
The Multidirectional Effect of Azelastine Hydrochloride on Cervical Cancer Cells


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Azelastine Induces Apoptosis in HeLa Cells
2.2. Azelastine Induces Caspase 3/7-Dependent Apoptosis
2.3. Azelastine Inhibits the Viability of HeLa Cells
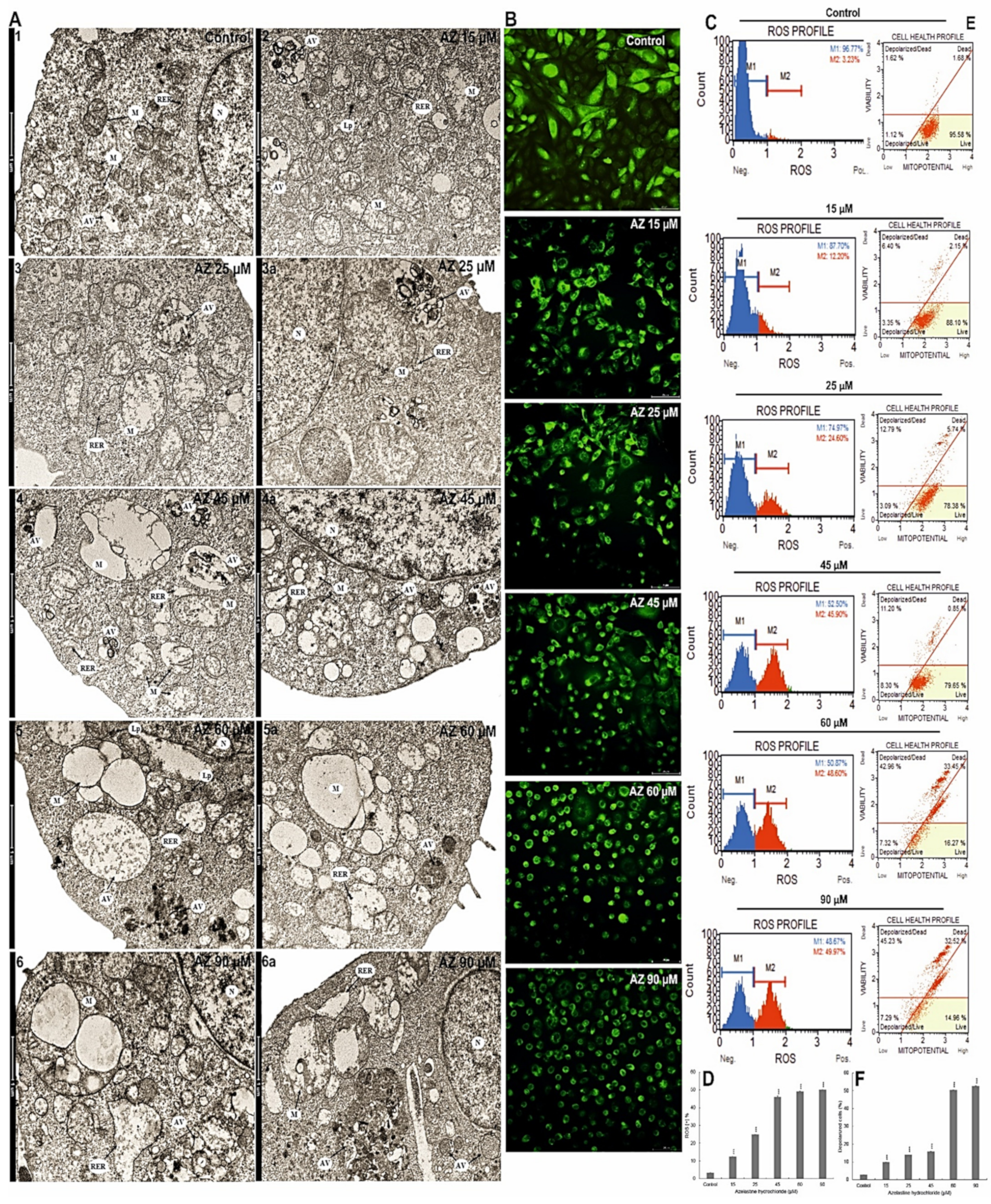
2.4. Azelastine Generates ROS Inducing Changes in Mitochondrial Structure and Induces Endoplasmic Reticulum Stress
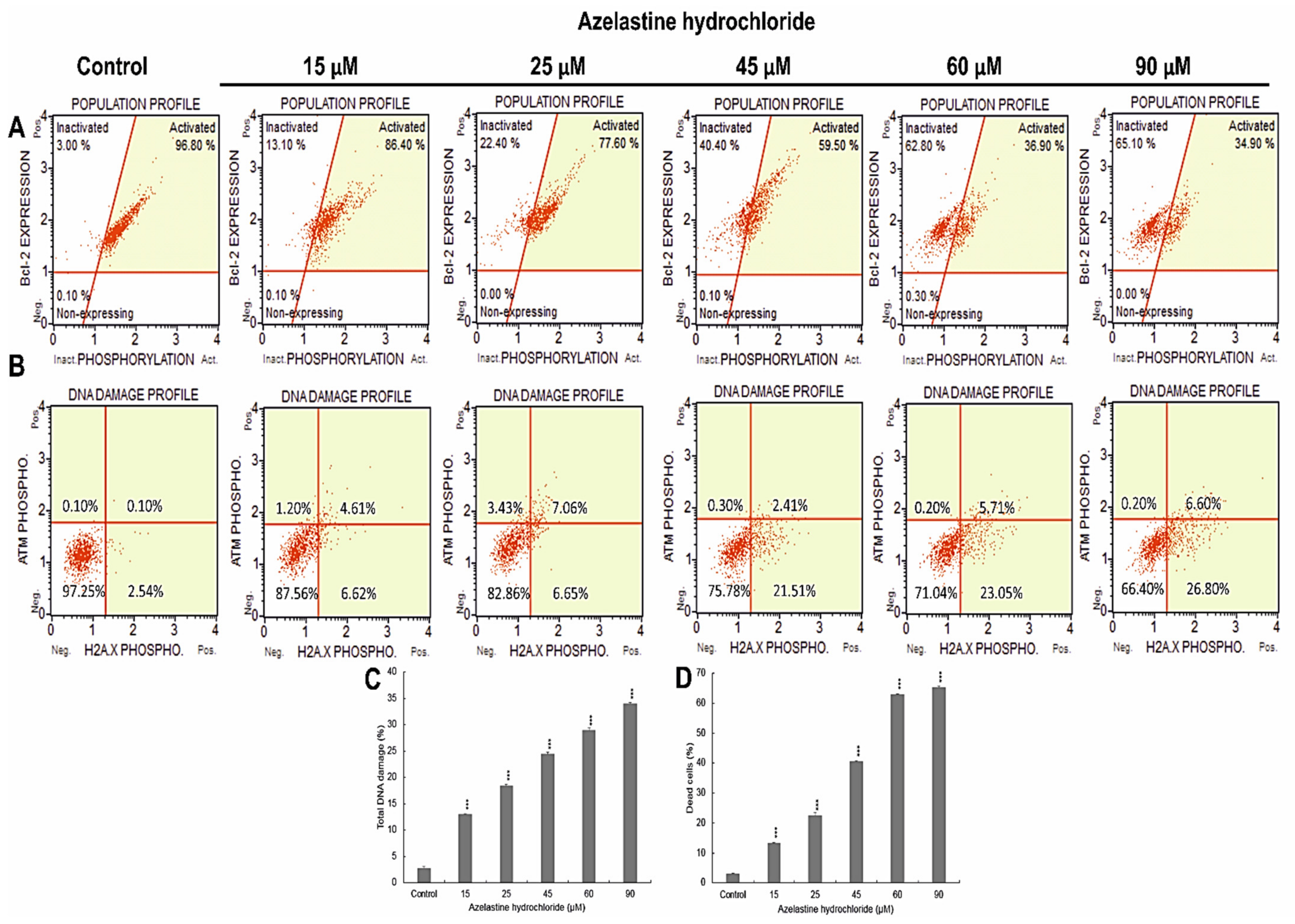
2.5. Azelastine Induces DNA Damage
2.6. Azelastine Inactivates the Bcl-2 Protein
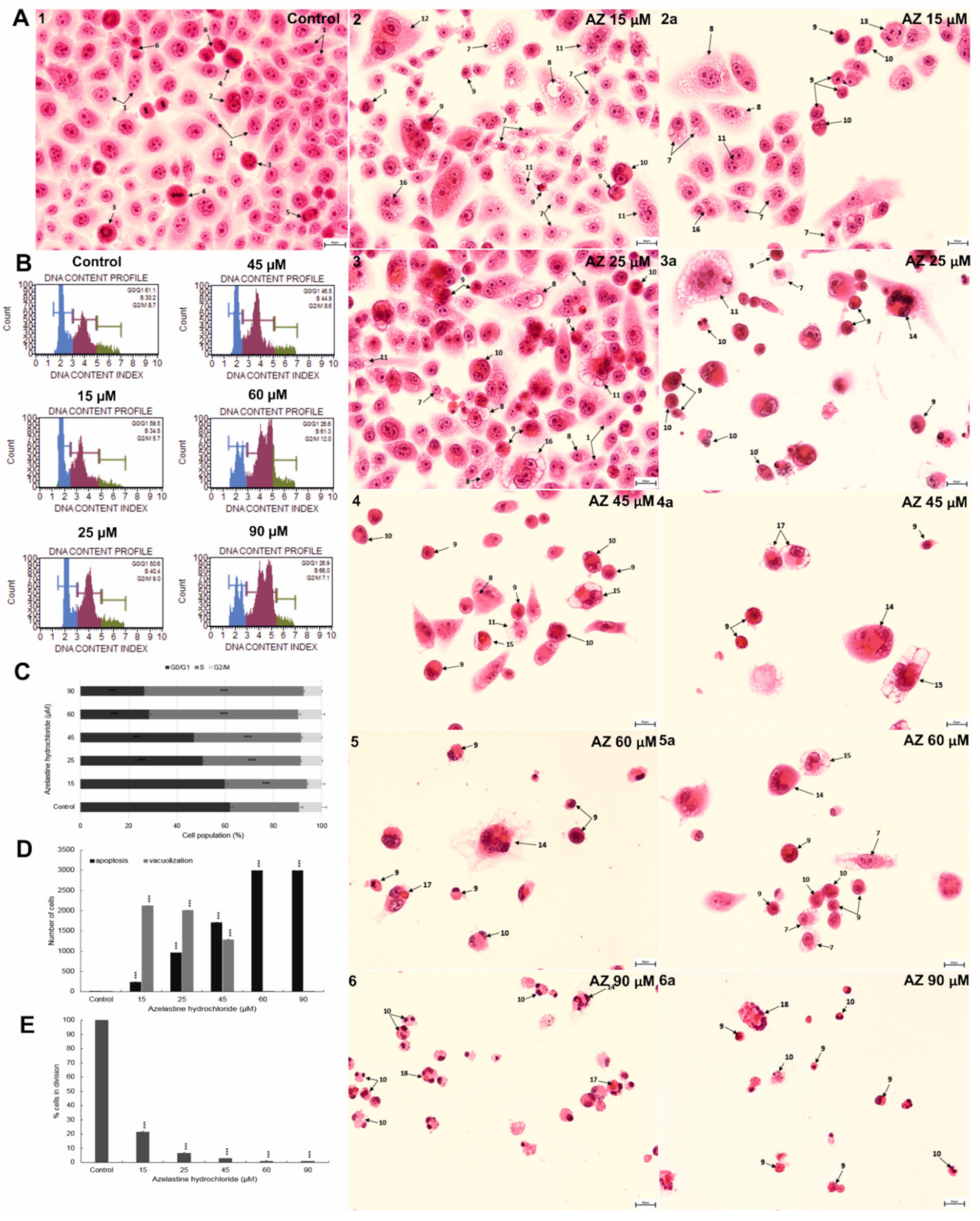
2.7. Azelastine Enhances Vacuolization and Apoptotic Changes in HeLa Cells—Morphological Evaluation
2.8. Azelastine Blocks Cells in S Phase and Reduces Mitotic Index
2.9. Azelastine Induces Mitotic Catastrophe
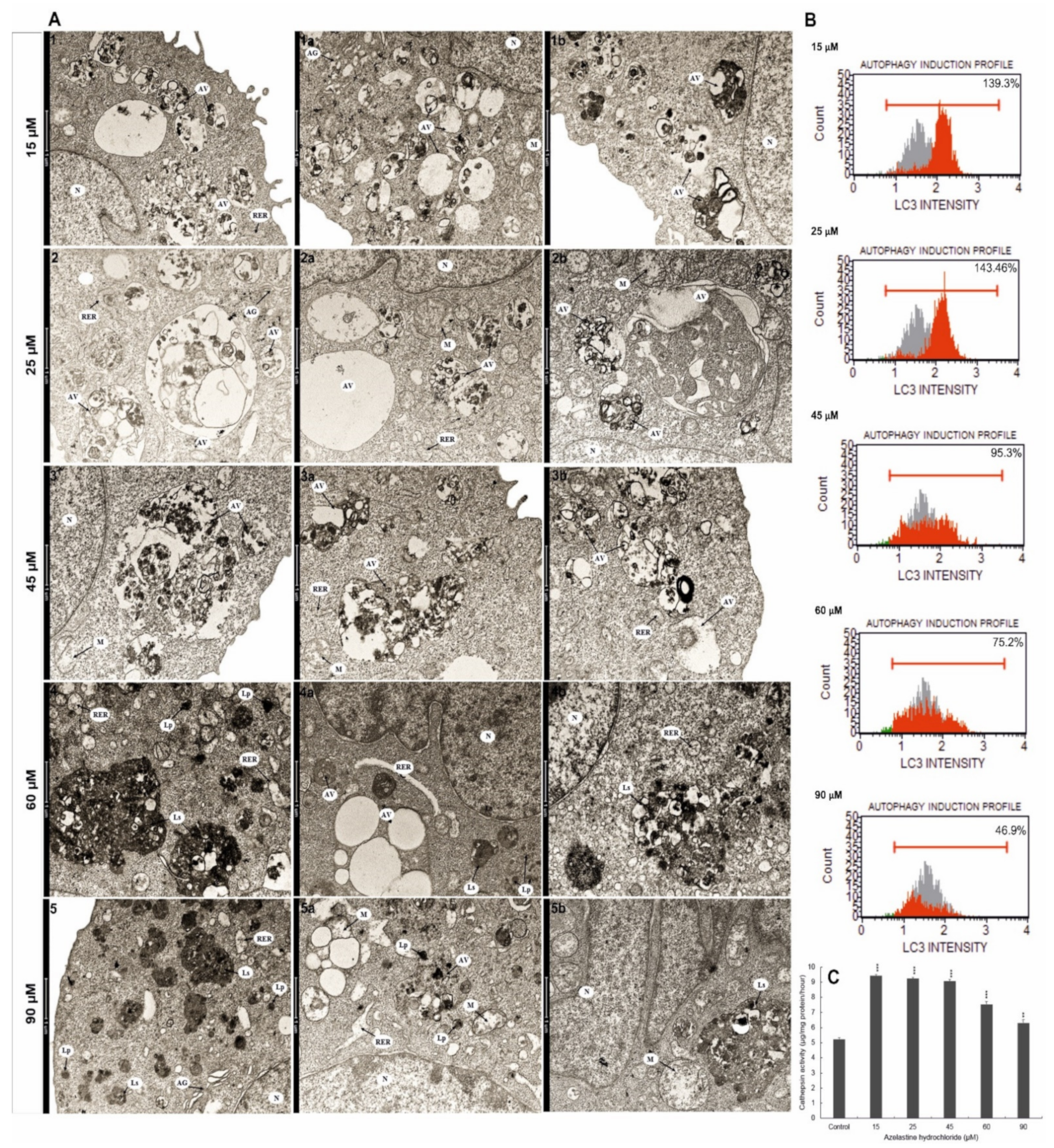
2.10. Azelastine Enhances Degradation Processes
2.11. Azelastine Activates Cathepsin D and L
2.12. Azelastine Induces Autophagy by Increasing LC3 Protein Levels
3. Discussion
4. Material and Methods
4.1. In Vitro Culture Conditions
4.2. Assessment of Cell Viability—MTT Test
4.3. Visualization of Apoptotic Cells under A Fluorescence Microscope
4.4. Detection of Apoptosis
4.5. Activity of Caspase-3/7
4.6. Analysis of Ultrastructural Changes
4.7. Measurement of the Mitochondrial Membrane Potential (Δψm)
4.8. Microscopic Evaluation of Changes in the Potential of Mitochondrial Membrane
4.9. Oxidative Stress Analysis
4.10. Assessment of Bcl-2 Protein Phosphorylation
4.11. DNA Damage Assessment
4.12. Cell Cycle Analysis
4.13. Visualization of Morphological Changes and Assessment of the Dividing Capacity of HeLa Cells
4.14. Evaluation of Cathepsin D and L Activity Levels
4.15. LC3-Antibody Detection
4.16. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Matsumoto, N.; Ebihara, M.; Oishi, S.; Fujimoto, Y.; Okada, T.; Imamura, T. Histamine H1 receptor antagonists selectively kill cisplatin-resistant human cancer cells. Sci. Rep. 2021, 11, 1492. [Google Scholar] [CrossRef] [PubMed]
- Chang, A. Chemotherapy, chemoresistance and the changing treatment landscape for NSCLC. Lung Cancer 2011, 71, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Xia, J.; Yu, X.; Song, X.; Li, G.; Mao, X.; Zhang, Y. Inhibiting the cytoplasmic location of HMGB1 reverses cisplatin resistance in human cervical cancer cells. Mol. Med. Rep. 2017, 15, 488–494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, H.; Zou, Z. Targeting autophagy to overcome drug resistance: Further developments. J. Hematol. Oncol. 2020, 13, 159. [Google Scholar] [CrossRef] [PubMed]
- Ellegaard, A.M.; Dehlendorff, C.; Vind, A.C.; Anand, A.; Cederkvist, L.; Petersen, N.H.T.; Nylandsted, J.; Stenvang, J.; Mellemgaard, A.; Osterlind, K.; et al. Repurposing Cationic Amphiphilic Antihistamines for Cancer Treatment. EBioMedicine 2016, 9, 130–139. [Google Scholar] [CrossRef] [Green Version]
- Greten, F.R.; Grivennikov, S.I. Inflammation and Cancer: Triggers, Mechanisms, and Consequences. Immunity 2019, 51, 27–41. [Google Scholar] [CrossRef]
- Verdoodt, F.; Dehlendorff, C.; Jaattela, M.; Strauss, R.; Pottegard, A.; Hallas, J.; Friis, S.; Kjaer, S.K. Antihistamines and Ovarian Cancer Survival: Nationwide Cohort Study and in Vitro Cell Viability Assay. J. Natl. Cancer Inst. 2020, 112, 964–967. [Google Scholar] [CrossRef]
- Kuzu, O.F.; Toprak, M.; Noory, M.A.; Robertson, G.P. Effect of lysosomotropic molecules on cellular homeostasis. Pharmacol. Res. 2017, 117, 177–184. [Google Scholar] [CrossRef]
- Serrano-Puebla, A.; Boya, P. Lysosomal membrane permeabilization as a cell death mechanism in cancer cells. Biochem. Soc. Trans. 2018, 46, 207–215. [Google Scholar] [CrossRef]
- Tatarkiewicz, J.; Rzodkiewicz, P.; Zochowska, M.; Staniszewska, A.; Bujalska-Zadrozny, M. New antihistamines—Perspectives in the treatment of some allergic and inflammatory disorders. Arch. Med. Sci. AMS 2019, 15, 537–553. [Google Scholar] [CrossRef]
- Bens, A.; Dehlendorff, C.; Friis, S.; Cronin-Fenton, D.; Jensen, M.B.; Ejlertsen, B.; Lash, T.L.; Kroman, N.; Mellemkjaer, L. The role of H1 antihistamines in contralateral breast cancer: A Danish nationwide cohort study. Br. J. Cancer 2020, 122, 1102–1108. [Google Scholar] [CrossRef] [PubMed]
- Mandola, A.; Nozawa, A.; Eiwegger, T. Histamine, histamine receptors, and anti-histamines in the context of allergic responses. LymphoSign J. 2019, 6, 35–51. [Google Scholar] [CrossRef]
- Criado, P.R.; Criado, R.F.J.; Maruta, C.W.; Fiho, C. Histamine, histamine receptors and antihistamines: New concepts. An. Bras. De Dermatol. 2010, 85, 195–210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thangam, E.B.; Jemima, E.A.; Singh, H.; Baig, M.S.; Khan, M.; Mathias, C.B.; Church, M.K.; Saluja, R. The Role of Histamine and Histamine Receptors in Mast Cell-Mediated Allergy and Inflammation: The Hunt for New Therapeutic Targets. Front. Immunol. 2018, 9, 1873. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, E.; Zhang, Y.; Fan, Z.; Cheng, L.; Han, S.; Che, H. Apigenin Inhibits Histamine-Induced Cervical Cancer Tumor Growth by Regulating Estrogen Receptor Expression. Molecules 2020, 25, 1960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Massari, N.A.; Nicoud, M.B.; Medina, V.A. Histamine receptors and cancer pharmacology: An update. Br. J. Pharmacol. 2020, 177, 516–538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Guadalupe Chavez-Lopez, M.; Perez-Carreon, J.I.; Zuniga-Garcia, V.; Diaz-Chavez, J.; Herrera, L.A.; Caro-Sanchez, C.H.; Acuna-Macias, I.; Gariglio, P.; Hernandez-Gallegos, E.; Chiliquinga, A.J.; et al. Astemizole-based anticancer therapy for hepatocellular carcinoma (HCC), and Eag1 channels as potential early-stage markers of HCC. Tumour Biol. J. Int. Soc. Oncodev. Biol. Med. 2015, 36, 6149–6158. [Google Scholar] [CrossRef]
- Garcia-Quiroz, J.; Camacho, J. Astemizole: An old anti-histamine as a new promising anti-cancer drug. Anti-Cancer Agents Med. Chem. 2011, 11, 307–314. [Google Scholar] [CrossRef]
- Jangi, S.M.; Diaz-Perez, J.L.; Ochoa-Lizarralde, B.; Martin-Ruiz, I.; Asumendi, A.; Perez-Yarza, G.; Gardeazabal, J.; Diaz-Ramon, J.L.; Boyano, M.D. H1 histamine receptor antagonists induce genotoxic and caspase-2-dependent apoptosis in human melanoma cells. Carcinogenesis 2006, 27, 1787–1796. [Google Scholar] [CrossRef]
- Wang, W.T.; Chen, Y.H.; Hsu, J.L.; Leu, W.J.; Yu, C.C.; Chan, S.H.; Ho, Y.F.; Hsu, L.C.; Guh, J.H. Terfenadine induces anti-proliferative and apoptotic activities in human hormone-refractory prostate cancer through histamine receptor-independent Mcl-1 cleavage and Bak up-regulation. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2014, 387, 33–45. [Google Scholar] [CrossRef]
- Fritz, I.; Wagner, P.; Olsson, H. Improved survival in several cancers with use of H1-antihistamines desloratadine and loratadine. Transl. Oncol. 2021, 14, 101029. [Google Scholar] [CrossRef] [PubMed]
- Fritz, I.; Wagner, P.; Broberg, P.; Einefors, R.; Olsson, H. Desloratadine and loratadine stand out among common H1-antihistamines for association with improved breast cancer survival. Acta Oncol. 2020, 59, 1103–1109. [Google Scholar] [CrossRef] [PubMed]
- Olsson, H.; Einefors, R.; Broberg, P. Second generation antihistmines after breast cancer diagnosis to improve prognosis both in patients with ER+ and ER- breast cancer. J. Clin. Oncol. 2015, 33, 3062. [Google Scholar] [CrossRef]
- Fritz, I.; Wagner, P.; Bottai, M.; Eriksson, H.; Ingvar, C.; Krakowski, I.; Nielsen, K.; Olsson, H. Desloratadine and loratadine use associated with improved melanoma survival. Allergy 2020, 75, 2096–2099. [Google Scholar] [CrossRef]
- Dobbeling, U.; Waeckerle-Men, Y.; Zabel, F.; Graf, N.; Kundig, T.M.; Johansen, P. The antihistamines clemastine and desloratadine inhibit STAT3 and c-Myc activities and induce apoptosis in cutaneous T-cell lymphoma cell lines. Exp. Dermatol. 2013, 22, 119–124. [Google Scholar] [CrossRef]
- Soule, B.P.; Simone, N.L.; DeGraff, W.G.; Choudhuri, R.; Cook, J.A.; Mitchell, J.B. Loratadine dysregulates cell cycle progression and enhances the effect of radiation in human tumor cell lines. Radiat. Oncol. 2010, 5, 8. [Google Scholar] [CrossRef] [Green Version]
- Procopiou, P.A.; Ford, A.J.; Gore, P.M.; Looker, B.E.; Hodgson, S.T.; Holmes, D.S.; Vile, S.; Clark, K.L.; Saunders, K.A.; Slack, R.J.; et al. Design of Phthalazinone Amide Histamine H1 Receptor Antagonists for Use in Rhinitis. ACS Med. Chem. Lett. 2017, 8, 577–581. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.; Corren, J. Review of azelastine nasal spray in the treatment of allergic and non-allergic rhinitis. Expert Opin. Pharmacother. 2007, 8, 701–709. [Google Scholar] [CrossRef]
- Ellis, A.K.; Zhu, Y.; Steacy, L.M.; Walker, T.; Day, J.H. A four-way, double-blind, randomized, placebo controlled study to determine the efficacy and speed of azelastine nasal spray, versus loratadine, and cetirizine in adult subjects with allergen-induced seasonal allergic rhinitis. Allergy Asthma Clin. Immunol. Off. J. Can. Soc. Allergy Clin. Immunol. 2013, 9, 16. [Google Scholar] [CrossRef] [Green Version]
- Jonathan, M.; Horbal, J.M.; Bernstein, J.A. Azelastine HCl: A Review of the Old and New Formulations. Clin. Med. Insights Ther. 2010, 2, 427–437. [Google Scholar]
- Konrat, R.; Papp, H.; Szijártó, V.; Gesell, T.; Gábor, N.; Madai, M.; Zeghbibc, S.; Kuczmog, A.; Lanszki, Z.; Helyes, Z.; et al. The Anti-histamine Azelastine, Identified by Computational Drug Repurposing, Inhibits SARS-CoV-2 Infection in Reconstituted Human Nasal Tissue In Vitro. Biorxiv 2020. [Google Scholar] [CrossRef]
- Blaess, M.; Kaiser, L.; Sommerfeld, O.; Csuk, R.; Deigner, H.P. Drugs, Metabolites, and Lung Accumulating Small Lysosomotropic Molecules: Multiple Targeting Impedes SARS-CoV-2 Infection and Progress to COVID-19. Int. J. Mol. Sci. 2021, 22, 1797. [Google Scholar] [CrossRef]
- Mourad, A.K.; Makhlouf, A.A.; Soliman, A.Y.; Mohamed, S.A. Phthalazines and phthalazine hybrids as antimicrobial agents: Synthesis and biological evaluation. J. Chem. Res. 2019, 44, 31–41. [Google Scholar] [CrossRef]
- Papadopoulos, N.G.; Aggelides, X.; Stamataki, S.; Prokopakis, E.; Katotomichelakis, M.; Xepapadaki, P. New Concepts in Pediatric Rhinitis; European Society of Pediatric Allergy and Immunology: Zurich, Switzerland, 2021; Volume 32, pp. 635–646. [Google Scholar] [CrossRef]
- Berger, W.; Sher, E.; Gawchik, S.; Fineman, S. Safety of a novel intranasal formulation of azelastine hydrochloride and fluticasone propionate in children: A randomized clinical trial. Allergy Asthma Proc. 2018, 39, 110–116. [Google Scholar] [CrossRef] [Green Version]
- Church, M.K. Allergy, Histamine and Antihistamines. In Handbook of Experimental Pharmacology; Springer: Berlin/Heidelberg, Germany, 2017; Volume 241, pp. 321–331. [Google Scholar] [CrossRef]
- Horak, F. Effectiveness of twice daily azelastine nasal spray in patients with seasonal allergic rhinitis. Ther. Clin. Risk Manag. 2008, 4, 1009–1022. [Google Scholar] [CrossRef] [Green Version]
- Saito, K.; Abe, N.; Toyama, H.; Ejima, Y.; Yamauchi, M.; Mushiake, H.; Kazama, I. Second-Generation Histamine H1 Receptor Antagonists Suppress Delayed Rectifier K(+)-Channel Currents in Murine Thymocytes. BioMed Res. Int. 2019, 2019, 6261951. [Google Scholar] [CrossRef] [Green Version]
- Fein, M.N.; Fischer, D.A.; O’Keefe, A.W.; Sussman, G.L. CSACI position statement: Newer generation H1-antihistamines are safer than first-generation H1-antihistamines and should be the first-line antihistamines for the treatment of allergic rhinitis and urticaria. Allergy Asthma Clin. Immunol. Off. J. Can. Soc. Allergy Clin. Immunol. 2019, 15, 61. [Google Scholar] [CrossRef] [Green Version]
- Marshal, G.D. Therapeutic options in allergic disease: Antihistamines as systemic antiallergic agents. J. Allergy Clin. Immunol. 2000, 106, 303–309. [Google Scholar] [CrossRef]
- Watts, A.M.; Cripps, A.W.; West, N.P.; Cox, A.J. Modulation of Allergic Inflammation in the Nasal Mucosa of Allergic Rhinitis Sufferers With Topical Pharmaceutical Agents. Front. Pharmacol. 2019, 10, 294. [Google Scholar] [CrossRef] [Green Version]
- Hill, S.J.; Ganellin, C.R.; Timmerman, H.; Schwartz, J.C.; Shankley, N.P.; Young, J.M.; Schunack, W.; Levi, R.; Haas, H.L. International Union of Pharmacology. XIII. Classification of histamine receptors. Phamacol. Rev. 1997, 49, 253–278. [Google Scholar]
- McNeely, W.; Wiseman, L.R. Intranasal azelastine. A review of its efficacy in the management of allergic rhinitis. Drugs 1998, 56, 91–114. [Google Scholar] [CrossRef]
- El-Nakeeb, M.A.; Abou-Shleib, H.M.; Khalil, A.M.; Omar, H.G.; El-Halfawy, O.M. In vitro antibacterial activity of some antihistaminics belonging to different groups against multi-drug resistant clinical isolates. Braz. J. Microbiol. 2011, 42, 980–991. [Google Scholar] [CrossRef] [Green Version]
- Peniche, A.G.; Osorio, E.Y.; Melby, P.C.; Travi, B.L. Efficacy of histamine H1 receptor antagonists azelastine and fexofenadine against cutaneous Leishmania major infection. PLoS Negl. Trop. Dis. 2020, 14, e0008482. [Google Scholar] [CrossRef]
- Hu, H.F.; Xu, W.W.; Li, Y.J.; He, Y.; Zhang, W.X.; Liao, L.; Zhang, Q.H.; Han, L.; Yin, X.F.; Zhao, X.X.; et al. Anti-allergic drug azelastine suppresses colon tumorigenesis by directly targeting ARF1 to inhibit IQGAP1-ERK-Drp1-mediated mitochondrial fission. Theranostics 2021, 11, 1828–1844. [Google Scholar] [CrossRef]
- Kim, J.Y.; Kim, K.S.; Kim, I.S.; Yoon, S. Histamine Receptor Antagonists, Loratadine and Azelastine, Sensitize P-gp-overexpressing Antimitotic Drug-resistant KBV20C Cells Through Different Molecular Mechanisms. Anticancer. Res. 2019, 39, 3767–3775. [Google Scholar] [CrossRef]
- Church, D.S.; Church, M.K. Pharmacology of antihistamines. World Allergy Organ. J. 2011, 4, 22–27. [Google Scholar] [CrossRef] [Green Version]
- Johnson, D.E.; Ostrowski, P.; Jaumouille, V.; Grinstein, S. The position of lysosomes within the cell determines their luminal pH. J. Cell Biol. 2016, 212, 677–692. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Tan, S.H.; Nicolas, V.; Bauvy, C.; Yang, N.D.; Zhang, J.; Xue, Y.; Codogno, P.; Shen, H.M. Activation of lysosomal function in the course of autophagy via mTORC1 suppression and autophagosome-lysosome fusion. Cell Res. 2013, 23, 508–523. [Google Scholar] [CrossRef] [Green Version]
- Bursch, W.; Hochegger, K.; Torok, L.; Marian, B.; Ellinger, A.; Hermann, R.S. Autophagic and apoptotic types of programmed cell death exhibit different fates of cytoskeletal filaments. J. Cell Sci. 2000, 113, 1189–1198. [Google Scholar] [CrossRef]
- Chen, Q.; Kang, J.; Fu, C. The independence of and associations among apoptosis, autophagy, and necrosis. Signal Transduct. Target. Ther. 2018, 3, 18. [Google Scholar] [CrossRef] [Green Version]
- Condello, M.; Pellegrini, E.; Caraglia, M.; Meschini, S. Targeting Autophagy to Overcome Human Diseases. Int. J. Mol. Sci. 2019, 20, 725. [Google Scholar] [CrossRef] [Green Version]
- Fimia, G.M.; Piacentini, M. Regulation of autophagy in mammals and its interplay with apoptosis. Cell. Mol. Life Sci. CMLS 2010, 67, 1581–1588. [Google Scholar] [CrossRef]
- Nicolau-Galmes, F.; Asumendi, A.; Alonso-Tejerina, E.; Perez-Yarza, G.; Jangi, S.M.; Gardeazabal, J.; Arroyo-Berdugo, Y.; Careaga, J.M.; Diaz-Ramon, J.L.; Apraiz, A.; et al. Terfenadine induces apoptosis and autophagy in melanoma cells through ROS-dependent and -independent mechanisms. Apoptosis Int. J. Program. Cell Death 2011, 16, 1253–1267. [Google Scholar] [CrossRef] [Green Version]
- Shubin, A.V.; Demidyuk, I.V.; Komissarov, A.A.; Rafieva, L.M.; Kostrov, S.V. Cytoplasmic vacuolization in cell death and survival. Oncotarget 2016, 7, 55863–55889. [Google Scholar] [CrossRef] [Green Version]
- Espinosa, E.; Zamora, P.; Feliu, J.; Gonzalez Baron, M. Classification of anticancer drugs—A new system based on therapeutic targets. Cancer Treat. Rev. 2003, 29, 515–523. [Google Scholar] [CrossRef]
- Mc Gee, M.M. Targeting the Mitotic Catastrophe Signaling Pathway in Cancer. Mediat. Inflamm. 2015, 2015, 146282. [Google Scholar] [CrossRef] [Green Version]
- Vakifahmetoglu, H.; Olsson, M.; Zhivotovsky, B. Death through a tragedy: Mitotic catastrophe. Cell Death Differ. 2008, 15, 1153–1162. [Google Scholar] [CrossRef] [Green Version]
- Gu, J.J.; Kaufman, G.P.; Mavis, C.; Czuczman, M.S.; Hernandez-Ilizaliturri, F.J. Mitotic catastrophe and cell cycle arrest are alternative cell death pathways executed by bortezomib in rituximab resistant B-cell lymphoma cells. Oncotarget 2017, 8, 12741–12753. [Google Scholar] [CrossRef] [Green Version]
- Chan, D.C. Mitochondria: Dynamic organelles in disease, aging, and development. Cell 2006, 125, 1241–1252. [Google Scholar] [CrossRef] [Green Version]
- Cornet-Masana, J.M.; Banus-Mulet, A.; Carbo, J.M.; Torrente, M.A.; Guijarro, F.; Cuesta-Casanovas, L.; Esteve, J.; Risueno, R.M. Dual lysosomal-mitochondrial targeting by antihistamines to eradicate leukaemic cells. EBioMedicine 2019, 47, 221–234. [Google Scholar] [CrossRef] [Green Version]
- Pathania, D.; Millard, M.; Neamati, N. Opportunities in discovery and delivery of anticancer drugs targeting mitochondria and cancer cell metabolism. Adv. Drug Deliv. Rev. 2009, 61, 1250–1275. [Google Scholar] [CrossRef] [PubMed]
- Hasnain, S.Z.; Prins, J.B.; McGuckin, M.A. Oxidative and endoplasmic reticulum stress in beta-cell dysfunction in diabetes. J. Mol. Endocrinol. 2016, 56, 33–54. [Google Scholar] [CrossRef] [Green Version]
- Malhotra, J.D.; Kaufman, R.J. Endoplasmic reticulum stress and oxidative stress: A vicious cycle or a double-edged sword? Antioxid. Redox Signal. 2007, 9, 2277–2293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, S.S.; Kaufman, R.J. Endoplasmic reticulum stress and oxidative stress in cell fate decision and human disease. Antioxid. Redox Signal. 2014, 21, 396–413. [Google Scholar] [CrossRef]
- Bolisetty, S.; Jaimes, E.A. Mitochondria and reactive oxygen species: Physiology and pathophysiology. Int. J. Mol. Sci. 2013, 14, 6306–6344. [Google Scholar] [CrossRef]
- Xiong, Y.; Yin, Q.; Li, J.; He, S. Oxidative Stress and Endoplasmic Reticulum Stress Are Involved in the Protective Effect of Alpha Lipoic Acid Against Heat Damage in Chicken Testes. Anim. Open Access J. MDPI 2020, 10, 384. [Google Scholar] [CrossRef] [Green Version]
- Sena, L.A.; Chandel, N.S. Physiological roles of mitochondrial reactive oxygen species. Mol. Cell 2012, 48, 158–167. [Google Scholar] [CrossRef] [Green Version]
- Rahmani, M.; Davis, E.M.; Crabtree, T.R.; Habibi, J.R.; Nguyen, T.K.; Dent, P.; Grant, S. The kinase inhibitor sorafenib induces cell death through a process involving induction of endoplasmic reticulum stress. Mol. Cell. Biol. 2007, 27, 5499–5513. [Google Scholar] [CrossRef] [Green Version]
- Schardt, J.A.; Weber, D.; Eyholzer, M.; Mueller, B.U.; Pabst, T. Activation of the unfolded protein response is associated with favorable prognosis in acute myeloid leukemia. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2009, 15, 3834–3841. [Google Scholar] [CrossRef] [Green Version]
- Doran, A.C.; Yurdagul, A., Jr.; Tabas, I. Efferocytosis in health and disease. Nat. Rev. Immunol. 2020, 20, 254–267. [Google Scholar] [CrossRef]
- Abdolmaleki, F.; Farahani, N.; Gheibi Hayat, S.M.; Pirro, M.; Bianconi, V.; Barreto, G.E.; Sahebkar, A. The Role of Efferocytosis in Autoimmune Diseases. Front. Immunol. 2018, 9, 1645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boada-Romero, E.; Martinez, J.; Heckmann, B.L.; Green, D.R. The clearance of dead cells by efferocytosis. Nat. Rev. Mol. Cell Biol. 2020, 21, 398–414. [Google Scholar] [CrossRef]
- Rajput, S.; Wilber, A. Roles of inflammation in cancer initiation, progression, and metastasis. Front. Biosci. 2010, 2, 176–183. [Google Scholar] [CrossRef] [Green Version]
- Gheibi Hayat, S.M.; Bianconi, V.; Pirro, M.; Sahebkar, A. Efferocytosis: Molecular mechanisms and pathophysiological perspectives. Immunol. Cell Biol. 2019, 97, 124–133. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Trybus, E.; Król, T.; Trybus, W. The Multidirectional Effect of Azelastine Hydrochloride on Cervical Cancer Cells. Int. J. Mol. Sci. 2022, 23, 5890. https://doi.org/10.3390/ijms23115890
Trybus E, Król T, Trybus W. The Multidirectional Effect of Azelastine Hydrochloride on Cervical Cancer Cells. International Journal of Molecular Sciences. 2022; 23(11):5890. https://doi.org/10.3390/ijms23115890
Chicago/Turabian StyleTrybus, Ewa, Teodora Król, and Wojciech Trybus. 2022. "The Multidirectional Effect of Azelastine Hydrochloride on Cervical Cancer Cells" International Journal of Molecular Sciences 23, no. 11: 5890. https://doi.org/10.3390/ijms23115890
APA StyleTrybus, E., Król, T., & Trybus, W. (2022). The Multidirectional Effect of Azelastine Hydrochloride on Cervical Cancer Cells. International Journal of Molecular Sciences, 23(11), 5890. https://doi.org/10.3390/ijms23115890