
Substituted Syndecan-2-Derived Mimetic Peptides Show Improved Antitumor Activity over the Parent Syndecan-2-Derived Peptide

 , ,
, ,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Syndecan-2 Mediated the Localization of Pro-MMP-7 by Interacting with Pro-Domain of MMP-7
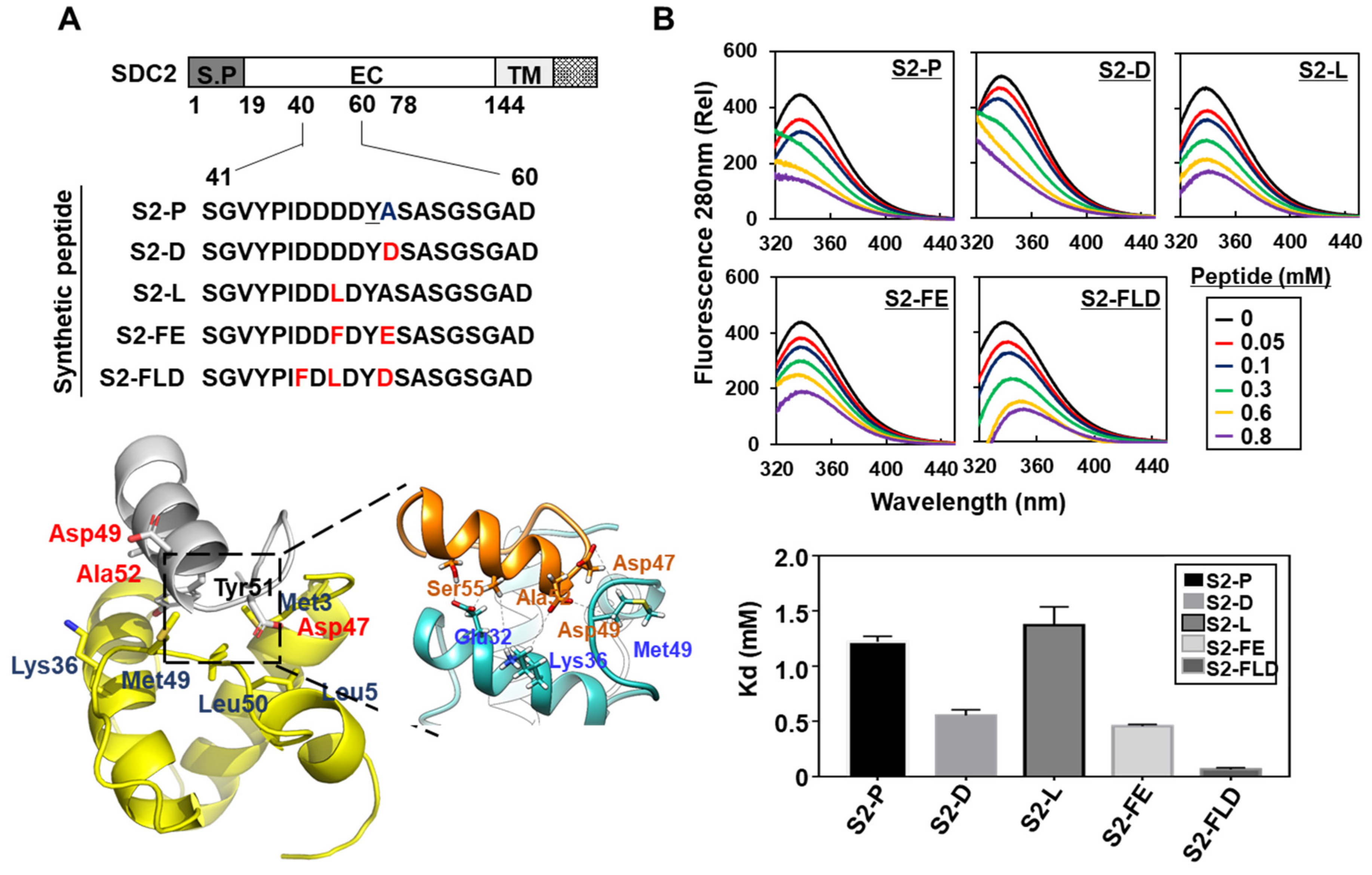
2.2. Synthetic Peptide Derived from Syndecan-2 Inhibited Its Interaction with the MMP-7 Pro-Domain
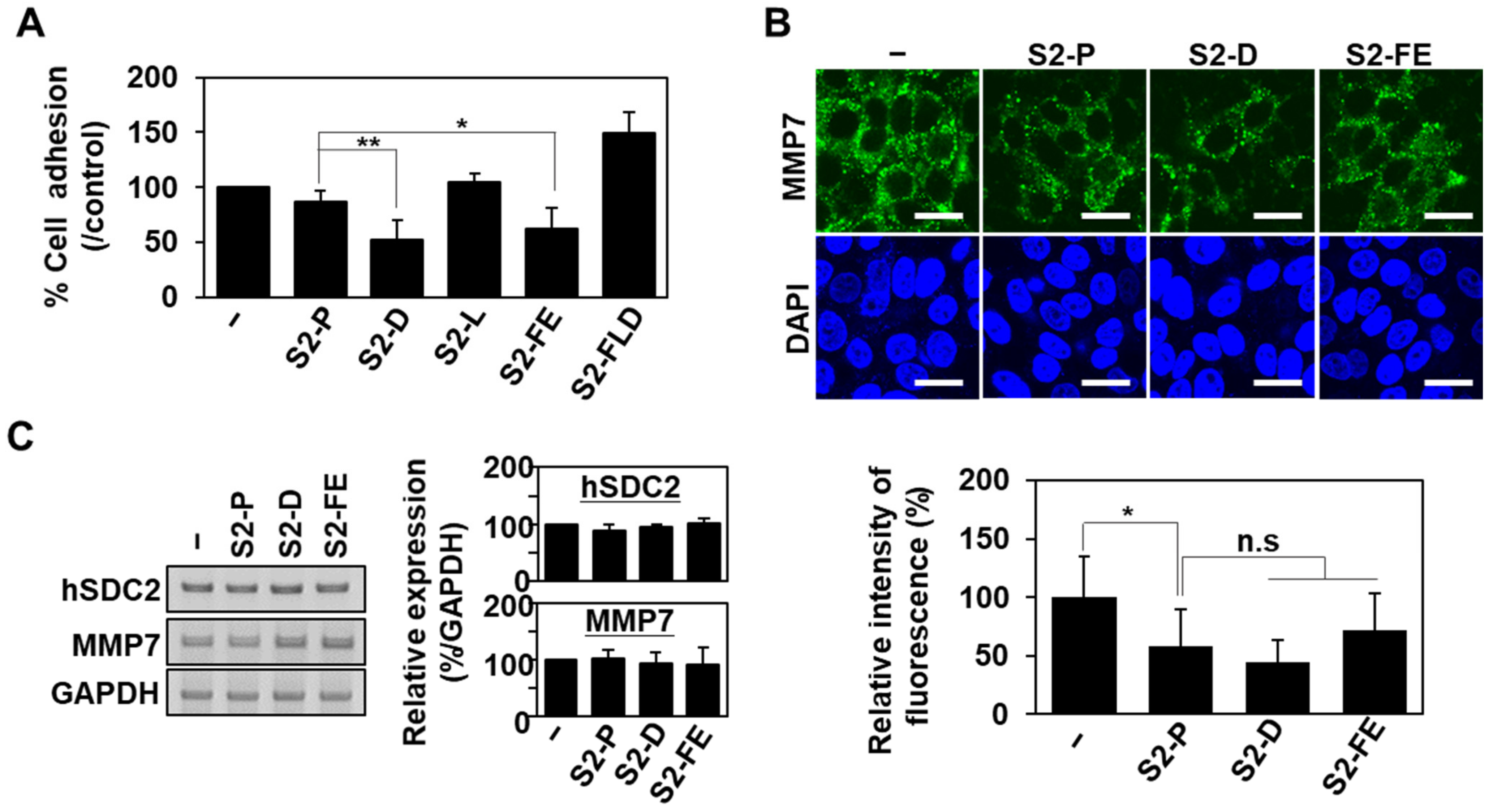
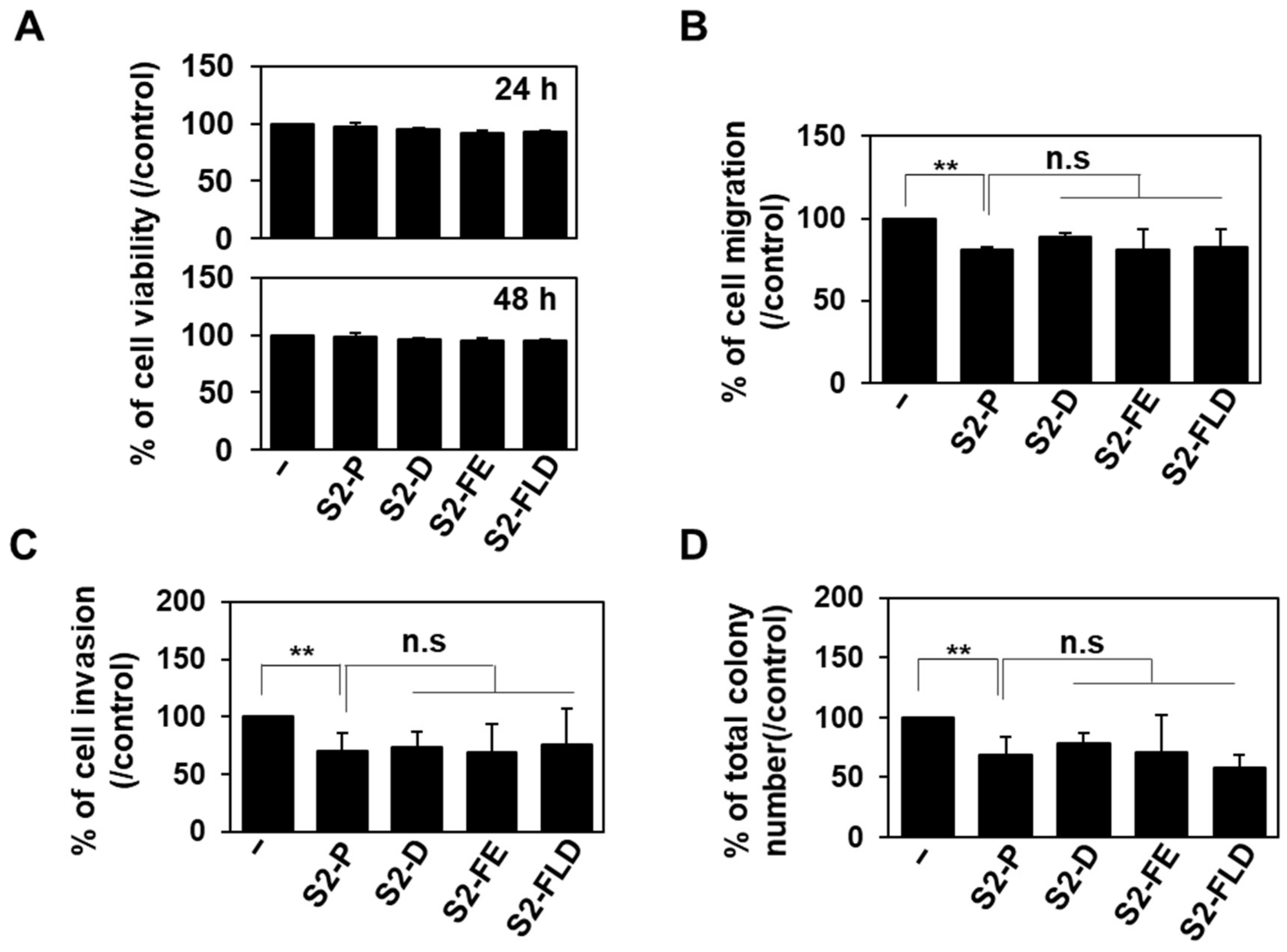
2.3. S2-D and S2-FE Show Anticancer Ability against Colon Cancer Cells
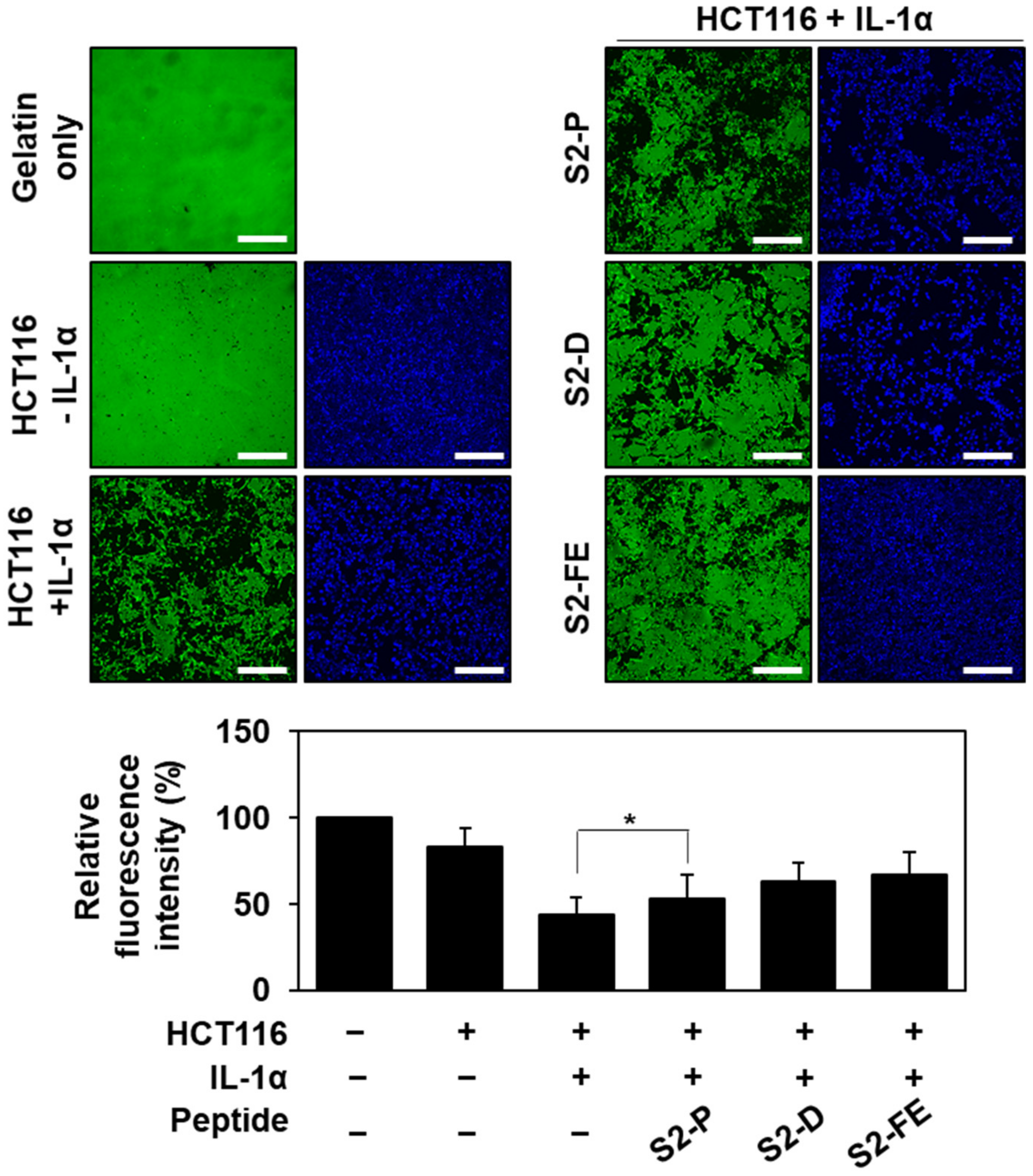
2.4. S2-D and S2-FE Inhibited MMP-7-Mediated ECM Degradation in Colon Cancer Cells
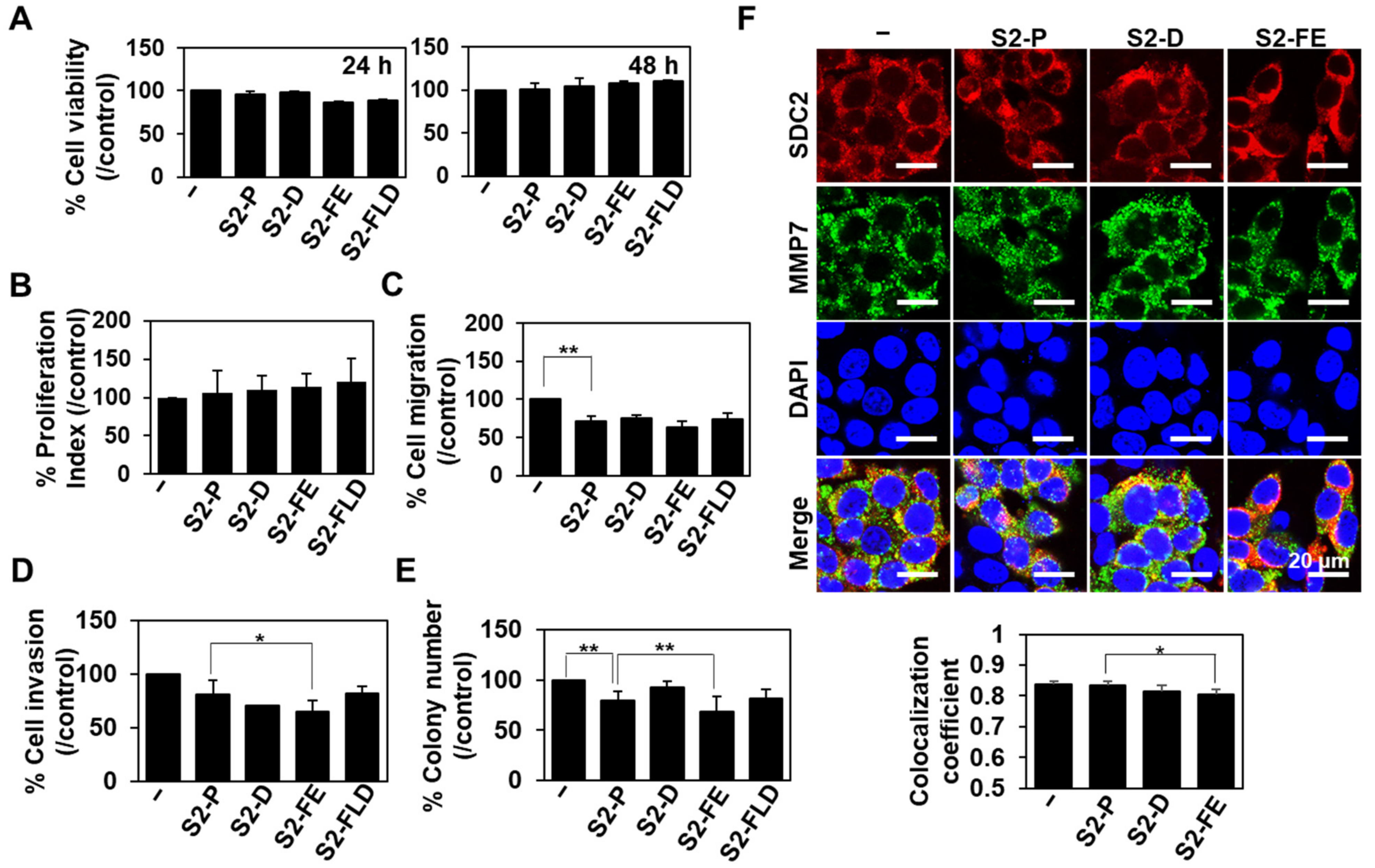
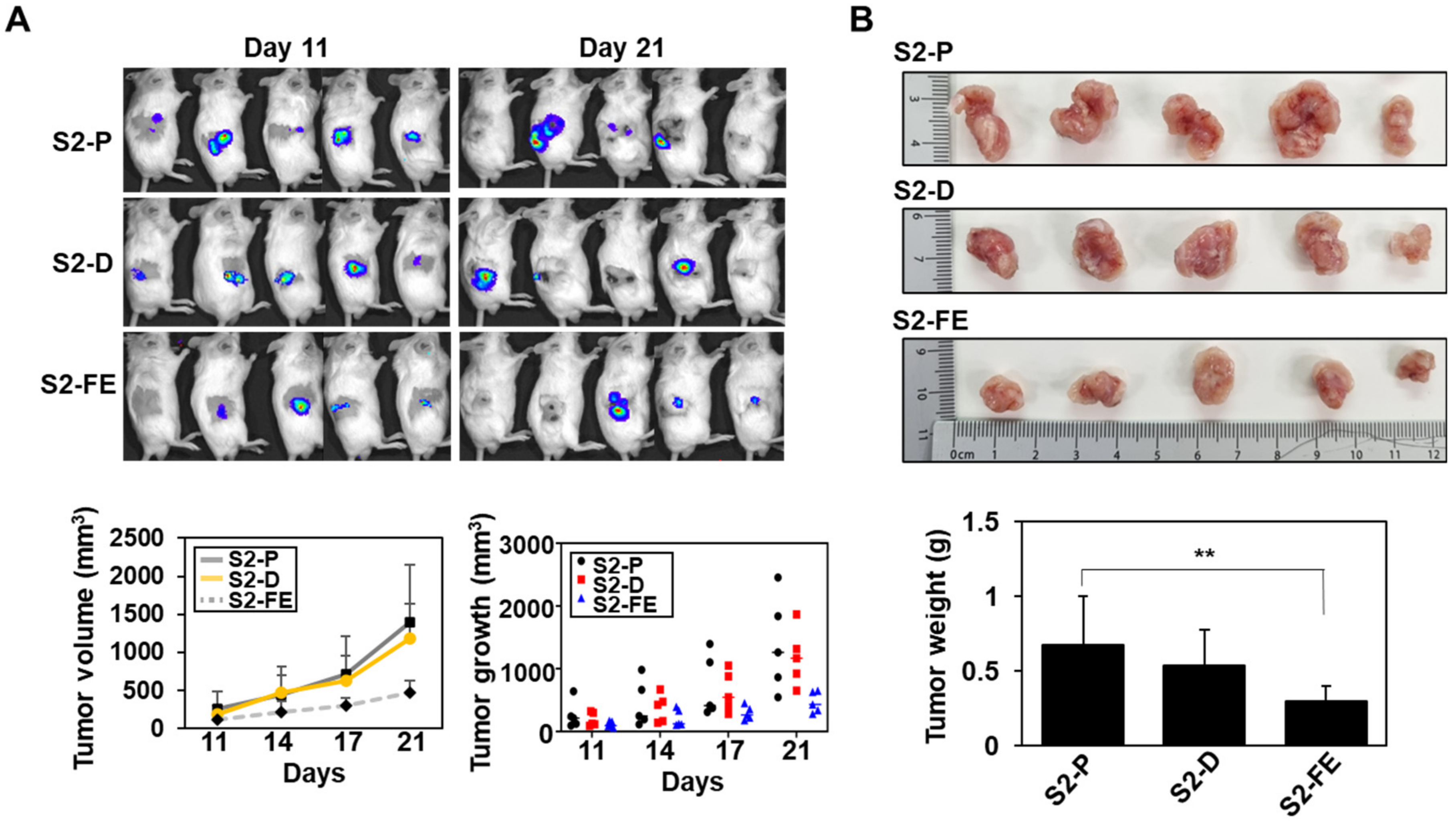
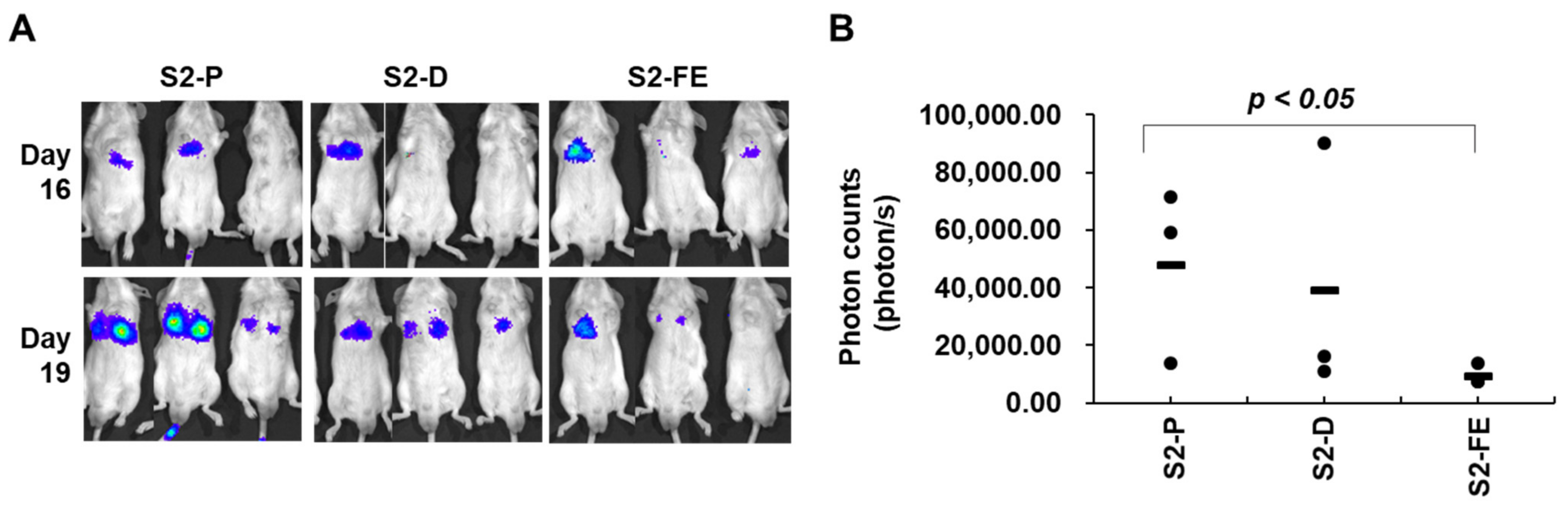
2.5. The S2-FE Peptide Was Sufficient for Reducing Primary Tumor Growth and Metastasis
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Cell Culture
4.3. Vector Construction and Generation of Stable Cell Lines
4.4. Peptide Synthesis
4.5. Expression and Purification of Recombinant Pro-Domain of MMP-7
4.6. Fluorescence Assay
4.7. MMP Enzyme Activity Assay
4.8. Monitoring of Cell Attachment
4.9. Immunofluorescence Analysis
4.10. RT-PCR
4.11. Cell Proliferation Assay
4.12. Cell Cycle Analysis
4.13. Invasion and Migration Assay
4.14. Anchorage-Independent Growth in Soft Agarose
4.15. Gelatin Degradation Assay
4.16. Mouse Model
4.17. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zimmermann, P.; David, G. The syndecans, tuners of transmembrane signaling. FASEB J. 1999, 13, 91–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carey, D.J. Syndecans: Multifunctional cell-surface co-receptors. Biochem. J. 1997, 327, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mytilinaiou, M.; Nikitovic, D.; Berdiaki, A.; Kostouras, A.; Papoutsidakis, A.; Tsatsakis, A.M.; Tzanakakis, G.N. Emerging roles of syndecan 2 in epithelial and mesenchymal cancer progression. IUBMB Life 2017, 69, 824–833. [Google Scholar] [CrossRef] [Green Version]
- Park, H.; Kim, Y.; Lim, Y.; Han, I.; Oh, E.S. Syndecan-2 mediates adhesion and proliferation of colon carcinoma cells. J. Biol. Chem. 2002, 277, 29730–29736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vicente, C.M.; Ricci, R.; Nader, H.B.; Toma, L. Syndecan-2 is upregulated in colorectal cancer cells through interactions with extracellular matrix produced by stromal fibroblasts. BMC Cell Biol. 2013, 14, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Ryu, H.Y.; Lee, J.; Yang, S.; Park, H.; Choi, S.; Jung, K.C.; Lee, S.T.; Seong, J.K.; Han, I.O.; Oh, E.S. Syndecan-2 functions as a docking receptor for pro-matrix metalloproteinase-7 in human colon cancer cells. J. Biol. Chem. 2009, 284, 35692–35701. [Google Scholar] [CrossRef] [Green Version]
- Hua, R.; Yu, J.; Yan, X.; Ni, Q.; Zhi, X.; Li, X.; Jiang, B.; Zhu, J. Syndecan-2 in colorectal cancer plays oncogenic role via epithelial-mesenchymal transition and MAPK pathway. Biomed. Pharmacother. 2020, 121, 109630. [Google Scholar] [CrossRef]
- Beauvais, D.M.; Rapraeger, A.C. Syndecans in tumor cell adhesion and signaling. Reprod. Biol. Endocrinol. 2004, 2, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Kwon, M.J.; Kim, Y.; Choi, Y.; Kim, S.H.; Park, S.; Han, I.; Kang, D.H.; Oh, E.S. The extracellular domain of syndecan-2 regulates the interaction of HCT116 human colon carcinoma cells with fibronectin. Biochem. Biophys. Res. Commun. 2013, 431, 415–420. [Google Scholar] [CrossRef]
- Choi, S.; Kim, Y.; Park, H.; Han, I.O.; Chung, E.; Lee, S.Y.; Kim, Y.B.; Lee, J.W.; Oh, E.S.; Yi, J.Y. Syndecan-2 overexpression regulates adhesion and migration through cooperation with integrin alpha2. Biochem. Biophys. Res. Commun. 2009, 38, 231–235. [Google Scholar] [CrossRef]
- Jang, B.; Yun, J.H.; Choi, S.; Park, J.; Shin, D.H.; Lee, S.T.; Lee, W.; Oh, E.S. Tyrosine 51 residue of the syndecan-2 extracellular domain is involved in the interaction with and activation of pro-matrix metalloproteinase-7. Sci. Rep. 2019, 9, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Kapoor, C.; Vaidya, S.; Wadhwan, V.; Hitesh; Kaur, G.; Pathak, A. Seesaw of matrix metalloproteinases (MMPs). J. Cancer Res. Ther. 2016, 12, 28–35. [Google Scholar] [CrossRef] [PubMed]
- Egeblad, M.; Werb, Z. New functions for the matrix metalloproteinases in cancer progression. Nat. Rev. Cancer 2002, 2, 161–174. [Google Scholar] [CrossRef] [PubMed]
- Jabłońska-Trypuć, A.; Matejczyk, M.; Rosochacki, S. Matrix metalloproteinases (MMPs), the main extracellular matrix (ECM) enzymes in collagen degradation, as a target for anticancer drugs. J. Enzyme Inhib. Med. Chem. 2016, 31, 177–183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cabral-Pacheco, G.A.; Garza-Veloz, I.; Castruita-De la Rosa, C.; Ramirez-Acuña, J.M.; Perez-Romero, B.A.; Guerrero-Rodriguez, J.F.; Martinez-Avila, N.; Martinez-Fierro, M.L. The roles of matrix metalloproteinases and their inhibitors in human diseases. Int. J. Mol. Sci. 2020, 21, 9739. [Google Scholar] [CrossRef]
- Sun, D.W.; Zhang, Y.Y.; Qi, Y.; Zhou, X.T.; Lv, G.Y. Prognostic significance of MMP-7 expression in colorectal cancer: A meta-analysis. Cancer Epidemiol. 2015, 39, 135–142. [Google Scholar] [CrossRef]
- Zucker, S.; Vacirca, J. Role of matrix metalloproteinases (MMPs) in colorectal cancer. Cancer Metastasis. Rev. 2004, 23, 101–117. [Google Scholar] [CrossRef]
- Murphy, G.; Stanton, H.; Cowell, S.; Butler, G.; Knäuper, V.; Atkinson, S.; Gavrilovic, J. Mechanisms for pro matrix metalloproteinase activation. APMIS 1999, 107, 38–44. [Google Scholar] [CrossRef]
- Ra, H.J.; Parks, W.C. Control of matrix metalloproteinase catalytic activity. Matrix Biol. 2007, 26, 587–596. [Google Scholar] [CrossRef] [Green Version]
- Grindel, B.J.; Martinez, J.R.; Tellman, T.V.; Harrington, D.A.; Zafar, H.; Nakhleh, L.; Chung, L.W.; Farach-Carson, M.C. Matrilysin/MMP-7 cleavage of perlecan/HSPG2 complexed with semaphorin 3A supports FAK-mediated stromal invasion by prostate cancer cells. Sci. Rep. 2018, 8, 1–14. [Google Scholar] [CrossRef]
- Tellman, T.V.; Cruz, L.A.; Grindel, B.J.; Farach-Carson, M.C. Cleavage of the perlecan-semaphorin 3A-plexin A1-neuropilin-1 (PSPN) complex by matrix metalloproteinase 7/Matrilysin triggers prostate cancer cell dyscohesion and migration. Int. J. Mol. Sci. 2021, 22, 3218. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.H.; Choi, E.Y.; Hyun, M.S.; Jang, B.I.; Kim, T.N.; Kim, S.W.; Song, S.K.; Kim, J.H.; Kim, J.R. Association of extracellular cleavage of E-cadherin mediated by MMP-7 with HGF-induced in vitro invasion in human stomach cancer cells. Eur. Surg. Res. 2007, 39, 208–215. [Google Scholar] [CrossRef] [PubMed]
- Davies, G.; Jiang, W.G.; Mason, M.D. Matrilysin mediates extracellular cleavage of E-cadherin from prostate cancer cells: A key mechanism in hepatocyte growth factor/scatter factor-induced cell-cell dissociation and in vitro invasion. Clin. Cancer Res. 2001, 7, 3289–3297. [Google Scholar] [PubMed]
- Jang, B.; Jung, H.; Chung, H.; Moon, B.I.; Oh, E.S. Syndecan-2 enhances E-cadherin shedding and fibroblast-like morphological changes by inducing MMP-7 expression in colon cancer cells. Biochem. Biophys. Res. Commun. 2016, 477, 47–53. [Google Scholar] [CrossRef] [PubMed]
- Nagase, H. Cell surface activation of progelatinase A (proMMP-2) and cell migration. Cell Res. 1998, 8, 179–186. [Google Scholar] [CrossRef] [Green Version]
- Guo, H.; Li, R.; Zucker, S.; Toole, B.P. EMMPRIN (CD147), an inducer of matrix metalloproteinase synthesis, also binds interstitial collagenase to the tumor cell surface. Cancer Res. 2000, 60, 888–891. [Google Scholar]
- Moll, U.M.; Lane, B.; Zucker, S.; Suzuki, K.; Nagase, H. Localization of collagenase at the basal plasma membrane of a human pancreatic carcinoma cell line. Cancer Res. 1990, 50, 6995–7002. [Google Scholar]
- Jang, B.; Jung, H.; Choi, S.; Lee, Y.H.; Lee, S.T.; Oh, E.S. Syndecan-2 cytoplasmic domain up-regulates matrix metalloproteinase-7 expression via the protein kinase Cγ-mediated FAK/ERK signaling pathway in colon cancer. J. Biol. Chem. 2017, 292, 16321–16332. [Google Scholar] [CrossRef] [Green Version]
- Pahari, S.; Li, G.; Murthy, A.K.; Liang, S.; Fragoza, R.; Yu, H.; Alexov, E. SAAMBE-3D: Predicting effect of mutations on protein–protein interactions. Int. J. Mol. Sci. 2020, 21, 2563. [Google Scholar] [CrossRef] [Green Version]
- Choi, S.; Choi, Y.; Jun, E.; Kim, I.S.; Kim, S.E.; Jung, S.A.; Oh, E.S. Shed syndecan-2 enhances tumorigenic activities of colon cancer cells. Oncotarget 2015, 6, 3874–3886. [Google Scholar] [CrossRef] [Green Version]
- Kwon, M.J.; Hong, E.; Choi, Y.; Kang, D.H.; Oh, E.S. Interleukin-1α promotes extracellular shedding of syndecan-2 via induction of matrix metalloproteinase-7 expression. Biochem. Biophys. Res. Commun. 2014, 446, 487–492. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.; Kim, J.Y.; Park, J.H.; Lee, S.T.; Han, I.O.; Oh, E.S. The matrix metalloproteinase-7 regulates the extracellular shedding of syndecan-2 from colon cancer cells. Biochem. Biophys. Res. Commun. 2012, 417, 1260–1264. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Fu, X.; Brown, P.D.; Crimmin, M.J.; Hoffman, R.M. Matrix metalloproteinase inhibitor BB-94 (batimastat) inhibits human colon tumor growth and spread in a patient-like orthotopic model in nude mice. Cancer Res. 1994, 54, 4726–4728. [Google Scholar] [PubMed]
- Rasmussen, H.S.; McCann, P.P. Matrix metalloproteinase inhibition as a novel anticancer strategy: A review with special focus on batimastat and marimastat. Pharmacol. Ther. 1997, 75, 69–75. [Google Scholar] [CrossRef]
- Winer, A.; Adams, S.; Mignatti, P. Matrix metalloproteinase inhibitors in cancer therapy: Turning past failures into future successes. Mol. Cancer Ther. 2018, 17, 1147–1155. [Google Scholar] [CrossRef] [Green Version]
- Fingleton, B. MMPs as therapeutic targets-still a viable option? Semin. Cell Dev. Biol. 2008, 19, 61–68. [Google Scholar] [CrossRef] [Green Version]
- Parsons, S.L.; Watson, S.A.; Steele, R.J. Phase I/II trial of batimastat, a matrix metalloproteinase inhibitor, in patients with malignant ascites. Eur. J. Surg. Oncol. 1997, 23, 526–531. [Google Scholar] [CrossRef]
- Macaulay, V.M.; O’Byrne, K.J.; Saunders, M.P.; Braybrooke, J.P.; Long, L.; Gleeson, F.; Mason, C.S.; Harris, A.L.; Brown, P.; Talbot, D.C. Phase I study of intrapleural batimastat (BB-94), a matrix metalloproteinase inhibitor, in the treatment of malignant pleural effusions. Clin. Cancer Res. 1999, 5, 513–520. [Google Scholar]
- Bramhall, S.R.; Hallissey, M.T.; Whiting, J.; Scholefield, J.; Tierney, G.; Stuart, R.C.; Hawkins, R.E.; McCulloch, P.; Maughan, T.; Brown, P.D.; et al. Marimastat as maintenance therapy for patients with advanced gastric cancer: A randomised trial. Br. J. Cancer 2002, 86, 1864–1870. [Google Scholar] [CrossRef]
- Sato, H.; Takino, T. Coordinate action of membrane-type matrix metalloproteinase-1 (MT1-MMP) and MMP-2 enhances pericellular proteolysis and invasion. Cancer Sci. 2010, 101, 843–847. [Google Scholar] [CrossRef] [Green Version]
- Sarkar, P.; Li, Z.; Ren, W.; Wang, S.; Shao, S.; Sun, J.; Ren, X.; Perkins, N.G.; Guo, Z.; Chang, C.A.; et al. Inhibiting matrix metalloproteinase-2 activation by perturbing protein-protein interactions using a cyclic peptide. J. Med. Chem. 2020, 63, 6979–6990. [Google Scholar] [CrossRef] [PubMed]
- Kwon, M.J.; Choi, Y.; Yun, J.H.; Lee, W.; Han, I.O.; Oh, E.S. A unique phenylalanine in the transmembrane domain strengthens homodimerization of the syndecan-2 transmembrane domain and functionally regulates syndecan-2. J. Biol. Chem. 2015, 290, 5772–5782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Werle, M.; Bernkop-Schnürch, A. Strategies to improve plasma half life time of peptide and protein drugs. Amino Acids 2006, 30, 351–367. [Google Scholar] [CrossRef] [PubMed]
- Vlieghe, P.; Lisowski, V.; Martinez, J.; Khrestchatisky, M. Synthetic therapeutic peptides: Science and market. Drug Discov. Today 2010, 15, 40–56. [Google Scholar] [CrossRef] [PubMed]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jang, B.; Kim, A.; Lee, Y.; Hwang, J.; Sung, J.-Y.; Jang, E.-J.; Kim, Y.-N.; Yun, J.-H.; Han, J.; Song, J.-J.; et al. Substituted Syndecan-2-Derived Mimetic Peptides Show Improved Antitumor Activity over the Parent Syndecan-2-Derived Peptide. Int. J. Mol. Sci. 2022, 23, 5888. https://doi.org/10.3390/ijms23115888
Jang B, Kim A, Lee Y, Hwang J, Sung J-Y, Jang E-J, Kim Y-N, Yun J-H, Han J, Song J-J, et al. Substituted Syndecan-2-Derived Mimetic Peptides Show Improved Antitumor Activity over the Parent Syndecan-2-Derived Peptide. International Journal of Molecular Sciences. 2022; 23(11):5888. https://doi.org/10.3390/ijms23115888
Chicago/Turabian StyleJang, Bohee, Ayoung Kim, Yejin Lee, Jisun Hwang, Jee-Young Sung, Eun-Ju Jang, Yong-Nyun Kim, Ji-Hye Yun, Jeongmin Han, Ji-Joon Song, and et al. 2022. "Substituted Syndecan-2-Derived Mimetic Peptides Show Improved Antitumor Activity over the Parent Syndecan-2-Derived Peptide" International Journal of Molecular Sciences 23, no. 11: 5888. https://doi.org/10.3390/ijms23115888
APA StyleJang, B., Kim, A., Lee, Y., Hwang, J., Sung, J. -Y., Jang, E. -J., Kim, Y. -N., Yun, J. -H., Han, J., Song, J. -J., Lee, W., & Oh, E. -S. (2022). Substituted Syndecan-2-Derived Mimetic Peptides Show Improved Antitumor Activity over the Parent Syndecan-2-Derived Peptide. International Journal of Molecular Sciences, 23(11), 5888. https://doi.org/10.3390/ijms23115888