J.A.; Troost, J.P.; Schurgers, L.; Kretzler, M.; Adler, S.G. Renal matrix Gla protein expression increases progressively with CKD and predicts renal outcome. Exp. Mol. Pathol. 2018, 105, 120–129. [Google Scholar] [CrossRef]
- Mizuiri, S.; Nishizawa, Y.; Yamashita, K.; Ono, K.; Naito, T.; Tanji, C.; Usui, K.; Doi, S.; Masaki, T.; Shigemoto, K. Relationship of matrix Gla protein and vitamin K with vascular calcification in hemodialysis patients. Ren. Fail. 2019, 41, 770–777. [Google Scholar] [CrossRef]
- Piscaer, I.; van den Ouweland, J.M.W.; Vermeersch, K.; Reynaert, N.L.; Franssen, F.M.E.; Keene, S.; Wouters, E.F.M.; Janssens, W.; Vermeer, C.; Janssen, R. Low Vitamin K Status Is Associated with Increased Elastin Degradation in Chronic Obstructive Pulmonary Disease. J. Clin. Med. 2019, 8, 1116. [Google Scholar] [CrossRef] [Green Version]
- Shepherd, C.; Reese, A.E.; Reynard, L.N.; Loughlin, J. Expression analysis of the osteoarthritis genetic susceptibility mapping to the matrix Gla protein gene MGP. Arthritis Res. Ther. 2019, 21, 149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, F.; Thijs, L.; Cauwenberghs, N.; Yang, W.; Zhang, Z.; Yu, C.; Kuznetsova, T.; Nawrot, T.; Struijker-Boudier, H.A.J.; Verhamme, P.; et al. Central Hemodynamics in Relation to Circulating Desphospho-Uncarboxylated Matrix Gla Protein: A Population Study. J. Am. Heart Assoc. 2019, 8, e011960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zwakenberg, S.R.; Burgess, S.; Sluijs, I.; Weiderpass, E.; Beulens, J.W.; van der Schouw, Y.T. Circulating phylloquinone, inactive Matrix Gla protein and coronary heart disease risk: A two-sample Mendelian Randomization study. Clin. Nutr. 2019, 39, 1131–1136. [Google Scholar] [CrossRef] [PubMed]
- Booth, S.L.; Suttie, J.W. Dietary intake and adequacy of vitamin K. J. Nutr. 1998, 128, 785–788. [Google Scholar] [CrossRef] [Green Version]
- Shearer, M.J.; Newman, P. Recent trends in the metabolism and cell biology of vitamin K with special reference to vitamin K cycling and MK-4 biosynthesis. J. Lipid Res. 2014, 55, 345–362. [Google Scholar] [CrossRef] [Green Version]
- Shearer, M.J.; Fu, X.; Booth, S.L. Vitamin K nutrition, metabolism, and requirements: Current concepts and future research. Adv. Nutr. 2012, 3, 182–195. [Google Scholar] [CrossRef] [Green Version]
- Watzka, M.; Geisen, C.; Bevans, C.G.; Sittinger, K.; Spohn, G.; Rost, S.; Seifried, E.; Müller, C.R.; Oldenburg, J. Thirteen novel VKORC1 mutations associated with oral anticoagulant resistance: Insights into improved patient diagnosis and treatment. J. Thromb. Haemost. 2011, 9, 109–118. [Google Scholar] [CrossRef]
- Hodroge, A.; Matagrin, B.; Moreau, C.; Fourel, I.; Hammed, A.; Benoit, E.; Lattard, V. VKORC1 mutations detected in patients resistant to vitamin K antagonists are not all associated with a resistant VKOR activity. J. Thromb. Haemost. 2012, 10, 2535–2543. [Google Scholar] [CrossRef]
- Tie, J.K.; Carneiro, J.D.; Jin, D.Y.; Martinhago, C.D.; Vermeer, C.; Stafford, D.W. Characterization of vitamin K-dependent carboxylase mutations that cause bleeding and nonbleeding disorders. Blood 2016, 127, 1847–1855. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, S.; Kraus, K.; Biswas, A.; Müller, J.; Buhl, A.; Forin, F.; Singer, H.; Höning, K.; Hornung, V.; Watzka, M.; et al. GGCX mutations show different responses to vitamin K thereby determining the severity of the hemorrhagic phenotype in VKCFD1 patients. J. Thromb. Haemost. 2021, 19, 1412–1424. [Google Scholar] [CrossRef]
- Hao, Z.; Jin, D.; Chen, X.; Schurgers, L.J.; Stafford, D.W.; Tie, J.K. γ-glutamyl carboxylase mutations differentially affect the biological function of vitamin K-dependent proteins. Blood 2021, 137, 533–543. [Google Scholar] [CrossRef] [PubMed]
- Card, D.J.; Gorska, R.; Harrington, D.J. Laboratory assessment of vitamin K status. J. Clin. Pathol. 2020, 73, 70–75. [Google Scholar] [CrossRef] [PubMed]
- Kondo, Y.; Kimura, O.; Shimosegawa, T. Significant biomarkers for the management of hepatocellular carcinoma. Clin. J. Gastroenterol. 2015, 8, 109–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schurgers, L.; Teunissen, K.J.; Knapen, M.H.; Kwaijtaal, M.; Van Diest, R.; Appels, A.; Reutelingsperger, C.P.; Cleutjens, J.P.; Vermeer, C. Novel conformation-specific antibodies against matrix γ-carboxyglutamic acid (Gla) protein: Undercarboxylated matrix Gla protein as marker for vascular calcification. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 1629–1633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, M.A.; Stenberg, L.M.; Persson, U.; Stenflo, J. Identification and purification of vitamin K-dependent proteins and peptides with monoclonal antibodies specific for gamma -carboxyglutamyl (Gla) residues. J. Biol. Chem. 2000, 275, 19795–19802. [Google Scholar] [CrossRef] [Green Version]
- Rishavy, M.A.; Hallgren, K.W.; Zhang, H.; Runge, K.W.; Berkner, K.L. Exon 2 Skipping Eliminates γ-Glutamyl Carboxylase Activity, Indicating a Partial Splicing Defect in a Patient with Vitamin K Clotting Factor Deficiency. J. Thromb. Haemost. 2019, 17, 1053–1063. [Google Scholar] [CrossRef]
- Berkner, K.L. Expression of recombinant vitamin K-dependent proteins in mammalian cells: Factors IX and VII. Methods Enzymol. 1993, 222, 450–477. [Google Scholar]
- Larson, P.J.; Camire, R.M.; Wong, D.; Fasano, N.C.; Monroe, D.M.; Tracy, P.B.; High, K.A. Structure/function analyses of recombinant variants of human factor Xa: Factor Xa incorporation into prothrombinase on the thrombin-activated platelet surface is not mimicked by synthetic phospholipid vesicles. Biochemistry 1998, 37, 5029–5038. [Google Scholar] [CrossRef]
- Ratcliffe, J.V.; Furie, B.; Furie, B.C. The importance of specific gamma-carboxyglutamic acid residues in prothrombin. Evaluation by site-specific mutagenesis. J. Biol. Chem. 1993, 268, 24339–24345. [Google Scholar] [CrossRef]
- Zhang, L.; Jhingan, A.; Castellino, F.J. Role of individual gamma-carboxyglutamic acid residues of activated human protein C in defining its in vitro anticoagulant activity. Blood 1992, 80, 942–952. [Google Scholar] [CrossRef]
- Price, P.A.; Williamson, M.K. Primary structure of bovine matrix Gla protein, a new vitamin K-dependent bone protein. J. Biol. Chem. 1985, 260, 14971–14975. [Google Scholar] [CrossRef]
- Rehder, D.S.; Gundberg, C.M.; Booth, S.L.; Borges, C.R. Gamma-carboxylation and fragmentation of osteocalcin in human serum defined by mass spectrometry. Mol. Cell. Proteom. 2015, 14, 1546–1555. [Google Scholar] [CrossRef] [PubMed] [Green Version]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Berkner, K.L.; Runge, K.W. Vitamin K-Dependent Protein Activation: Normal Gamma-Glutamyl Carboxylation and Disruption in Disease. Int. J. Mol. Sci. 2022, 23, 5759. https://doi.org/10.3390/ijms23105759
Berkner KL, Runge KW. Vitamin K-Dependent Protein Activation: Normal Gamma-Glutamyl Carboxylation and Disruption in Disease. International Journal of Molecular Sciences. 2022; 23(10):5759. https://doi.org/10.3390/ijms23105759
Chicago/Turabian StyleBerkner, Kathleen L., and Kurt W. Runge. 2022. "Vitamin K-Dependent Protein Activation: Normal Gamma-Glutamyl Carboxylation and Disruption in Disease" International Journal of Molecular Sciences 23, no. 10: 5759. https://doi.org/10.3390/ijms23105759
APA StyleBerkner, K. L., & Runge, K. W. (2022). Vitamin K-Dependent Protein Activation: Normal Gamma-Glutamyl Carboxylation and Disruption in Disease. International Journal of Molecular Sciences, 23(10), 5759. https://doi.org/10.3390/ijms23105759