
PD-1/PD-L1, MDSC Pathways, and Checkpoint Inhibitor Therapy in Ph(-) Myeloproliferative Neoplasm: A Review


Abstract
:1. Introduction
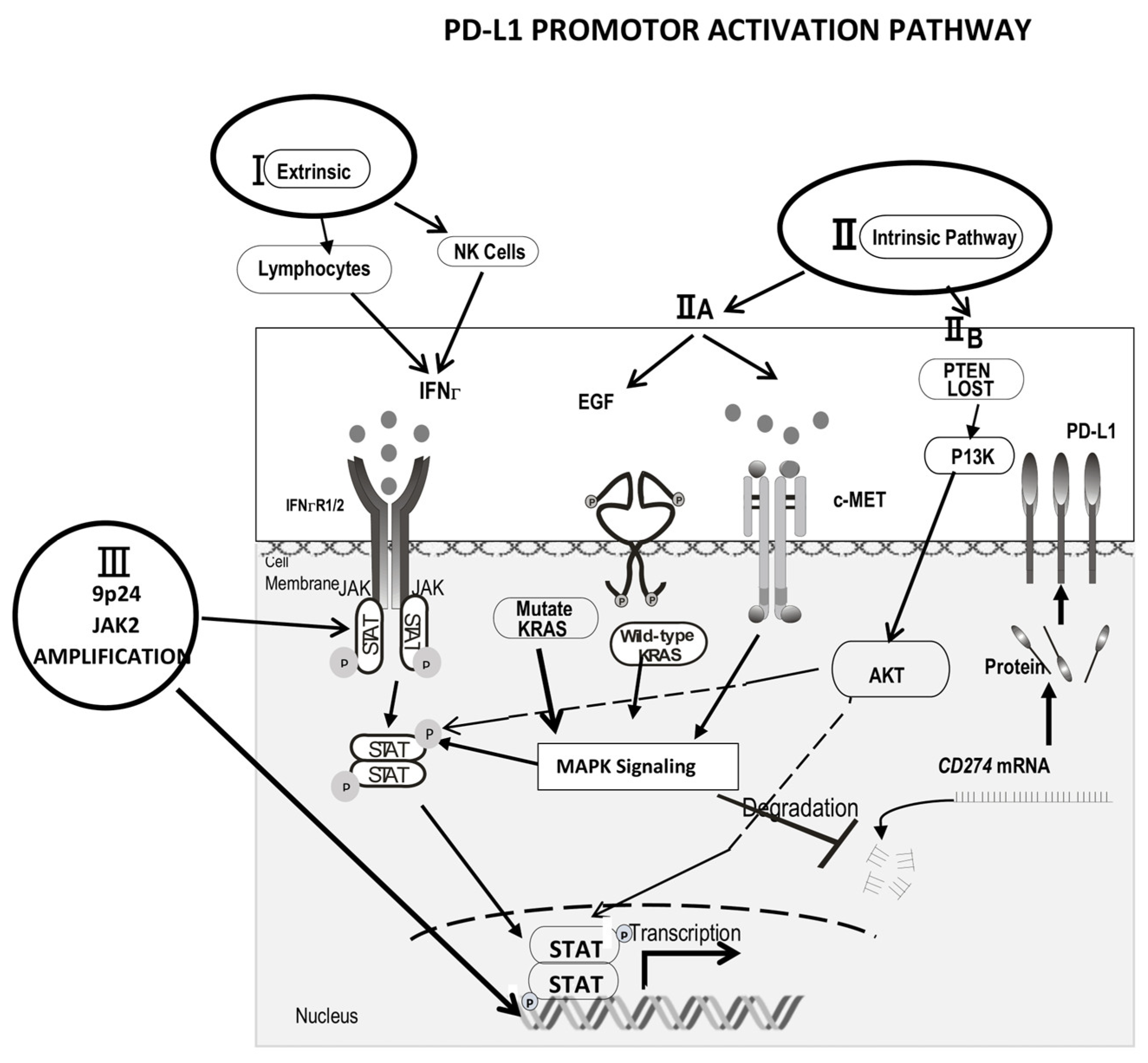
2. Mechanisms of Increased PD-L1 Expression
2.1. The Extrinsic (IFNγ) Pathway
2.2. The Intrinsic Mechanism (Oncogene Growth Factor Pathway)
2.3. 9p24.1 Gene Amplicon Amplification
3. PD-1 and PD-L1 Interaction with MDSCs
3.1. MDSC (Myeloid Suppressor Cell) Function
- (1)
- The expression of NOX2, iNOS, and Arginase 1. In contrast to T cells, which present immune-checkpoint molecules on their surface (including cytotoxic T-lymphocyte-associated antigen 4 (CTLA-4), lymphocyte-activation gene 3 (LAG-3), programmed cell death protein 1 (PD-1), T-cell immunoglobulin and mucin-domain containing 3 (TIM-3), and B and T lymphocyte attenuator (BTLA)) [43], MDSCs mainly exert their mechanism as an immune modulator through inducing the expression of NADPH oxidase (NOX2) and inducible nitric oxide synthase (iNOS), which generates different ROS compounds and nitric oxide (NO). It is known that the product of H2O2 and NO, i.e., peroxynitrite (ONOO−), can nitrate the 22 tyrosine residues of the T-cell receptor (TCR), after which, the receptor no longer recognizes antigen peptides; thus, the TCR signaling pathway is inhibited [44,45]. MDSCs also express arginase I [46], which inhibits and possibly deletes tumor-specific cytotoxic T cells (CTL): CD3ς is lost in T cells exposed to arginase I [47,48]. Overall, granulocyte MDSCs (G-MDSCs) have higher arginase-1, MPO, and ROS activities [49], and monocytic MDSCs (M-MDSCs) are mediated by arginase-1, NO, and different soluble factors [50]. Several other suppressive mechanisms from MDSCs have also been suggested: the secretion of TGF-ß [51,52], induction of regulatory T cells [53,54,55], depletion of cysteine [56], and up-regulation of cyclo-oxygenase 2 (cox2) and prostaglandin E2 (PGE2) [47].
- (2)
- MDSCs can show an up-regulation of PD-L1 [57] and CD 80 [58] on their surface to exert immunosuppression. PD-L1 is activated through IFNΓ-stat 1 activation [59,60,61,62]. IFNΓ is highly expressed in cells of the tumor tissues, and, through phosphorated STAT 1, it binds to a unique IRF-binding sequence element in vitro and chromatin in vivo in the cd274 promoter to activate PD-L1 transcription. In addition to IFNΓ, IL-10, VEGF, and hypoxia are other novel critical modulators of PD-L1 expression in MDSCs [63,64]. The expression of CD80, normally found on dendritic cells or macrophages, was increased in MDSCs in patients with malignant melanoma, and it was also up-regulated on MDSCs in a murine ovarian cancer model; its ligation of CTLA-4 through CD80 on Tregs is crucial for T cell suppression [54]. Treg cells are also able to stimulate B7-H1 expression in myeloid-derived suppressor cells [65], so MDSCs and Tregs co-operatively enhance each other’s immune-suppression functions.
- (3)
- Up-regulated myeloid cell receptor tyrosine kinases (RTKs) TYRO3, AXL, and MERTK, and their ligands Gas 6 and Protein S. These RTKs are the physiological pathways used to suppress innate immune responses, including in the tumor microenvironment. Myeloid-derived suppressor cells (MDSCs) can up-regulate TYRO3, AXL, and MERTK and their ligands to exert immunosuppressive functions [66].
- (4)
- Accumulation and expansion of MDSCs. These processes are controlled by a network of transcription factors and regulators to expand immature myeloid cells and ensure the pathologic activation of these immature cells. These expansions and activations are indispensable for MDSC accumulation [67]. STAT3, IRF8, C/EBPβ, RB1, and adenosine receptors A2b NLRP3 are the important transcription factors for MDSC expansion, and NF-kß, the STAT1 pathway, the STAT 6 pathway, PGE2 and COX 2, and ER stress are the transcriptional factors for MDSC activation. STAT3 was the first transcription factor implicated in MDSC expansion [67]. S100A9 and S100A8 also expand and activate MDSCs, and forced S100A9 can induce MDSCs and thereby induce clinical myelodysplastic syndrome [67,68,69,70].
3.2. MDSC and PD-1 Inhibitor Therapy
3.3. Anti-MDSC Therapy
- (1)
- Depleting MDSCs
- (2)
- Blockade of MDSC Migration
- (3)
- Attenuating MDSC Immunosuppressive Functions
- (4)
- Inducing MDSC Differentiation
- (5)
- Inhibition of PD-L1 and VISTA Expression on MDSCs
4. Perspectives and Future Directions
- (A)
- Review of Onco-immuno micro-environmental studies in MPN
- (1)
- Inflammatory cytokines and ROS formation.
- (2)
- Increased MDSC. In the inflamed micro-environment of MPN, the production of many inflammatory cytokines along with elevated S100A9 results in the accumulation of MDSC in MPN [157]. The mechanism of increased MDSC could be due to (1) the inflammatory cytokine stem cell growth factor (SCF) leading to the accumulation of MDSC [158]; (2) increased S100A9 levels, which inhibited dendric cell maturation and then increased MDSC [70], (3) the cytokine release of GM-CSF, VEGF, PGE2/COX2 (prostaglandin E2/cyclooxygenase-2), and interferon (IFN)-γ. These factors are responsible for MDSC accumulation and C5a, which facilitates MDSC infiltration into tumors and enhances their suppressive abilities [159].
- (3)
- Immune dysfunction in MPNs No differences between healthy donors and MPN patients were found in Th cells (T help) polarization at baseline level [160]. The frequency of thymus-derived regulatory T cells (Tregs) has also been studied in MPNs, and conflicting results have been published [161,162,163]. Natural killer (NK) cells in MPN also have a decreased function and numbers [159,164]. Ramano et al. [165] further studied MPN according to the JAK2 and CALR mutation status and reported that patients carrying the JAK2 (V617F) mutation had a reduction in Th17, myeloid-dendric cells (DCs), and effector Tregs, as well as increased ILC1 (hypofunctional lymphocytes) and cytokine-producing Tregs. The CALR-mutated patients revealed high ILC3 levels, reduced Th1, and a reduced capacity of monocytes to mature into fully committed DCs in vitro. Their Tregs were also less effective in inhibiting the proliferation of autologous effector T cells due to an increased proliferative status induced by CALR mutation. Triple-negative patients presented a reduced amount of total circulating CD3, effector Tregs, and Th1, as well as increased ILC1. Keohane et al. [160] reported that CD4+, CD127low, CD25high, and FOXP3+ T regulatory cells are reduced in MPN patients compared to healthy controls. They also reported that this decrease is even more pronounced following JAK2 inhibitor therapy. After 6 months of treatment, the number of T helper (Th)-17 cells increased, and there was a blockade of pro-inflammatory cytokine production, which explained why ruxolitinib therapy increased the incidence of infections. Interestingly, in CALR mutant MPN, a mutant-specific sequence generated by a frameshift mutation has been reported as a neoantigen for CD4+ T cells, but this response was reduced in cells derived from MPN patients harboring a CALR mutation [166,167,168]. Immune checkpoint inhibitor (CPI) therapy enhances shared neoantigen-induced T cell immunity directed against mutated calreticulin in myeloproliferative neoplasms [168].
- (B)
- Review of CPI studies in MPN. There have been very few studies on CPI therapy in MPN, especially in poor prognostic entities such as primary myelofibrosis (PMF), post-ET MF, or post-PV MF. Three NCI-sponsored clinical trials related to CPI therapy (NCT03065400, NCT02421354, and NCT02871323) were listed in 2021(clinicaltrials.gov, accessed on 1 October 2018). NCT02421354 [10], using nivolumab, was terminated due to a low efficacy. NCT02871323 was withdrawn because of a low enrollment, while NCT01822509 assessed ipilimumab in comparison with nivolumab in phase 1 studies. Another two clinical trials were NCT03566446 (Phase I), a CALRLong36 peptide (exon 9 mut) vaccine trial, and NCT04051307, a PD-L1Long [18,19,20,21,22,23,24,25] ArgLong2 [153,154,155,156,157,158,159,160,161,162,163,164,165,166,167,168,169,170,171,172,173,174,175,176,177,178,179,180,181,182,183,184,185,186,187,188] vaccine trial; both studies were vaccine trials based on mutated calreticulin-induced T cell immunity [166,167]. No clinical trials using CPI in the treatment of myelofibrosis or MPN have been listed in 2022 (clinicaltrials.gov, accessed on 1 October 2018).Only one small-scale study of CPI therapy in Ph(-) MPN [10] has been reported so far, in which, it was shown to be ineffective, and no clinical trial of CPI in the treatment of MPN has been listed in 2022 among the NIH clinical trials. MDSCs are considered as important to CPI therapy resistance [73,80,169,170,171]. Therefore, the failure of CPI therapy in Ph(-) MPN is most likely related to MDSCs.
- (C)
- Future Directions. Therefore, from reviewing inflamed microenvironments and deranged immune dysfunction in MPN resulting in the overactive MDSC and being resistant to CPI therapy in solid tumor history, targeting myeloid-derived suppressor cells is a promising strategy to overcome resistance to immune checkpoint inhibitors. Table 1 lists all of the agents that may reduce MDSC numbers, cause them to differentiate into dendric cells, or reduce their PD-L1 levels. The inhibition of VISTA expression levels on MDSCs also represents future work to improve CPI therapy for the treatment of MPN diseases.These listed agents that decrease MDSCs could be added to CPI to improve the CPI therapy efficacy in Ph(-) MPN.
- (a)
- Ruxolitinib in combination with CPIIn November 2011, ruxolitinib became the first Janus kinase (JAK) inhibitor approved by the United States FDA to treat myelofibrosis [172]. Recently, ruxolitinib was approved to treat steroid-refractory graft-versus-host disease (GvHD). Many JAK inhibitors have been developed, each with varying activities against other kinases and differential effects on the immune system [173]. In multiple myeloma (MM), PD-L1 expression is increased in plasma cells from patients with MM compared with that from healthy donors, and its expression is associated with a resistance to a variety of anti-MM treatments. The reported inhibition of the PD-L1/PD-1 pathway in plasma cells by ruxolitinib [174] and decreased PMC-MDSC (LOX-1 expression) in Hodgkin’s lymphoma treated with a PD-1 inhibitor plus ruxolitinib [175] suggest that ruxolitinib may be a suitable candidate to be added to CPI therapy in the treatment of MPN, because ruxolitinib is expected to reduce PD-L1 expression in MDSCs. We also found that MDSCs in MPN show an increased PD-L1 expression [176], and ruxolitinib added to the CPI therapy may thus be a promising treatment option.
- (b)
- IMID (Immunomodulatory imide drug) therapy in combination with CPIIMID therapy has been reported to have a 30% response rate in treating patients with myelofibrosis, and, in some cases of anemia, patients have changed from transfusion-dependent to transfusion-independent states [177,178]. In a study by Görgün, et al., both newly diagnosed multiple myeloma and relapsed myeloma cells present increased PD-L1 mRNA expression on the myeloma and MDSC cells compared to those from healthy individuals. PD1/PD-L1 blockade abrogated BM-stroma cell (BMSC)-induced MM growth, and lenalidomide decreased PD-L1 expression on myeloma and MDSC cells. The combined blockade of PD1/PD-L1 with lenalidomide further inhibited BMSC-induced tumor growth and decreased PD-L1 on the MDSC and myeloma cells, further improving T cell immunity [179]. Therefore, IMID in combination with CPI may also be worth exploring.
- (c)
- BTK inhibitors with CPIBTK inhibitors effectively treat B-cell malignancy, especially chronic lymphocytic leukemia [180]. MDSCs express BTK, and treatment with BTK inhibitors significantly inhibits MDSCs by impairing nitric oxide production and cell migration. In addition, ibrutinib was found to inhibit the in vitro generation of human MDSCs [89] and induce MDSCs to mature to dendric cells in mouse breast cancer models [115]. Reports have also shown a role for BTK in Toll-like receptor (TLR) signaling in myeloid cells [181], which is of interest because TLR signaling has been implicated in MDSC generation and function [182,183]. Therefore, BTK inhibitors in combination with CPI should be considered in MPN.
- (D)
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- The Nobel Prize in Physiology or Medicine. Available online: https://www.nobelprize.org/prizes/medicine/2018/press-release (accessed on 1 October 2018).
- Leach, D.R.; Krummel, M.F.; Allison, J.P. Enhancement of antitumor immunity by CTLA-4 blockade. Science 1996, 271, 1734–1736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- FDA Approves YERVOY” (Ipilimumab) for the Treatment of Patients with Newly Diagnosed or Previously-Treated Unresectable or Metastatic Melanoma. Available online: https://news.bms.com/news/details/2011/FDA-Approves-YERVOY-ipilimumab-for-the-Treatment-of-Patients-with-Newly-Diagnosed-or-Previously-Treated-Unresectable-or-Metastatic-Melanoma-the-Deadliest-Form-of-Skin-Cancer/default.aspx (accessed on 25 March 2011).
- Ishida, Y.; Agata, Y.; Shibahara, K.; Honjo, T. Induced expression of PD-1, a novel member of the immunoglobulin gene superfamily, upon programmed cell death. EMBO J. 1992, 11, 3887–3895. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, H.; Nose, M.; Hiai, H.; Minato, N.; Honjo, T. Development of lupus-like autoimmune diseases by disruption of the PD-1 gene encoding an ITIM motif-carrying immunoreceptor. Immunity 1999, 11, 141–151. [Google Scholar] [CrossRef] [Green Version]
- Dong, H.; Zhu, G.; Tamada, K.; Chen, L. B7-H1, a third member of the B7 family, co-stimulates T-cell proliferation and interleukin-10 secretion. Nat. Med. 1999, 5, 1365–1369. [Google Scholar] [CrossRef] [PubMed]
- Freeman, G.J.; Long, A.J.; Iwai, Y.; Bourque, K.; Chernova, T.; Nishimura, H.; Fitz, L.J.; Malenkovich, N.; Okazaki, T.; Byrne, M.C.; et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J. Exp. Med. 2000, 192, 1027–1034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaddepally, R.K.; Kharel, P.; Pandey, R.; Garje, R.; Chandra, A.B. Review of indications of FDA-approved immune checkpoint inhibitors per NCCN guidelines with the level of evidence. Cancers 2020, 12, 738. [Google Scholar] [CrossRef] [Green Version]
- Kythreotou, A.; Siddique, A.; Mauri, F.A.; Bower, M.; Pinato, D.J. PD-L1. J. Clin. Pathol. 2018, 71, 189–194. [Google Scholar] [CrossRef]
- Hobbs, G.S.; Bozkus, C.C.; Wadleigh, M.; Sandy, L.; Doughtery, M.; Johnson, K.; Sanchez, G.; Foster, J.E.; Macrae, M.; Som, T.T.; et al. Results of a Phase II study of PD-1 inhibition in advanced myeloproliferative neoplasms. Blood 2020, 135, 1–60. [Google Scholar] [CrossRef]
- Wang, J.-C.; Chen, C.; Kundra, A.; Kodali, S.; Pandey, A.; Wong, C.; Cheung, T.; Gotlieb, V.; Joseph, G.; Tribie, S. Programmed cell death receptor (PD-1) ligand (PD-L1) expression in philadelphia chromosome-negative myeloproliferative neoplasms. Leuk. Res. 2019, 79, 52–59. [Google Scholar] [CrossRef]
- Prestipino, A.; Emhardt, A.J.; Aumann, K.; Sullivan, D.O.; Gorantla, S.P.; Duquesne, S.; Melchinger, W.; Braun, L.; Vuckovic, S.; Boerries, M.; et al. Oncogenic JAK2V617F causes PD-L1 expression, mediating immune escape in myeloproliferative neoplasms. Sci. Transl. Med. 2018, 10, eaam7729. [Google Scholar] [CrossRef] [Green Version]
- Stutvoet, T.S.; Kol, A.; de Vries, E.G.; de Bruyn, M.; Fehrmann, R.S.; Terwisscha van Scheltinga, A.G.; de Jong, S. MAPK pathway activity plays a key role in PD-L1 expression of lung adenocarcinoma cells. J. Pathol. 2019, 249, 52–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Diaz, A.; Shin, D.S.; Moreno, B.H.; Saco, J.; Escuin-Ordinas, H.; Rodriguez, G.A.; Zaretsky, J.M.; Sun, L.; Hugo, W.; Wang, X.; et al. Interferon receptor signaling pathways regulating PD-L1 and PD-L2 expression. Cell Rep. 2017, 19, 1189–1201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Green, M.R.; Monti, S.; Rodig, S.J.; Juszczynski, P.; Currie, T.; O’Donnell, E.; Chapuy, B.; Takeyama, K.; Neuberg, D.; Golub, T.R.; et al. Integrative analysis reveals selective 9p24.1 amplification, increased PD-1 ligand expression, and further induction via JAK2 in nodular sclerosing Hodgkin lymphoma and primary mediastinal large B-cell lymphoma. Blood 2010, 116, 3268–3277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Concha-Benavente, F.; Srivastava, R.M.; Trivedi, S.; Lei, Y.; Chandran, U.; Seethala, R.R.; Freeman, G.J.; Ferris, R.L. Identification of the cell-intrinsic and -extrinsic pathways downstream of EGFR and IFNγ that induce PD-L1 expression in head and neck cancer. Cancer Res. 2016, 76, 1031–1043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Isaacs, A.; Lindenmann, J. Virus interference. I. The interferon. Proc. R. Soc. Lond. B Biol. Sci. 1957, 147, 258–267. [Google Scholar]
- Domanski, P.; Colamonici, O.R. The type-I interferon receptor. The long and short of it. Cytokine Growth Factor Rev. 1996, 7, 143–151. [Google Scholar] [CrossRef]
- Novick, D.; Cohen, B.; Rubinstein, M. The human interferon α/β receptor: Characterization and molecular cloning. Cell 1994, 77, 391–400. [Google Scholar] [CrossRef]
- Velazquez, L.; Fellous, M.; Stark, G.R.; Pellegrini, S. A protein tyrosine kinase in the interferon α/β signaling pathway. Cell 1992, 70, 313–322. [Google Scholar] [CrossRef]
- Darnell, J.E., Jr.; Kerr, I.M.; Stark, G.R. Jak-STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science 1994, 264, 1415–1421. [Google Scholar] [CrossRef] [Green Version]
- Kessler, D.S.; Levy, D.E.; Darnell Jr, J.E. Two interferon-induced nuclear factors bind a single promoter element in interferon-stimulated genes. Proc. Natl. Acad. Sci. USA 1988, 85, 8521–8525. [Google Scholar] [CrossRef] [Green Version]
- Decker, T.; Lew, D.J.; Mirkovitch, J.; Darnell Jr, J.E. Cytoplasmic activation of GAF, an IFN-gamma- regulated DNA-binding factor. EMBO J. 1991, 10, 927–932. [Google Scholar] [CrossRef] [PubMed]
- Lew, D.J.; Decker, T.; Strehlow, I.; Darnell Jr, J.E. Overlapping elements in the guanylate-binding protein gene promoter mediate transcriptional induction by alpha and gamma interferons. Mol. Cell. Biol. 1991, 11, 182–191. [Google Scholar] [PubMed] [Green Version]
- Shin, D.; Garcia-Diaz, A.; Zaretsky, J.; Escuin-Ordinas, H.; Hu-Lieskovan, S.; Palaskas, N.J.; Hugo, W.; Komenan, M.S.; Chmielowski, B.; Cherry, G.; et al. Innate resistance of PD-1 blockade through loss of function mutations in JAK resulting in inability to express PD-L1 upon interferon exposure. J. Immunother. Cancer 2015, 3, 311. [Google Scholar] [CrossRef] [Green Version]
- Gao, J.; Shi, L.Z.; Zhao, H.; Chen, J.; Xiong, L.; He, Q.; Chen, T.; Roszik, J.; Bernatchez, C.; Woodman, S.E. Loss of IFN-γ pathway genes in tumor cells as a mechanism of resistance to anti-CTLA-4 therapy. Cell 2016, 167, 397–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zaretsky, J.M.; Garcia-Diaz, A.; Shin, D.S.; Escuin-Ordinas, H.; Hugo, W.; Hu-Lieskovan, S.; Torrejon, D.Y.; Abril-Rodriguez, G.; Sandoval, S.; Barthly, L.; et al. Mutations associated with acquired resistance to PD-1 blockade in melanoma. N. Engl. J. Med. 2016, 375, 819–829. [Google Scholar] [CrossRef]
- Zitvogel, L.; Kepp, O.; Kroemer, G. Immune parameters affecting the efficacy of chemotherapeutic regimens. Nat. Rev. Clin. Oncol. 2011, 8, 151–160. [Google Scholar] [CrossRef]
- Galon, J.; Angell, H.K.; Bedognetti, D.; Marincola, F.M. The continuum of cancer immunosurveillance: Prognostic, predictive, and mechanistic signatures. Immunity 2013, 39, 11–26. [Google Scholar] [CrossRef] [Green Version]
- TKitano, A.; Ono, M.; Yoshida, M.; Noguchi, E.; Shimomura, A.; Shimoi, T.; Kodaira, M.; Yunokawa, M.; Yonemori, K.; Shimizu, C. Tumour-infiltrating lymphocytes are correlated with higher expression levels of PD-1 and PD-L1 in early breast cancer. ESMO Open 2017, 2, e000150. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, S.; Ogawa, T.; Sano, R.; Takahara, T.; Inukai, D.; Akira, S.; Tsuchida, H.; Yoshikawa, K.; Ueda, R.; Tsuzuki, T. Immune-checkpoint molecules on regulatory T-cells as a potential therapeutic target in head and neck squamous cell cancers. Cancer Sci. 2020, 111, 1943–1957. [Google Scholar] [CrossRef]
- He, Y.; Rozeboom, L.; Rivard, C.J.; Ellison, K.; Dziadziuszko, R.; Yu, H.; Zhou, C.; Hirsch, F.R. PD-1, PD-L1 protein expression in non-small cell lung cancer and their relationship with tumor-infiltrating lymphocytes. Med. Sci. Monit. 2017, 23, 1208–1216. [Google Scholar] [CrossRef] [Green Version]
- McBride, S.M.; Rothenberg, S.M.; Faquin, W.C.; Chan, A.W.; Clark, J.R.; Ellisen, L.W.; Wirth, L.J. Mutation frequency in 15 common cancer genes in high-risk head and neck squamous cell carcinoma. Head Neck 2014, 36, 1181–1188. [Google Scholar] [CrossRef] [PubMed]
- Stransky, N.; Egloff, A.M.; Tward, A.D.; Kostic, A.D.; Cibulskis, K.; Sivachenko, A.; Kryukov, G.V.; Lawrence, M.S.; Sougnez, C.; McKenna, A.; et al. The mutational landscape of head and neck squamous cell carcinoma. Science 2011, 333, 1157–1160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sumimoto, H.; Takano, A.; Teramoto, K.; Daigo, Y. RAS-mitogen-activated protein kinase signal is required for enhanced PD-L1 expression in human lung cancers. PLoS ONE 2016, 11, e0166626. [Google Scholar] [CrossRef] [PubMed]
- Azuma, K.; Ota, K.; Kawahara, A.; Hattori, S.; Iwama, E.; Harada, T.; Matsumoto, K.; Takayama, K.; Takamori, S.; Kage, M.; et al. Association of PD-L1 overexpression with activating EGFR mutations in surgically resected nonsmall-cell lung cancer. Ann. Oncol. 2014, 25, 1935–1940. [Google Scholar] [CrossRef]
- Marzeca, M.; Zhanga, Q.; Goradiaa, A.; Puthiyaveettil; Raghunatha, N.; Liua, X.; Paesslera, M.; Wanga, H.Y.; Wysockab, M.; Chengc, M. Oncogenic kinase NPM/ALK induces through STAT3 expression of immunosuppressive protein CD274 (PD-L1, B7-H1). Proc. Natl. Acad. Sci. USA 2008, 105, 20852–20857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parsa, A.T.; Waldron, J.S.; Panner, A.; Crane, C.A.; Parney, I.F.; Barry, J.J.; Cachola, K.E.; Murray, J.C.; Tihan, T.; Jensen, M.C.; et al. Loss of tumor suppressor PTEN function increases B7-H1 expression and im-munoresistance in glioma. Nat. Med. 2007, 13, 84–88. [Google Scholar] [CrossRef]
- Xu, C.; Fillmore, C.M.; Koyama, S.; Wu, H.; Zhao, Y.; Chen, Z.; Herter-Sprie, G.S.; Akbay, E.A.; Tchaicha, J.H.; Altabef, A.; et al. Loss of Lkb1 and Pten leads to lung squamous cell carcinoma with elevated PD-L1 expression. Cancer Cell 2014, 25, 590–604. [Google Scholar] [CrossRef] [Green Version]
- Ikeda, S.; Okamoto, T.; Okano, S.; Umemoto, Y.; Tagawa, T.; Morodomi, Y.; Kohno, M.; Shimamatsu, S.; Kitahara, H.; Suzuki, Y.; et al. PD-L1 is upregulated by simultaneous amplification of the PD-L1 and JAK2 genes in non-small cell lung cancer. J. Thorac. Oncol. 2016, 11, 62–71. [Google Scholar] [CrossRef]
- Howitt, B.E.; Sun, H.H.; Roemer, M.G.; Kelley, A.; Chapuy, B.; Aviki, E.; Pak, C.; Connelly, C.; Gjini, E.; Shi, Y.; et al. Genetic basis for PD-L1 expression in squamous cell carcinomas of the cervix and vulva. JAMA Oncol. 2016, 2, 518–522. [Google Scholar] [CrossRef]
- Barrett, M.T.; Anderson, K.S.; Lenkiewicz, E.; Andreozzi, M.; Cunliffe, H.E.; Klassen, C.L.; Dueck, A.C.; McCullough, A.E.; Reddy, S.K.; Ramanathan, R.K.; et al. Genomic amplification of 9p24.1 targeting JAK2, PD-L1, and PD-L2 is enriched in high-risk triple negative breast cance. Oncotarget 2015, 6, 26483–26493. [Google Scholar] [CrossRef] [Green Version]
- Bai, J.; Gao, Z.; Li, X.; Dong, L.; Han, W.; Nie, J. Regulation of PD-1/PD-L1 pathway and resistance to PD-1/PD-L1 blockade. Oncotarget 2017, 8, 110693–110707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corzo, C.A.; Cotter, M.J.; Cheng, P.; Cheng, F.; Kusmartsev, S.; Sotomayor, E.; Padhya, T.; McCaffrey, T.V.; McCaffrey, J.C.; Gabrilovich, D.I. Mechanism regulating reactive oxygen species in tumor-induced myeloid-derived suppressor cells. J. Immunol. 2009, 182, 5693–5701. [Google Scholar] [CrossRef] [PubMed]
- Umansky, V.; Blattner, C.; Gebhardt, C.; Utikal, J. The role of Myeloid-Derived Suppressor Cells (MDSC) in cancer progression. Vaccines 2016, 4, 36. [Google Scholar] [CrossRef]
- Liu, Y.; Yu, Y.; Yang, S.; Zeng, B.; Zhang, Z.; Jiao, G.; Zhang, Y.; Cai, L.; Yang, R. Regulation of arginase I activity and expression by both PD-1 and CTLA-4 on the myeloid-derived suppressor cells. Cancer Immunol. Immunother. 2009, 58, 687–697. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, P.C.; Hernandez, C.P.; Quiceno, D.; Dubinett, S.M.; Zabaleta, J.; Ochoa, J.B.; Gilbert, J.; Ochoa, A.C. Arginase I in myeloid suppressor cells is induced by COX–2 in lung carcinoma. J. Exp. Med. 2005, 202, 931–939. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez, P.C.; Quiceno, D.G.; Zabaleta, J.; Ortiz, B.; Zea, A.H.; Piazuelo, M.B.; Delgado, A.; Correa, P.; Brayer, J.; Sotomayor, E.M. Arginase I production in the tumor microenvironment by mature myeloid cells inhibits T-cell receptor expression and antigen-speciWc T-cell responses. Cancer Res. 2004, 64, 5839–5849. [Google Scholar] [CrossRef] [Green Version]
- Youn, J.I.; Collazo, M.; Shalova, I.N.; Biswas, S.K.; Gabrilovich, D.I. Characterization of the nature of granulocytic myeloid-derived suppressor cells in tumor-bearing mice. J. Leukoc. Biol. 2012, 91, 167–181. [Google Scholar] [CrossRef] [Green Version]
- Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Bronte, V. Coordinated regulation of myeloid cells by tumours. Nat. Rev. Immunol. 2012, 12, 253–268. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Han, Y.; Guo, Q.; Zhang, M.; Cao, X. Cancer-expanded myeloid-derived suppressor cells induce anergy of NK cells through membrane-bound TGF-beta 1. J. Immunol. 2009, 182, 240–249. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Huang, J.; Ren, X.; Gorska, A.E.; Chytil, A.; Aakre, M.; Carbone, D.P.; Matrisian, L.M.; Richmond, A.; Lin, P.C.; et al. Abrogation of TGF beta signaling in mammary carcinomas recruits Gr-1+CD11b+ myeloid cells that promote metastasis. Cancer Cell 2008, 13, 23–35. [Google Scholar] [CrossRef] [Green Version]
- Huang, B.; Pan, P.-Y.; Li, Q.; Sato, A.I.; Levy, D.E.; Bromberg, J.; Divino, C.M.; Chen, S.-H. Gr-1+CD115+ immature myeloid suppressor cells mediate the development of tumor-induced T regulatory cells and T-cell anergy in tumor-bearing host. Cancer Res. 2006, 66, 1123–1131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, P.-Y.; Ma, G.; Weber, K.J.; Ozao-Choy, J.; Wang, G.; Yin, B.; Divino, C.M.; Chen, S.-H. Immune stimulatory receptor CD40 is required for T-cell suppression and T regulatory cell activation mediated by myeloid-derived suppressor cells in cancer. Cancer Res. 2010, 70, 99–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serafini, P.; Mgebroff, S.; Noonan, K.; Borrello, I. Myeloid-derived suppressor cells promote cross-tolerance in B-cell lymphoma by expanding regulatory T cells. Cancer Res. 2008, 68, 5439–5449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Srivastava, M.K.; Sinha, P.; Clements, V.K.; Rodriguez, P.; Ostrand-Rosenberg, S. Myeloid-derived suppressor cells inhibit T-cell activation by depleting cystine and cysteine. Cancer Res. 2010, 70, 68–77. [Google Scholar] [CrossRef] [Green Version]
- Lu, C.; Redd, P.S.; Lee, J.R.; Savage, N.; Liu, K. The expression profiles and regulation of PD-L1 in tumor-induced myeloid-derived suppressor cells. Oncoimmunology 2016, 5, e1247135. [Google Scholar] [CrossRef] [Green Version]
- Yang, R.; Cai, Z.; Zhang, Y.; Yutzy, W.H.T.; Roby, K.F.; Roden, R.B. CD80 in immune suppression by mouse ovarian carcinoma-associated Gr-1 +CD11b+ myeloid cells. Cancer Res. 2006, 66, 6807–6815. [Google Scholar] [CrossRef] [Green Version]
- Fang, W.; Zhang, J.; Hong, S.; Zhan, J.; Chen, N.; Qin, T.; Qin, T.; Tang, Y.; Zhang, Y.; Kang, S.; et al. EBV-driven LMP1 and IFN-gamma up-regu- late PD-L1 in nasopharyngeal carcinoma: Implications for oncotar- geted therapy. Oncotarget 2014, 5, 12189–12202. [Google Scholar] [CrossRef]
- Lee, S.J.; Jang, B.C.; Lee, S.W.; Yang, Y.I.; Suh, S.I.; Park, Y.M.; Oh, S.; Shin, J.G.; Yao, S.; Chen, L.; et al. Interferon regulatory factor-1 is prerequisite to the constitutive expression and IFN-gamma-induced upregulation of B7-H1 (CD274). FEBS Lett. 2006, 580, 755–762. [Google Scholar] [CrossRef] [Green Version]
- Taube, J.M.; Anders, R.A.; Young, G.D.; Xu, H.; Sharma, R.; McMiller, T.L.; Chen, S.; Klein, A.P.; Pardoll, D.M.; Topalian, S.L.; et al. Colocalization of inflammatory response with B7-h1 expression in human melanocytic lesions supports an adaptive resistance mechanism of immune escape. Sci. Transl. Med. 2012, 4, 127ra37. [Google Scholar] [CrossRef] [Green Version]
- Mandai, M.; Hamanishi, J.; Abiko, K.; Matsumura, N.; Baba, T.; Konishi, I. Dual faces of IFN gamma in cancer progression: A role of pd-l1 induc- tion in the determination of pro- and antitumor immunity. Clin. Cancer Res. 2016, 22, 2329–2334. [Google Scholar] [CrossRef] [Green Version]
- Noman, M.Z.; Desantis, G.; Janji, B.; Hasmim, M.; Karray, S.; Dessen, P.; Bronte, V.; Chouaib, S.J. PD-L1 is a novel direct target of HIF-1α, and its blockade under hypoxia enhanced MDSC-mediated T cell activation. Exp. Med. 2014, 211, 781–790. [Google Scholar] [CrossRef] [PubMed]
- Curiel, T.J.; Wei, S.; Dong, H.; Alvarez, X.; Cheng, P.; Mottram, P.; Krzysiek, R.; Knutson, K.L.; Daniel, B.; Zimmermann, M.C.; et al. Blockade of B7-H1 improves myeloid dendritic cell-mediated antitumor immunity. Nat. Med. 2003, 9, 562–567. [Google Scholar] [CrossRef] [PubMed]
- Fujimura, T.; Ring, S.; Umansky, V.; Mahnke, K.; Enk, A.H. Regulatory T cells stimulate B7-H1 expression in myeloid-derived suppressor cells in ret melanomas. J. Investig. Dermatol. 2012, 132, 1239–1246. [Google Scholar] [CrossRef] [Green Version]
- Holtzhausen, A.; Harris, W.; Ubil, E.; Hunter, D.M.; Zhao, J.; Zhang, Y.; Zhang, D.; Liu, Q.; Wang, X.; Graham, D.K.; et al. TAM family receptor kinase inhibition reverses MDSC-mediated suppression and augments Anti-PD-1 therapy in melanoma. Cancer Immunol. Res. 2019, 7, 1672–1686. [Google Scholar] [CrossRef]
- Condamine, T.; Mastio, J.; Gabrilovich, D.I. Transcriptional regulation of myeloid-derived suppressor cells. J. Leukoc. Biol. 2015, 98, 913–922. [Google Scholar] [CrossRef]
- Hsu, K.; Chung, Y.M.; Endoh, Y.; Geczy, C.L. TLR9 ligands induce S100A8 in macrophages via a STAT3-dependent pathway which requires IL-10 and PGE2. PLoS ONE 2014, 9, e103629. [Google Scholar] [CrossRef]
- Cheng, P.; Eksioglu, E.A.; Chen, X.; Kandell, W.; Le Trinh, T.; Cen, L.; Qi, J.; Sallman, D.A.; Zhang, Y.; Tu, N.; et al. S100A9-induced overexpression of PD-1/PD-L1 contributes to ineffective hematopoiesis in myelodysplastic syndromes. Leukemia 2019, 33, 2034–2046. [Google Scholar] [CrossRef] [PubMed]
- Cheng, P.; Corzo, C.A.; Luetteke, N.; Yu, B.; Nagaraj, S.; Bui, M.M.; Ortiz, M.; Nacken, W.; Sorg, C.; Vogl, T.; et al. Inhibition of dendritic cell differentiation and accumulation of myeloid-derived suppressor cells in cancer is regulated by S100A9 protein. J. Exp. Med. 2008, 205, 2235–2249. [Google Scholar] [CrossRef]
- Hou, A.; Hou, K.; Huang, Q.; Lei, Y.; Chen, W. Targeting myeloid-derived suppressor cell, a promising strategy to overcome resistance to immune checkpoint inhibitors. Front. Immunol. 2020, 11, 783. [Google Scholar] [CrossRef]
- Weber, R.; Fleming, V.; Hu, X.; Nagibin, V.; Groth, C.; Altevogt, P.; Utikal, J.; Umansky, V. Myeloid-derived suppressor cells hinder the anti-cancer activity of immune checkpoint inhibitors. Front. Immunol. 2018, 9, 1310. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.; Skora, A.D.; Li, Z.; Liu, Q.; Tam, A.J.; Blosser, R.L.; Diaz, L.A., Jr.; Papadopoulos, N.; Kinzler, K.W.; Vogelstein, B.; et al. Eradication of metastatic mouse cancers resistant to immune checkpoint blockade by suppression of myeloid-derived cells. Proc. Natl. Acad. Sci. USA 2014, 111, 11774–11779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meyer, C.; Cagnon, L.; Costa-Nunes, C.M.; Baumgaertner, P.; Montandon, N.; Leyvraz, L.; Michielin, O.; Romano, E.; Speiser, D.E. Frequencies of circulating MDSC correlate with clinical outcome of melanoma patients treated with ipilimumab. Cancer Immunol. Immunother. 2014, 63, 247–257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sade-Feldman, M.; Kanterman, J.; Klieger, Y.; Ish-Shalom, E.; Olga, M.; Saragovi, A.; Shtainberg, H.; Lotem, M.; Baniyash, M. Clinical significance of circulating CD33+CD11b+HLA-DR- myeloid cells in patients with stage iv melanoma treated with ipilimumab. Clin. Cancer Res. 2016, 22, 5661–5672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martens, A.; Wistuba-Hamprecht, K.; Foppen, M.G.; Yuan, J.; Postow, M.A.; Wong, P.; Romano, E.; Khammari, A.; Dreno, B.; Capone, M. Baseline peripheral blood biomarkers associated with clinical outcome of advanced melanoma patients treated with ipilimumab. Clin. Cancer Res. 2016, 22, 2908–2918. [Google Scholar] [CrossRef] [Green Version]
- Gebhardt, C.; Sevko, A.; Jiang, H.; Lichtenberger, R.; Reith, M.; Tarnanidis, K.; Holland-Letz, T.; Umansky, L.; Beckhove, P.; Sucker, A.; et al. Myeloid cells and related chronic inflammatory factors as novel predictive markers in melanoma treatment with ipilimumab. Clin. Cancer Res. 2015, 21, 5453–5459. [Google Scholar] [CrossRef] [Green Version]
- Weide, B.; Martens, A.; Zelba, H.; Stutz, C.; Derhovanessian, E.; Di Giacomo, A.M.; Maio, M.; Sucker, A.; Schilling, B.; Schadendorf, D.; et al. Myeloid-derived suppressor cells predict survival of patients with advanced melanoma: Comparison with regulatory T cells and NY-ESO-1- or melan-A-specific T cells. Clin. Cancer Res. 2014, 20, 1601–1609. [Google Scholar] [CrossRef] [Green Version]
- Soda, H.; Ogawara, D.; Fukuda, Y.; Tomono, H.; Okuno, D.; Koga, S.; Taniguchi, H.; Yoshida, M.; Harada, T.; Umemura, A.; et al. Dynamics of blood neutrophil—Related indices during nivolumab treatment may be associated with response to salvage chemotherapy for non—Small cell lung cancer: A hypothesis—Generating study. Thorac. Cancer 2019, 10, 341–346. [Google Scholar] [CrossRef]
- Feng, J.; Chen, S.; Li, S.; Wu, B.; Lu, J.; Tan, L.; Li, J.; Song, Y.; Shi, G.; Shi, Y.G.; et al. The association between monocytic myeloid-derived suppressor cells levels and the anti-tumor efficacy of anti-PD-1 therapy in NSCLC patients. Transl. Oncol. 2020, 13, 100865. [Google Scholar] [CrossRef]
- Law, A.M.K.; Valdes-Mora, F.; Gallego-Ortega, D. Myeloid-derived suppressor cells as a therapeutic target for cancer. Cells 2020, 9, 561. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, E. Gemcitabine selectively eliminates splenic Gr-1+/CD11b+ myeloid suppressor cells in tumor-bearing animals and enhances antitumor immune activity. Clin. Cancer Res. 2005, 11, 6713–6721. [Google Scholar] [CrossRef] [Green Version]
- Sevko, A.; Michels, T.; Vrohlings, M.; Umansky, L.; Beckhove, P.; Kato, M.; Shurin, G.V.; Shurin, M.R.; Umansky, V. Antitumor effect of paclitaxel is mediated by inhibition of myeloid-derived suppressor cells chronic inflammation in the spontaneous melanoma model. J. Immunol. 2013, 190, 2464–2471. [Google Scholar] [CrossRef] [PubMed]
- Eriksson, E. Gemcitabine reduces MDSCs, tregs andTGFbeta-1while restoring the teff/treg ratio in patients with pancreatic cancer. J. Transl. Med. 2016, 14, 282. [Google Scholar] [CrossRef] [PubMed]
- Vincent, J. 5-Fluorouraci lselectively kills tumor-associated myeloid-derived suppressor cells resulting in enhanced T cell-dependent antitumor immunity. Cancer Res. 2010, 70, 3052–3061. [Google Scholar] [CrossRef] [Green Version]
- Fleming, V. Targeting myeloid-derived suppressor cells to bypass tumor-induced immunosuppression. Front. Immunol. 2018, 9, 398. [Google Scholar] [CrossRef] [PubMed]
- Ko, J.S. Sunitinib mediates reversal of myeloid-derived suppressor cell accumulation in renal cell carcinoma patients. Clin. Cancer Res. 2009, 15, 2148–2157. [Google Scholar] [CrossRef] [Green Version]
- Kodera, Y. Sunitinib inhibits lymphatic endothelial cell functions and lymphnode metastasis in a breast cancer model through inhibition of vascular endothelial growth factor receptor 3. Breast Cancer Res. 2011, 13, R66. [Google Scholar] [CrossRef] [Green Version]
- Stiff, A.; Trikha, P.; Wesolowski, R.; Kendra, K.; Hsu, V.; Uppati, S.; McMichael, E.; Duggan, M.; Campbell, A.; Keller, K.; et al. Myeloid-derived suppressor cells express bruton’s tyrosine kinase and can be depleted in tumor-bearing hosts by ibrutinib treatment. Cancer Res. 2016, 76, 2125–2136. [Google Scholar] [CrossRef] [Green Version]
- Bluttner, C. CCR5(+) myeloid-derived suppressor cells are enriched and activated in melanoma lesions. Cancer Res. 2018, 78, 157–167. [Google Scholar] [CrossRef] [Green Version]
- Robinson, S.C. A chemokine receptor antagonist inhibits experimental breast tumor growth. Cancer Res. 2003, 63, 8360–8365. [Google Scholar]
- Velasco-Velazquez, M. CCR5 antagonist blocks metastasis of basal breast cancer cells. Cancer Res. 2012, 72, 3839–3850. [Google Scholar] [CrossRef] [Green Version]
- Holmgaard, R.B. Targeting myeloid-derived suppressor cells with colony stimulating factor-1 receptor blockade can reverse immune resistance to immunotherapy in indoleamine 2,3-dioxygenase-expressing tumors. E Bio. Med. 2016, 6, 50–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Y. CSF1/CSF1Rblockade reprograms tumor-infiltrating macrophages and improves response to T-cell checkpoint immunotherapy in pancreatic cancer models. Cancer Res. 2014, 74, 5057–5069. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richardsen, E. Macrophage-colony stimulating factor (CSF1) predicts breast cancer progression and mortality. Anticancer Res. 2015, 35, 865–874. [Google Scholar] [PubMed]
- Sluijter, M. Inhibition of. CSF-1R supports T-cell mediated melanoma therapy. PLoS ONE 2014, 9, e104230. [Google Scholar] [CrossRef] [PubMed]
- Mok, S. Inhibition of CSF-1receptorimprovestheantitumorefficacyofadoptivecelltransferimmunotherapy. Cancer Res. 2014, 74, 153–161. [Google Scholar] [CrossRef] [Green Version]
- Obermajer, N.; Muthuswamy, R.; Lesnock, J.; Edwards, R.P.; Kalinski, P. Positive feedback between PGE2 and COX2 redirects the differentiation of human dendritic cells toward stable myeloid-derived suppressor cells. Blood 2011, 118, 5498–5505. [Google Scholar] [CrossRef]
- Obermajer, N.; Kalinski, P. Generation of myeloid-derived suppressor cells using prostaglandin E2. Transplant. Res. 2012, 1, 15. [Google Scholar] [CrossRef] [Green Version]
- Mao, Y.; Poschke, I.; Wennerberg, E.; Pico de Coaña, Y.; Egyhazi Brage, S.; Schultz, I.; Hansson, J.; Masucci, G.; Lundqvist, A.; Kiessling, R. Melanoma- educated CD14+ cells acquire a myeloid-derived suppressor cell phenotype through COX-2-dependent mechanisms. Cancer Res. 2013, 73, 3877–3887. [Google Scholar] [CrossRef] [Green Version]
- Mao, Y.; Sarhan, D.; Steven, A.; Seliger, B.; Kiessling, R.; Lundqvist, A. Inhibition of tumor-derived prostaglandin-e2 blocks the induction of myeloid-derived suppressor cells and recovers natural killer cell activity. Clin. Cancer Res. 2014, 20, 4096–4106. [Google Scholar] [CrossRef] [Green Version]
- Fujita, M.; Kohanbash, G.; Fellows-Mayle, W.; Hamilton, R.L.; Komohara, Y.; Decker, S.A.; Ohlfest, J.R.; Okada, H. COX-2 blockade suppresses gliomagenesis by inhibiting myeloid-derived suppressor cells. Cancer Res. 2011, 71, 2664–2674. [Google Scholar] [CrossRef] [Green Version]
- Veltman, J.D.; Lambers, M.E.; van Nimwegen, M.; Hendriks, R.W.; Hoogsteden, H.C.; Aerts, J.G.; Hegmans, J.P. COX-2 inhibition improves immunotherapy and is associated with decreased numbers of myeloid-derived suppressor cells in mesothelioma. Celecoxib influences MDSC function. BMC Cancer 2010, 10, 464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ochoa, A.C. Arginase, prostaglandins and myeloid-derived suppressor cells in renalcellcarcinoma. Clin. Cancer Res. 2007, 13, 721s–726s. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zelenay, S. Cyclooxygenase-dependent tumor growth through evasion of immunity. Cell 2015, 162, 1257–1270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohl, K.; Tenbrock, K. Reactive oxygen species as regulators of MDSC-mediated immune suppression. Front. Immunol. 2018, 9, 2499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.Y. Bardoxolone methyl(CDDO-Me)as a therapeutic agent: An update on its pharmacokinetic and pharmacodynamic properties. Drug Des. Devel. Ther. 2014, 8, 2075–2088. [Google Scholar]
- Hiramoto, K. Myeloid lineage-specific deletion of antioxidant system enhances tumor metastasis. Cancer Prev. Res. 2014, 7, 835–844. [Google Scholar] [CrossRef] [Green Version]
- Nagaraj, S. Anti-inflammatory triterpenoid blocks immunesuppressive function of MDSCs and improves immune response in cancer. Clin. Cancer Res. 2010, 16, 1812–1823. [Google Scholar] [CrossRef] [Green Version]
- Kusmartsev, S. Reversalofmyeloidcell-mediated immunosuppression in patients with metastatic renal cell carcinoma. Clin. Cancer Res. 2008, 14, 8270–8278. [Google Scholar] [CrossRef] [Green Version]
- Mirza, N. All-trans-retinoicacidimprovesdifferentiationofmyeloidcellsandimmuneresponseincancer patients. Cancer Res. 2006, 66, 9299–9307. [Google Scholar] [CrossRef] [Green Version]
- Iclozan, C. Therapeutic regulation of myeloid-derived suppressor cells and immune response to cancer vaccine in patients with extensive stage small cell lung cancer. Cancer Immunol. Immunother. 2013, 62, 909–918. [Google Scholar] [CrossRef] [Green Version]
- Mikyskova, R. DNA demethylatingagent5-azacytidine inhibits myeloid-derivedsuppressor cells induced by tumor growth and cyclophosphamide treatment. J. Leukoc. Biol. 2014, 95, 743–753. [Google Scholar] [CrossRef] [PubMed]
- Kodumudi, K.N. Anovel chemoimmunomodulating property of. docetaxel: Suppression of myeloid-derived suppressor cells in tumor bearers. Clin. Cancer Res. 2010, 16, 4583–4594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varikuti, S.; Singh, B.; Volpedo, G.; Ahirwar, D.K.; Jha, B.K.; Saljoughian, N.; Viana, A.G.; Verma, C.; Hamza, O.; Halsey, G.; et al. Ibrutinib treatment inhibits breast cancer progression and metastasis by inducing conversion of myeloid-derived suppressor cells to dendritic cells. Br. J. Cancer 2020, 122, 1005–1013. [Google Scholar] [CrossRef] [PubMed]
- Huang, A.; Zhang, B.; Yan, W.; Wang, B.; Wei, H.; Zhang, F.; Wu, L.; Fan, K.; Guo, Y. Myeloid-derived suppressor cells regu- late immune response in patients with chronic hepatitis B virus in- fection through PD-1-induced IL-10. J. Immunol. 2014, 193, 5461–5469. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.; Bueso-Ramos, C.; DiNardo, C.; Estecio, M.R.; Davanlou, M.; Geng, Q.R.; Fang, Z.; Nguyen, M.; Pierce, S.; Wei, Y.; et al. Expression of PD-L1, PD-L2, PD-1 and CTLA4 in myelodysplastic syndromes is enhanced by treatment with hypomethylating agents. Leukemia 2014, 28, 1280–1288. [Google Scholar] [CrossRef]
- Boddu, P.; Kantarjian, H.; Garcia-Manero, G.; Allison, J.; Sharma, P.; Daver, N. The emerging role of immune checkpoint ased approa- ches in AML and MDS. Leuk. Lymphoma. 2018, 59, 790–802. [Google Scholar] [CrossRef]
- Wang, L.; Rubinstein, R.; Lines, J.L.; Wasiuk, A.; Ahonen, C.; Guo, Y.; Lu, L.F.; Gondek, D.; Wang, Y.; Fava, R.A.; et al. Vista, a novel mouse ig superfamily ligand that negatively regulates t cell responses. J. Exp. Med. 2011, 208, 577–592. [Google Scholar] [CrossRef]
- Flies, D.B.; Wang, S.; Xu, H.; Chen, L. Cutting edge: A monoclonal antibody specific for the programmed death-1 homolog prevents graft-versus-host disease in mouse models. J. Immunol. 2011, 187, 1537–1541. [Google Scholar] [CrossRef]
- Wang, L.; Jia, B.; Claxton, D.F.; Ehmann, W.C.; Rybka, W.B.; Mineishi, S.; Naik, S.; Khawaja, M.R.; Sivik, J.; Han, J.; et al. HVISTA is highly expressed on MDSCs mediates an inhibition of T cell response in patients with AML. Oncoimmunology 2018, 7, e1469594. [Google Scholar] [CrossRef]
- Le Mercier, I.; Chen, W.; Lines, J.L.; Day, M.; Li, J.; Sergent, P.; Noelle, R.J.; Wang, L. Vista regulates the development of protective antitumor immunity. Cancer Res. 2014, 74, 1933–1944. [Google Scholar] [CrossRef] [Green Version]
- Lines, J.L.; Pantazi, E.; Mak, J.; Sempere, L.F.; Wang, L.; O’Connell, S.; Ceeraz, S.; Suriawinata, A.A.; Yan, S.; Ernstoff, M.S.; et al. Vista is an immune checkpoint molecule for human t cells. Cancer Res. 2014, 74, 1924–1932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le Mercier, I.; Lines, J.L.; Noelle, R.J. Beyond ctla-4 and pd-1, the generation z of negative checkpoint regulators. Front. Immunol. 2015, 6, 418. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Allavena, P.; Sica, A.; Balkwill, F. Cancer-related inflammation. Nature 2008, 454, 436–444. [Google Scholar] [CrossRef] [PubMed]
- Hasselbalch, H.C. Chronic inflammation as a promotor of mutagenesis in essential thrombocythemia, polycythemia vera and myelofibrosis. A human inflammation model for cancer development? Leuk. Res. 2013, 37, 214–220. [Google Scholar] [CrossRef]
- Andersen, M.; Sajid, Z.; Pedersen, R.K.; Gudmand-Hoeyer, J.; Ellervik, C.; Skov, V.; Kjær, L.; Pallisgaard, N.; Kruse, T.A.; Thomassen, M.; et al. Mathematical modelling as a proof of concept for MPNs as a human inflammation model for cancer development. PLoS ONE 2017, 12, e0183620. [Google Scholar] [CrossRef] [Green Version]
- Hermouet, S.; Godard, A.; Pineau, D.; Corre, I.; Raher, S.; Lippert, E.; Jacques, Y. Abnormal production of interleukin (IL)-11 and IL-8 in polycythaemia vera. Cytokine 2002, 20, 178–183. [Google Scholar] [CrossRef]
- Panteli, K.E.; Hatzimichael, E.C.; Bouranta, P.K.; Katsaraki, A.; Seferiadis, K.; Stebbing, J.; Bourantas, K.L. Serum interleukin (IL)-1, IL-2, SIL-2Ra, IL-6 and thrombopoietin levels in patients with chronic myeloproliferative diseases. Br. J. Haematol. 2005, 130, 709–715. [Google Scholar] [CrossRef]
- Alonci, A.; Allegra, A.; Bellomo, G.; Penna, G.; D’Angelo, A.; Quartarone, E.; Musolino, C. Evaluation of circulating endothelial cells, VEGF and VEGFR2 serum levels in patients with chronic myeloproliferative diseases. Hematol. Oncol. 2008, 26, 235–239. [Google Scholar] [CrossRef]
- Allegra, A.; Alonci, A.; Bellomo, G.; D’Angelo, A.; Granata, A.; Russo, S.; Quartarone, E.; Musolino, C. Evaluation of interleukin-17 serum levels in patients with chronic myeloproliferative diseases. Tumori 2009, 95, 404–405. [Google Scholar] [CrossRef]
- Pourcelot, E.; Trocme, C.; Mondet, J.; Bailly, S.; Toussaint, B.; Mossuz, P. Cytokine profiles in polycythemia vera and essential thrombocythemia patients: Clinical implications. Exp. Hematol. 2014, 42, 360–368. [Google Scholar] [CrossRef]
- Boissinot, M.; Cleyrat, C.; Vilaine, M.; Jacques, Y.; Corre, I.; Hermouet, S. Anti-inflammatory cytokines hepatocyte growth factor and interleukin-11 are over-expressed in polycythemia vera and contribute to the growth of clonal erythroblasts independently of JAK2V617F. Oncogene 2011, 30, 990–1001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vener, C.; Novembrino, C.; Catena, F.B.; Fracchiolla, N.S.; Gianelli, U.; Savi, F.; Radaelli, F.; Fermo, E.; Cortelezzi, A.; Lonati, S.; et al. Oxidative stress is increased in primary and post-polycythemia vera myelofibrosis. Exp. Hematol. 2010, 38, 1058–1065. [Google Scholar] [CrossRef] [PubMed]
- Marty, C.; Lacout, C.; Droin, N.; Le Couédic, J.P.; Ribrag, V.; Solary, E.; Vainchenker, W.; Villeval, J.L.; Plo, I. A role for reactive oxygen species in JAK2 V617F myeloproliferative neoplasm progression. Leukemia 2013, 27, 2187–2195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hermouet, S.; Hasselbalch, H.C.; Cokic, V. Mediators of inflammation in myeloproliferative neoplasms: State of the art. Mediat. Inflamm. 2015, 2015, 964613. [Google Scholar] [CrossRef]
- Thiele, J.; Kvasnicka, H.M.; Dietrich, H.; Stein, G.; Hann, M.; Kaminski, A.; Rathjen, N.; Metz, K.A.; Beelen, D.W.; Ditschkowski, M.; et al. Dynamics of bone marrow changes in patients with chronic idiopathic myelofibrosis following allogeneic stem cell transplantation. Histol. Histopathol. 2005, 20, 879–889. [Google Scholar]
- Mantovani, A.; Garlanda, C.; Allavena, P. Molecular pathways and targets in cancer-related inflammation. Ann. Med. 2010, 42, 161–170. [Google Scholar] [CrossRef]
- Lussana, F.; Rambaldi, A.J. Inflammation and myeloproliferative neoplasms. J. Autoimmun. 2017, 85, 58–63. [Google Scholar] [CrossRef]
- Jutzi, J.S.; Pahl, H.L. The hen or the egg: Inflammatory aspects of murine MPN models. Mediat. Inflamm. 2015, 2015, 101987. [Google Scholar] [CrossRef] [Green Version]
- Goyette, J.; Geczy, C.L. Inflammation-associated S100 proteins: New mechanisms that regulate function. Amino. Acids. 2011, 41, 821–842. [Google Scholar] [CrossRef]
- Turovskaya, O.; Foell, D.; Sinha, P.; Vogl, T.; Newlin, R.; Nayak, J.; Nguyen, M.; Olsson, A.; Nawroth, P.P.; Bierhaus, A.; et al. RAGE, carboxylated gly-cans and S100A8/A9 play essential roles in colitis-associated car- cinogenesis. Carcinogenesis 2008, 29, 2035–2043. [Google Scholar] [CrossRef]
- Goldszmid, R.S.; Trinchieri, G. The price of immunity. Nat. Immunol. 2012, 13, 932–938. [Google Scholar] [CrossRef] [PubMed]
- Newton, K.; Dixit, V.M. Signaling in innate immunity and inflammation. Cold Spring Harb. Perspect. Biol. 2012, 4, a006049. [Google Scholar] [CrossRef] [PubMed]
- So, E.Y.; Ouchi, T. The application of toll like receptors for cancer therapy. Int. J. Biol. Sci. 2010, 6, 675–681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoebe, K.; Du, X.; Georgel, P.; Janssen, E.; Tabeta, K.; Kim, S.O.; Goode, J.; Lin, P.; Mann, N.; Mudd, S.; et al. Identification of Lps2 as a key transducer of MyD88-independent TIR signalling. Nature 2003, 424, 743–748. [Google Scholar] [CrossRef]
- Hemmati, S.; Haque, T.; Gritsman, K. Inflammatory signaling pathways in preleukemic and leukemic stem cells. Front. Oncol. 2017, 7, 265. [Google Scholar] [CrossRef] [Green Version]
- Allegra, A.; Pioggia, G.; Tonacci, A.; Casciaro, M.; Musolino, C.; Gangemi, S. Synergic crosstalk between inflammation, oxidative tress, and genomic alterations in BCR-ABL-negative myeloproliferative neoplasm. Antioxidants 2020, 9, 1037. [Google Scholar] [CrossRef]
- Bjørn, M.E.; Hasselbalch, H.C. The role of reactive oxygen species in myelofibrosis and related neoplasms. Mediat. Inflamm. 2015, 2015, 648090. [Google Scholar] [CrossRef] [Green Version]
- Dupont, S.; Masse, A.; James, C.; Teyssandier, I.; Lecluse, Y.; Larbret, F.; Ugo, V.; Saulnier, P.; Koscielny, S.; Le Couédic, J.P.; et al. The JAK2 617V4F mutation triggers erythropoietin hypersensitivity and terminal erythroid amplification in primary cells from patients with polycythemia vera. Blood 2007, 110, 1013–1021. [Google Scholar] [CrossRef]
- Godfrey, A.L.; Chen, E.; Pagano, F.; Ortmann, C.A.; Silber, Y.; Bellosillo, B.; Guglielmelli, P.; Harrison, C.N.; Reilly, J.T.; Stegelmann, F.; et al. JAK2V617F homozygosity arises commonly and recurrently in PV and ET, but PV is characterized by expansion of a dominant homozygous subclone. Blood 2012, 120, 2704–2707. [Google Scholar] [CrossRef]
- Tiedt, R.; Hao-Shen, H.; Sobas, M.A.; Looser, R.; Dirnhofer, S.; Schwaller, J.; Skoda, R.C. Ratio of mutant JAK2-V617F to wild-type Jak2 determines the MPD phenotypes in transgenic mice. Blood 2008, 111, 3931–3940. [Google Scholar] [CrossRef]
- Kleppe, M.; Kwak, M.; Koppikar, P.; Riester, M.; Keller, M.; Bastian, L.; Hricik, T.; Bhagwat, N.; McKenney, A.S.; Papalexi, E.; et al. JAK-STAT pathway activation in malignant and nonmalignant cells contributes to MPN pathogenesis and therapeutic response. Cancer Discov. 2015, 5, 316–331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cook, E.K.; Luo, M.; Rauh, M.J. Clonal hematopoiesis and inflammation: Partners in leukemogenesis and comorbidity. Exp. Hematol. 2020, 83, 85–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, G.; Nair, K.; Ramachandran, P.; Chen, C.; Wong, C.; Joseph, G.; Gotlieb, V.; Liaukovich, M.; Wang, J.C. TLR, RAGE and HMGB1 in the inflammatory response in Ph(-) myeloproliferative neoplasm. Blood 2020, 136, 31–32. [Google Scholar] [CrossRef]
- Kovačić, M.; Mitrović-Ajtić, O.; Beleslin-Čokić, B.; Djikić, D.; Subotički, T.; Diklić, M.; Leković, D.; Gotić, M.; Mossuz, P.; Čokić, V.P. TLR4 and RAGE conversely mediate pro-inflammatory S100A8/9-mediated inhibition of proliferation-linked signaling in myeloproliferative neoplasms. Cell. Oncol. 2018, 41, 541–553. [Google Scholar] [CrossRef]
- Wang, J.C.; Kundra, A.; Andrei, M.; Baptiste, S.; Chen, C.; Wong, C.; Sindhu, H. Myeloid-derived suppressor cells in patients with myeloproliferative neoplasm. Leuk. Res. 2016, 43, 39–43. [Google Scholar] [CrossRef]
- Pan, P.Y.; Wang, G.X.; Yin, B.; Ozao, J.; Ku, T.; Divino, C.M.; Chen, S.H. Reversion of immune tolerance in advanced malignancy: Modulation of myeloid derived suppressor cell development by block- ade of SCF function. Blood 2008, 111, 219–228. [Google Scholar] [CrossRef]
- Briard, D.; Brouty-Boye, D.; Giron-Michel, J.; Azzarone, B.; Jasmin, C.; Le Bousse-Kerdiles, C. Impaired NK cell differentiation of blood-derived CD34C progenitors from patients with myeloid metaplasia with mye- lofibrosis. Clin. Immunol. 2003, 106, 201–212. [Google Scholar] [CrossRef]
- Keohane, C.; Kordasti, S.; Seidl, T.; Perez Abellan, P.; Thomas, N.S.B.; Harrison, C.N.; McLornan, D.P.; Mufti, G.J. JAK Inhibition induces silencing of T helper cytokine secretion and a profound reduction in t regulatory cells. Br. J. Haematol. 2015, 171, 60–73. [Google Scholar] [CrossRef] [Green Version]
- Massa, M.; Campanelli, R.; Fois, G.; Villani, L.; Bonetti, E.; Catarsi, P.; Poletto, V.; Viarengo, G.; De Amici, M.; Rosti, V.; et al. Reduced frequency of cir- culating CD4CCD25brightCD127lowFOXP3C regulatory T cells in primary myelofibrosis. Blood 2016, 128, 1660–1662. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.C.; Sindhu, H.; Chen, C.; Kundra, A.; Kafeel, M.I.; Wong, C.; Lichter, S. Immune derangements in patients with myelofibrosis: The role of Treg, Th17, and sIL2Ra. PLoS ONE 2015, 10, e0116723. [Google Scholar] [CrossRef]
- Riley, C.H.; Jensen, M.K.; Brimnes, M.K.; Hasselbalch, H.C.; Bjerrum, O.W.; Straten, P.T.; Svane, I.M. Increase in circulating CD4CCD25CFoxp3C T cells in patients with Philadelphia-negative chronic myeloprolif- erative neoplasms during treatment with IFN-a. Blood 2011, 118, 2170–2173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Froom, P.; Aghai, E.; Kinarty, A.; Lahat, N. Decreased natural killer (NK) activity in patients with myeloproliferative disorders. Cancer 1989, 64, 1038–1040. [Google Scholar] [CrossRef]
- Romano, M.; Sollazzo, D.; Trabanelli, S.; Barone, M.; Polverelli, N.; Perricone, M.; Forte, D.; Luatti, S.; Cavo, M.; Vianelli, N.; et al. Mutations in JAK2 and calreticulin genes are associated with specific alterations of the immune system in myelofibrosis. OncoImmunology 2017, 6, e1345402. [Google Scholar] [CrossRef] [Green Version]
- Holmstrom, M.O.; Martinenaite, E.; Ahmad, S.M.; Met, Ö.; Friese, C.; Kjær, L.; Riley, C.H.; Thor Straten, P.; Svane, I.M.; Hasselbalch, H.C.; et al. The calreticulin (CALR) exon 9 mutations are promising targets for cancer immune therapy. Leukemia 2018, 32, 429–437. [Google Scholar] [CrossRef] [PubMed]
- Holmstrom, M.O.; Ahmad, S.M.; Klausen, U.; Bendtsen, S.K.; Martinenaite, E.; Riley, C.H.; Svane, I.M.; Kjær, L.; Skov, V.; Ellervik, C.; et al. High frequencies of circulating memory T cells specific for calreticulin exon 9 mutations in healthy individuals. Blood Cancer J. 2019, 9, 8. [Google Scholar] [CrossRef]
- Cimen Bozkus, C.; Roudko, V.; Finnigan, J.P.; Mascarenhas, J.; Hoffman, R.; Iancu-Rubin, C.; Bhardwaj, N. Immune checkpoint blockade enhances shared neoantigen-induced T-cell immunity directed against mutated calreticulin in myeloproliferative neo-plasms. Cancer Discov. 2019, 9, 1192–1207. [Google Scholar] [CrossRef]
- de Haas, N.; de Koning, C.; Spilgies, L.; de Vries, I.J.; Hato, S.V. Improving cancer immunotherapy by targeting the STATe of MDSCs. Oncoimmunology 2016, 5, e1196312. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez-Pascual, J.; Ayuso-Sacido, A.; Belda-Iniesta, C. Drug resistance in cancer immunotherapy: New strategies to improve checkpoint inhibitor therapies. Cancer Drug Resist. 2019, 2, 980–993. [Google Scholar] [CrossRef] [Green Version]
- Koustas, E.; Sarantis, P.; Papavassiliou, A.G.; Karamouzis, M.V. The resistance mechanisms of checkpoint inhibitors in solid tumors. Biomolecules 2020, 10, 666. [Google Scholar] [CrossRef]
- Verstovsek, S.; Mesa, R.A.; Gotlib, J.; Levy, R.S.; Gupta, V.; DiPersio, J.F.; Catalano, J.V.; Deininger, M.; Miller, C.; Silver, R.T.; et al. A double-blind, placebo-controlled trial of ruxolitinib for myelofibrosis. N. Engl. J. Med. 2012, 366, 799–807. [Google Scholar] [CrossRef] [Green Version]
- Giles, H.; Pratt, G. Janus kinase 2 (JAK2) inhibitors in the treatment of multiple myeloma: Modulating the myeloma immune microenvironment. Br. J. Haematol. 2021, 192, 420–422. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Li, M.; Ng, N.; Yu, E.; Bujarski, S.; Yin, Z.; Wen, M.; Hekmati, T.; Field, D.; Wang, J.; et al. Ruxolitinib reverses checkpoint inhibition by reducing programmed cell death ligand-1 (PD-L1) expression and increases anti-tumour effects of T cells in multiple myeloma. Br. J. Haematol. 2021, 192, 568–576. [Google Scholar] [CrossRef] [PubMed]
- Bachanova, V.; Hegerova, L.; Cao, Q.; Janakiram, M.; Maakaron, J.; Ayyappan, S.; Weisdorf, D.J.; Zak, J.; Farooq, U.; Kenkre, V.P. Ruxolitinib plus nivolumab in patients with R/R hodgkin lymphoma after failure of check-point inhibitors: Preliminary report on safety and efficacy. Blood 2021, 138, 230. [Google Scholar] [CrossRef]
- Wang, J.; Chen, C.; Gotlieb, V.; Nalghranyan, S.; Wong, C.; Yeo, I. Elevated levels of PD-L1 on MDSCs in patients with Ph(-) myeloproliferative neoplasm. Blood 2021, 138, 4591. [Google Scholar] [CrossRef]
- Tefferi, A.; Cortes, J.; Verstovsek, S.; Mesa, R.A.; Thomas, D.; Lasho, T.L.; Hogan, W.J.; Litzow, M.R.; Allred, J.B.; Jones, D.; et al. Lena-lidomide therapy in myelofibrosis with myeloid metaplasia. Blood 2006, 108, 1158–1164. [Google Scholar] [CrossRef] [Green Version]
- Quintás-Cardama, A.; Kantarjian, H.M.; Manshouri, T.; Thomas, D.; Cortes, J.; Ravandi, F.; Garcia-Manero, G.; Ferrajoli, A.; Bueso-Ramos, C.; Verstovsek, S. Lenalidomide plus prednisone results in durable clinical, histopathologic, and molecular responses in patients with myelofibrosis. J. Clin. Oncol. 2009, 27, 4760–4766. [Google Scholar] [CrossRef] [Green Version]
- Görgün, G.; Samur, M.K.; Cowens, K.B.; Paula, S.; Bianchi, G.; Anderson, J.E.; White, R.E.; Singh, A.; Ohguchi, H.; Suzuki, R.; et al. Lenalidomide enhances immune checkpoint blockade-induced immune response in multiple myeloma. Clin. Cancer Res. 2015, 21, 4607–4618. [Google Scholar] [CrossRef] [Green Version]
- Shanafelt, T.D.; Wang, X.V.; Kay, N.E.; Hanson, C.A.; O’Brien, S.; Barrientos, J.; Jelinek, D.F.; Braggio, E.; Leis, J.F.; Zhang, C.C.; et al. Ibrutinib-rituximab or chemoimmunotherapy for chronic lymphocytic leukemia. N. Engl. J. Med. 2019, 381, 432–443. [Google Scholar] [CrossRef]
- Lee, K.G.; Xu, S.; Kang, Z.H.; Huo, J.; Huang, M.; Liu, D.; Takeuchi, O.; Akira, S.; Lam, K.P. Bruton’s tyrosine kinase phosphorylates Toll-like receptor 3 to initiate antiviral response. Proc. Natl. Acad. Sci. USA 2012, 109, 5791–5796. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Zhan, Z.; Li, D.; Xu, L.; Ma, F.; Zhang, P.; Yao, H.; Cao, X. Intracellular MHC class II molecules promote TLR-triggered innate immune responses by maintain-ing activation of the kinase Btk. Nat. Immunol. 2011, 12, 416–424. [Google Scholar] [CrossRef]
- Bunt, S.K.; Clements, V.K.; Hanson, E.M.; Sinha, P.; Ostrand-Rosenberg, S. Inflammation enhances myeloid-derived suppressor cell cross-talk by signaling through Toll-like receptor 4. J. Leukoc. Biol. 2009, 85, 996–1004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gill, H.; Yacoub, A.; Pettit, K.M.; Bradley, T.; Gerds, A.T.; Tatarczuch, M.; Shortt, J.; Curtin, N.J.; Rossetti, J.M.; Burbury, K.; et al. A phase 2 study of the LSD1 inhibitor Img-7289 (bomedemstat) for the treatment of advanced myelofibrosis. Blood 2021, 138, 139. [Google Scholar] [CrossRef]
- Kremyanskaya, M.; Mascarenhas, J.; Palandri, F.; Vannucchi, A.; Verstovsek, S.; Harrison, C.N.; Bose, P.; Schiller, G.J.; Rampal, R.; Drummond, M.W.; et al. Pelabresib (CPI-0610) monotherapy in patients with myelofibrosis—Update of clinical and translational data from the ongoing manifest trial. Blood 2021, 138, 141. [Google Scholar] [CrossRef]
- Verstovsek, S.; Salama, M.E.; Mascarenhas, J.; Talpaz, M.; Mesa, R.A.; Vannucchi, A.; Rampal, R.; Oh, S.T.; Olteanu, H.; Chiu, A.; et al. Disease-modifying potential of BET inhibitor pelabresib (CPI-0610) as demonstrated by improvements in bone marrow function and clinical activity in patients with myelofibrosis—Preliminary data. Blood 2021, 138, 2568. [Google Scholar] [CrossRef]
- Mascarenhas, J.; Kosiorek, H.; Bhave, R.; Palmer, J.; Kuykendall, A.; Mesa, R.; Rampal, R.; Gerds, A.; Yacoub, A.; Pettit, K.M.; et al. Treatment of Myelofibrosis Patients with the TGF-β 1/3 Inhibitor AVID200 (MPN-RC 118) Induces a Profound Effect on Platelet Production. Blood 2021, 138, 142. [Google Scholar] [CrossRef]
- Ehrchen, J.M.; Sunderkötter, C.; Foell, D.; Vogl, T.; Roth, J. The endogenous toll-like receptor 4 agonist S100A8/S100A9 (calprotectin) as innate amplifier of infection, autoimmunity, and cancer. J. Leukoc. Biol. 2009, 86, 557–566. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Anti-MDSC in Cancer | |||
|---|---|---|---|
| Agent | |||
| 1 | Depleting MDSC |
| 2. inhibit VEGF, angioenesis, STAT 3 of tumor microenvironment: sunnitinib etc. |
| 2 | Blocking MDSC recruitment |
| 3. NLRP pathway inhibitor |
| 3 | Attenuating the immunosuppressive mechanisms of MDSC | 1. COX2 inhibit PGE2 then inhibit arginase | 2. Triterpenoid activate Nrf2 to reduce ROS formation |
| 4 | Induction of Differentiation of MDSC | 1. ATRA induced differentiation of MDSC in both mice and patients in various cancer types, such as renal cell carcinoma |
|
| 5 | Decreasing VISTA, PD-L1 expression on the MDSC | PD-1, PD-L1 antibody and anti-Vista antibody | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, J.-C.; Sun, L. PD-1/PD-L1, MDSC Pathways, and Checkpoint Inhibitor Therapy in Ph(-) Myeloproliferative Neoplasm: A Review. Int. J. Mol. Sci. 2022, 23, 5837. https://doi.org/10.3390/ijms23105837
Wang J-C, Sun L. PD-1/PD-L1, MDSC Pathways, and Checkpoint Inhibitor Therapy in Ph(-) Myeloproliferative Neoplasm: A Review. International Journal of Molecular Sciences. 2022; 23(10):5837. https://doi.org/10.3390/ijms23105837
Chicago/Turabian StyleWang, Jen-Chin, and Lishi Sun. 2022. "PD-1/PD-L1, MDSC Pathways, and Checkpoint Inhibitor Therapy in Ph(-) Myeloproliferative Neoplasm: A Review" International Journal of Molecular Sciences 23, no. 10: 5837. https://doi.org/10.3390/ijms23105837
APA StyleWang, J. -C., & Sun, L. (2022). PD-1/PD-L1, MDSC Pathways, and Checkpoint Inhibitor Therapy in Ph(-) Myeloproliferative Neoplasm: A Review. International Journal of Molecular Sciences, 23(10), 5837. https://doi.org/10.3390/ijms23105837