
Maternal Pyrroloquinoline Quinone Supplementation Improves Offspring Liver Bioactive Lipid Profiles throughout the Lifespan and Protects against the Development of Adult NAFLD

 ,
, 
 ,
,  ,
,  and
and
Abstract
:1. Introduction
2. Results
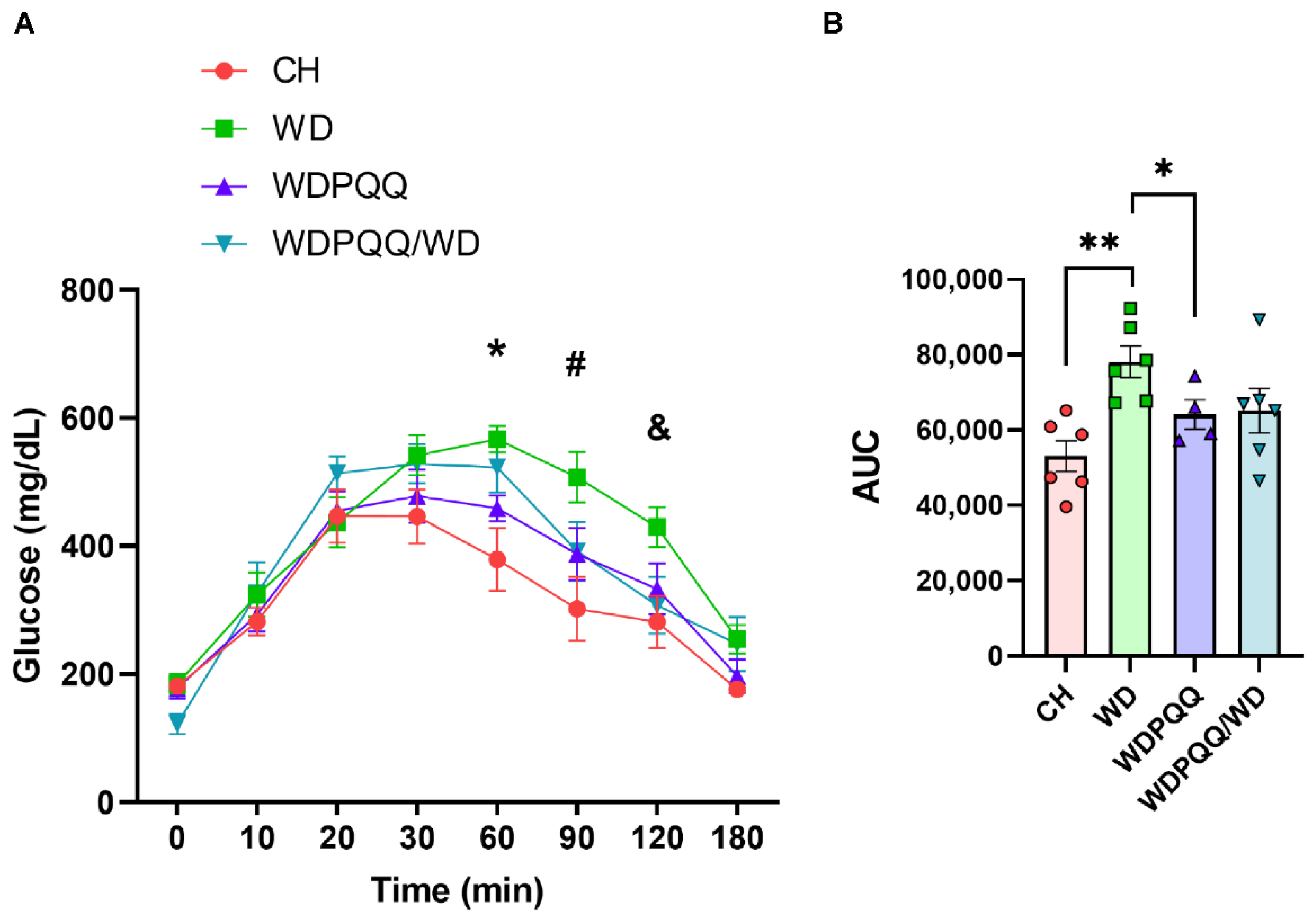
2.1. PQQ Protects Offspring of Obese Dams from Weight Gain, Even When Only Exposed to PQQ during Gestation and Lactation
2.2. PQQ Reduces Hepatic Steatosis, Beginning in Fetal Life, and Improves Lipid Metabolism in Adults
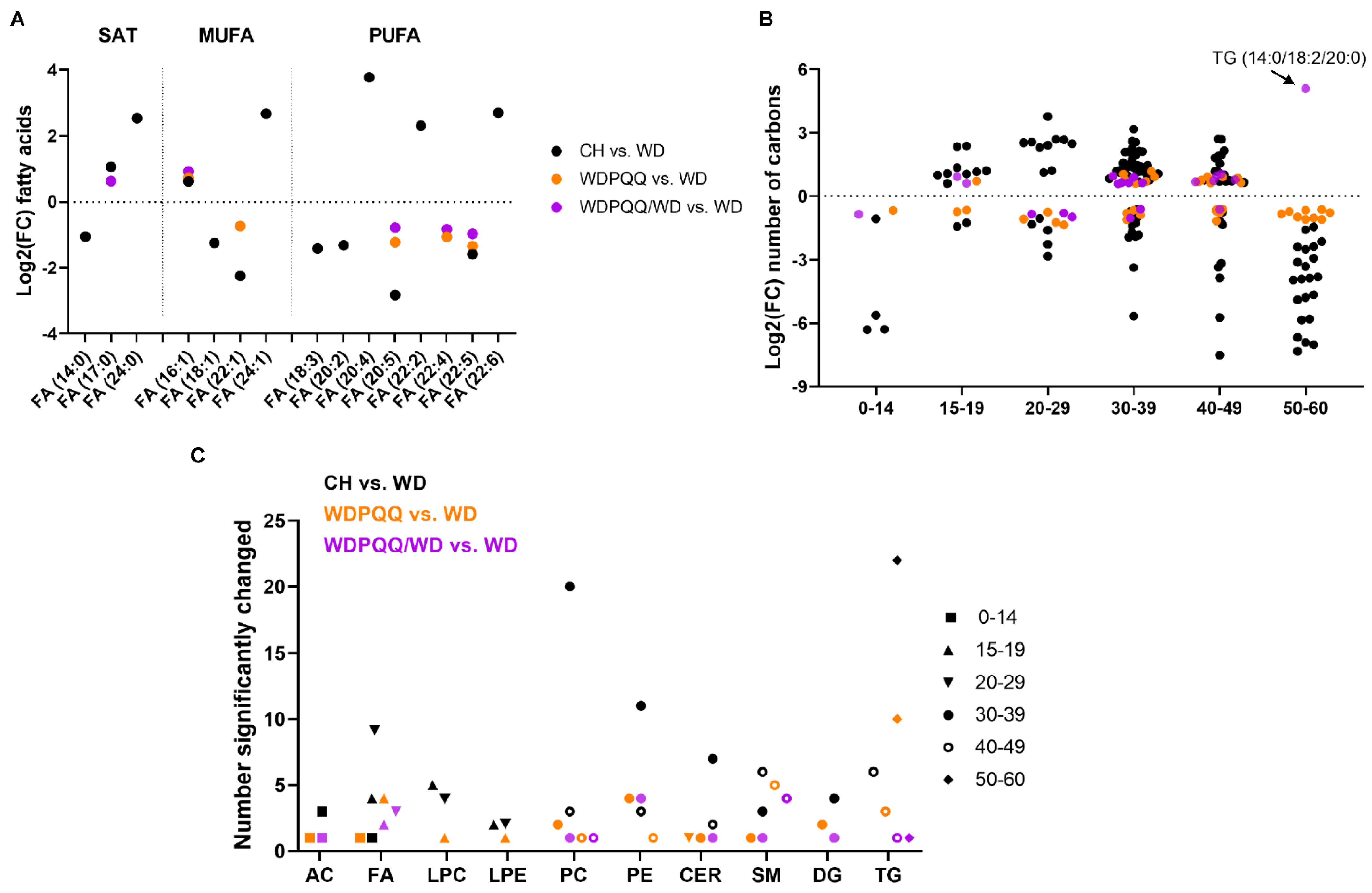
2.3. Maternal PQQ Alters Lipid Profiles in WD-Fed Offspring
3. Discussion
3.1. PQQ Improves Mitochondrial FAO
3.2. Long-Duration PQQ Exposure Alters Saturation of Hepatic Lipids
3.3. PQQ Supplementation Elevates Abundance of VLC FAs in Liver
3.4. PQQ Alters Abundance of Short-Chain Acylcarnitines and Long-Chain Glycerolipids in Early Life
3.5. PQQ Supplementation Attenuates WD-Induced Increase in PC/PE
4. Materials and Methods
4.1. Animals and Diets
4.2. Fluorescence Lifetime Imaging Microscopy (FLIM)
4.3. Lipidomics
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Doycheva, I.; Watt, K.D.; Alkhouri, N. Nonalcoholic fatty liver disease in adolescents and young adults: The next frontier in the epidemic. Hepatology 2017, 65, 2100–2109. [Google Scholar] [CrossRef] [Green Version]
- Younossi, Z.M.; Koenig, A.B.; Abdelatif, D.; Fazel, Y.; Henry, L.; Wymer, M. Global epidemiology of nonalcoholic fatty liver disease-Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 2016, 64, 73–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matteoni, C.A.; Younossi, Z.M.; Gramlich, T.; Boparai, N.; Liu, Y.C.; McCullough, A.J. Nonalcoholic fatty liver disease: A spectrum of clinical and pathological severity. Gastroenterology 1999, 116, 1413–1419. [Google Scholar] [CrossRef]
- Wesolowski, S.R.; El Kasmi, K.C.; Jonscher, K.R.; Friedman, J.E. Developmental origins of NAFLD: A womb with a clue. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 81–96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mandala, A.; Janssen, R.C.; Palle, S.; Short, K.R.; Friedman, J.E. Pediatric non-alcoholic fatty liver disease: Nutritional origins and potential molecular mechanisms. Nutrients 2020, 12, 3166. [Google Scholar] [CrossRef]
- Schwimmer, J.B.; Behling, C.; Newbury, R.; Deutsch, R.; Nievergelt, C.; Schork, N.J.; Lavine, J.E. Histopathology of pediatric nonalcoholic fatty liver disease. Hepatology 2005, 42, 641–649. [Google Scholar] [CrossRef]
- Vajro, P.; Lenta, S.; Socha, P.; Dhawan, A.; McKiernan, P.; Baumann, U.; Durmaz, O.; Lacaille, F.; McLin, V.; Nobili, V. Diagnosis of nonalcoholic fatty liver disease in children and adolescents: Position paper of the ESPGHAN Hepatology Committee. J. Pediatr. Gastroenterol. Nutr. 2012, 54, 700–713. [Google Scholar] [CrossRef]
- Boney, C.M.; Verma, A.; Tucker, R.; Vohr, B.R. Metabolic syndrome in childhood: Association with birth weight, maternal obesity, and gestational diabetes mellitus. Pediatrics 2005, 115, e290–e296. [Google Scholar] [CrossRef] [Green Version]
- Schack-Nielsen, L.; Michaelsen, K.F.; Gamborg, M.; Mortensen, E.L.; Sørensen, T.I. Gestational weight gain in relation to offspring body mass index and obesity from infancy through adulthood. Int. J. Obes. (Lond.) 2010, 34, 67–74. [Google Scholar] [CrossRef] [Green Version]
- Oben, J.A.; Mouralidarane, A.; Samuelsson, A.M.; Matthews, P.J.; Morgan, M.L.; McKee, C.; Soeda, J.; Fernandez-Twinn, D.S.; Martin-Gronert, M.S.; Ozanne, S.E.; et al. Maternal obesity during pregnancy and lactation programs the development of offspring non-alcoholic fatty liver disease in mice. J. Hepatol. 2010, 52, 913–920. [Google Scholar] [CrossRef]
- McCurdy, C.E.; Bishop, J.M.; Williams, S.M.; Grayson, B.E.; Smith, M.S.; Friedman, J.E.; Grove, K.L. Maternal high-fat diet triggers lipotoxicity in the fetal livers of nonhuman primates. J. Clin. Investig. 2009, 119, 323–335. [Google Scholar] [CrossRef] [Green Version]
- Nash, M.J.; Dobrinskikh, E.; Newsom, S.A.; Messaoudi, I.; Janssen, R.C.; Aagaard, K.M.; McCurdy, C.E.; Gannon, M.; Kievit, P.; Friedman, J.E.; et al. Maternal Western diet exposure increases periportal fibrosis beginning in utero in nonhuman primate offspring. JCI Insight 2021, 6, e154093. [Google Scholar] [CrossRef]
- Wesolowski, S.R.; Mulligan, C.M.; Janssen, R.C.; Baker, P.R., II; Bergman, B.C.; D’Alessandro, A.; Nemkov, T.; Maclean, K.N.; Jiang, H.; Dean, T.A.; et al. Switching obese mothers to a healthy diet improves fetal hypoxemia, hepatic metabolites, and lipotoxicity in non-human primates. Mol. Metab. 2018, 18, 25–41. [Google Scholar] [CrossRef]
- Cohen, C.C.; Francis, E.C.; Perng, W.; Sauder, K.A.; Scherzinger, A.; Sundaram, S.S.; Shankar, K.; Dabelea, D. Exposure to maternal fuels during pregnancy and offspring hepatic fat in early childhood: The healthy start study. Pediatr. Obes. 2022, e12902. [Google Scholar] [CrossRef]
- Cohen, C.C.; Perng, W.; Sauder, K.A.; Ringham, B.M.; Bellatorre, A.; Scherzinger, A.; Stanislawski, M.A.; Lange, L.A.; Shankar, K.; Dabelea, D. Associations of nutrient intake changes during childhood with adolescent hepatic fat: The Exploring Perinatal Outcomes among Children Study. J. Pediatr. 2021, 237, 50–58.e53. [Google Scholar] [CrossRef]
- Marí, M.; Fernández-Checa, J.C. Sphingolipid signalling and liver diseases. Liver Int. 2007, 27, 440–450. [Google Scholar] [CrossRef] [Green Version]
- Garris, C.S.; Blaho, V.A.; Hla, T.; Han, M.H. Sphingosine-1-phosphate receptor 1 signalling in T cells: Trafficking and beyond. Immunology 2014, 142, 347–353. [Google Scholar] [CrossRef]
- Sun, K.; Zhang, Y.; D’Alessandro, A.; Nemkov, T.; Song, A.; Wu, H.; Liu, H.; Adebiyi, M.; Huang, A.; Wen, Y.E.; et al. Sphingosine-1-phosphate promotes erythrocyte glycolysis and oxygen release for adaptation to high-altitude hypoxia. Nat. Commun. 2016, 7, 12086. [Google Scholar] [CrossRef]
- Johnson, D.R.; Decker, E.A. The role of oxygen in lipid oxidation reactions: A review. Annu. Rev. Food Sci. Technol. 2015, 6, 171–190. [Google Scholar] [CrossRef]
- Chavez, J.A.; Summers, S.A. A ceramide-centric view of insulin resistance. Cell Metab. 2012, 15, 585–594. [Google Scholar] [CrossRef] [Green Version]
- Turpin, S.M.; Nicholls, H.T.; Willmes, D.M.; Mourier, A.; Brodesser, S.; Wunderlich, C.M.; Mauer, J.; Xu, E.; Hammerschmidt, P.; Brönneke, H.S.; et al. Obesity-induced CerS6-dependent C16:0 ceramide production promotes weight gain and glucose intolerance. Cell Metab. 2014, 20, 678–686. [Google Scholar] [CrossRef] [Green Version]
- Luukkonen, P.K.; Zhou, Y.; Sädevirta, S.; Leivonen, M.; Arola, J.; Orešič, M.; Hyötyläinen, T.; Yki-Järvinen, H. Hepatic ceramides dissociate steatosis and insulin resistance in patients with non-alcoholic fatty liver disease. J. Hepatol. 2016, 64, 1167–1175. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.R.; Lee, E.J.; Shin, K.O.; Kim, M.H.; Pewzner-Jung, Y.; Lee, Y.M.; Park, J.W.; Futerman, A.H.; Park, W.-J. Hepatic triglyceride accumulation via endoplasmic reticulum stress-induced SREBP-1 activation is regulated by ceramide synthases. Exp. Mol. Med. 2019, 51, 1–16. [Google Scholar] [CrossRef]
- Park, W.J.; Park, J.W.; Merrill, A.H.; Storch, J.; Pewzner-Jung, Y.; Futerman, A.H. Hepatic fatty acid uptake is regulated by the sphingolipid acyl chain length. Biochim. Biophys. Acta 2014, 1841, 1754–1766. [Google Scholar] [CrossRef] [Green Version]
- Saroha, A.; Pewzner-Jung, Y.; Ferreira, N.S.; Sharma, P.; Jouan, Y.; Kelly, S.L.; Feldmesser, E.; Merrill, A.H., Jr.; Trottein, F.; Paget, C.; et al. Critical role for very-long chain sphingolipids in invariant natural killer T cell development and homeostasis. Front. Immunol. 2017, 8, 1386. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.R.; Volpert, G.; Shin, K.O.; Kim, S.Y.; Shin, S.H.; Lee, Y.; Sung, S.H.; Lee, Y.M.; Ahn, J.-H.; Pewzner-Jung, Y.; et al. Ablation of ceramide synthase 2 exacerbates dextran sodium sulphate-induced colitis in mice due to increased intestinal permeability. J. Cell Mol. Med. 2017, 21, 3565–3578. [Google Scholar] [CrossRef] [Green Version]
- D’Alessandro, A.; Hay, A.; Dzieciatkowska, M.; Brown, B.C.; Morrison, E.J.; Hansen, K.C.; Zimring, J.C. Protein-L-isoaspartate O-methyltransferase is required for in vivo control of oxidative damage in red blood cells. Haematologica 2021, 106, 2726–2739. [Google Scholar] [CrossRef]
- van der Veen, J.N.; Kennelly, J.P.; Wan, S.; Vance, J.E.; Vance, D.E.; Jacobs, R.L. The critical role of phosphatidylcholine and phosphatidylethanolamine metabolism in health and disease. Biochim. Biophys. Acta Biomembr. 2017, 1859, 1558–1572. [Google Scholar] [CrossRef]
- Smidt, C.R.; Steinberg, F.M.; Rucker, R.B. Physiologic importance of pyrroloquinoline quinone. Proc. Soc. Exp. Biol. Med. 1991, 197, 19–26. [Google Scholar] [CrossRef]
- Ouchi, A.; Nakano, M.; Nagaoka, S.; Mukai, K. Kinetic study of the antioxidant activity of pyrroloquinolinequinol (PQQH(2), a reduced form of pyrroloquinolinequinone) in micellar solution. J. Agric. Food Chem. 2009, 57, 450–456. [Google Scholar] [CrossRef]
- Jonscher, K.R.; Stewart, M.S.; Alfonso-Garcia, A.; DeFelice, B.C.; Wang, X.X.; Luo, Y.; Levi, M.; Heerwagen, M.J.; Janssen, R.C.; de la Houssaye, B.A.; et al. Early PQQ supplementation has persistent long-term protective effects on developmental programming of hepatic lipotoxicity and inflammation in obese mice. FASEB J. 2017, 31, 1434–1448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedman, J.E.; Dobrinskikh, E.; Alfonso-Garcia, A.; Fast, A.; Janssen, R.C.; Soderborg, T.K.; Anderson, A.L.; Reisz, J.A.; D’Alessandro, A.; Frank, D.N.; et al. Pyrroloquinoline quinone prevents developmental programming of microbial dysbiosis and macrophage polarization to attenuate liver fibrosis in offspring of obese mice. Hepatol. Commun. 2018, 2, 313–328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jelenik, T.; Kaul, K.; Séquaris, G.; Flögel, U.; Phielix, E.; Kotzka, J.; Knebel, B.; Fahlbusch, P.; Hörbelt, T.; Lehr, S.; et al. Mechanisms of insulin resistance in primary and secondary nonalcoholic fatty liver. Diabetes 2017, 66, 2241–2253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Srinivasan, M.; Katewa, S.D.; Palaniyappan, A.; Pandya, J.D.; Patel, M.S. Maternal high-fat diet consumption results in fetal malprogramming predisposing to the onset of metabolic syndrome-like phenotype in adulthood. Am. J. Physiol. Endocrinol. Metab. 2006, 291, E792–E799. [Google Scholar] [CrossRef]
- Desai, M.; Jellyman, J.K.; Han, G.; Beall, M.; Lane, R.H.; Ross, M.G. Maternal obesity and high-fat diet program offspring metabolic syndrome. Am. J. Obs. Gynecol. 2014, 211, 237.e1–237.e13. [Google Scholar] [CrossRef] [Green Version]
- Brumbaugh, D.E.; Friedman, J.E. Developmental origins of nonalcoholic fatty liver disease. Pediatr. Res. 2014, 75, 140–147. [Google Scholar] [CrossRef] [Green Version]
- Charron, M.J.; Williams, L.; Seki, Y.; Du, X.Q.; Chaurasia, B.; Saghatelian, A.; Summers, S.A.; Katz, E.B.; Vuguin, P.M.; Reznik, S.E. Antioxidant effects of N-acetylcysteine prevent programmed metabolic disease in mice. Diabetes 2020, 69, 1650–1661. [Google Scholar] [CrossRef]
- Huang, Y.; Gao, S.; Jun, G.; Zhao, R.; Yang, X. Supplementing the maternal diet of rats with butyrate enhances mitochondrial biogenesis in the skeletal muscles of weaned offspring. Br. J. Nutr. 2017, 117, 12–20. [Google Scholar] [CrossRef] [Green Version]
- Lu, H.; Su, S.; Ajuwon, K.M. Butyrate supplementation to gestating sows and piglets induces muscle and adipose tissue oxidative genes and improves growth performance. J. Anim. Sci. 2012, 90 (Suppl. 4), 430–432. [Google Scholar] [CrossRef] [Green Version]
- Lin, Y.; Fang, Z.F.; Che, L.Q.; Xu, S.Y.; Wu, D.; Wu, C.M.; Wu, X.Q. Use of sodium butyrate as an alternative to dietary fiber: Effects on the embryonic development and anti-oxidative capacity of rats. PLoS ONE 2014, 9, e97838. [Google Scholar] [CrossRef]
- Jonscher, K.R.; Rucker, R.B. Chapter 13—Pyrroloquinoline quinone: Its profile, effects on the liver and implications for health and disease prevention. In Dietary Interventions in Liver Disease; Watson, R.R., Preedy, V.R., Eds.; Academic Press: New York, NY, USA, 2019; pp. 157–173. [Google Scholar]
- Steinberg, F.M.; Gershwin, M.E.; Rucker, R.B. Dietary pyrroloquinoline quinone: Growth and immune response in BALB/c mice. J. Nutr. 1994, 124, 744–753. [Google Scholar] [CrossRef]
- Steinberg, F.; Stites, T.E.; Anderson, P.; Storms, D.; Chan, I.; Eghbali, S.; Rucker, R. Pyrroloquinoline quinone improves growth and reproductive performance in mice fed chemically defined diets. Exp. Biol. Med. (Maywood) 2003, 228, 160–166. [Google Scholar] [CrossRef]
- Ipsen, D.H.; Lykkesfeldt, J.; Tveden-Nyborg, P. Molecular mechanisms of hepatic lipid accumulation in non-alcoholic fatty liver disease. Cell Mol. Life Sci. 2018, 75, 3313–3327. [Google Scholar] [CrossRef] [Green Version]
- Loomba, R.; Friedman, S.L.; Shulman, G.I. Mechanisms and disease consequences of nonalcoholic fatty liver disease. Cell 2021, 184, 2537–2564. [Google Scholar] [CrossRef]
- Wang, Z.; Li, Y.; Wang, Y.; Zhao, K.; Chi, Y.; Wang, B. Pyrroloquinoline quinine protects HK-2 cells against high glucose-induced oxidative stress and apoptosis through Sirt3 and PI3K/Akt/FoxO3a signaling pathway. Biochem. Biophys. Res. Commun. 2019, 508, 398–404. [Google Scholar] [CrossRef]
- Kersten, S.; Stienstra, R. The role and regulation of the peroxisome proliferator activated receptor alpha in human liver. Biochimie 2017, 136, 75–84. [Google Scholar] [CrossRef]
- Puri, P.; Baillie, R.A.; Wiest, M.M.; Mirshahi, F.; Choudhury, J.; Cheung, O.; Sargeant, C.; Contos, M.J.; Sanyal, A.J. A lipidomic analysis of nonalcoholic fatty liver disease. Hepatology 2007, 46, 1081–1090. [Google Scholar] [CrossRef]
- de Almeida, I.T.; Cortez-Pinto, H.; Fidalgo, G.; Rodrigues, D.; Camilo, M.E. Plasma total and free fatty acids composition in human non-alcoholic steatohepatitis. Clin. Nutr. 2002, 21, 219–223. [Google Scholar] [CrossRef]
- Larter, C.Z.; Yeh, M.M.; Haigh, W.G.; Williams, J.; Brown, S.; Bell-Anderson, K.S.; Lee, S.P.; Farrell, G.C. Hepatic free fatty acids accumulate in experimental steatohepatitis: Role of adaptive pathways. J. Hepatol. 2008, 48, 638–647. [Google Scholar] [CrossRef]
- Burri, L.; Berge, K.; Wibrand, K.; Berge, R.K.; Barger, J.L. Differential effects of krill oil and fish oil on the hepatic transcriptome in mice. Front. Genet. 2011, 2, 45. [Google Scholar] [CrossRef] [Green Version]
- Ferramosca, A.; Zara, V. Modulation of hepatic steatosis by dietary fatty acids. World J. Gastroenterol. 2014, 20, 1746–1755. [Google Scholar] [CrossRef]
- Ren, C.; Hou, L.; Liu, B.; Yang, G.P.; Wang, Y.Y.; Shi, Q.Z. Distinct structures of coordination polymers incorporating flexible triazole-based ligand: Topological diversities, crystal structures and property studies. Dalton Trans. 2011, 40, 793–804. [Google Scholar] [CrossRef]
- Albracht-Schulte, K.; Kalupahana, N.S.; Ramalingam, L.; Wang, S.; Rahman, S.M.; Robert-McComb, J.; Moustaid-Moussa, N. Omega-3 fatty acids in obesity and metabolic syndrome: A mechanistic update. J. Nutr. Biochem. 2018, 58, 1–16. [Google Scholar] [CrossRef]
- Albracht-Schulte, K.; Gonzalez, S.; Jackson, A.; Wilson, S.; Ramalingam, L.; Kalupahana, N.S.; Moustaid-Moussa, N. Eicosapentaenoic acid improves hepatic metabolism and reduces inflammation independent of obesity in high-fat-fed mice and in HepG2 cells. Nutrients 2019, 11, 599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalupahana, N.S.; Claycombe, K.; Newman, S.J.; Stewart, T.; Siriwardhana, N.; Matthan, N.; Lichtenstein, A.H.; Moustaid-Moussa, N. Eicosapentaenoic acid prevents and reverses insulin resistance in high-fat diet-induced obese mice via modulation of adipose tissue inflammation. J. Nutr. 2010, 140, 1915–1922. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heerwagen, M.J.; Stewart, M.S.; de la Houssaye, B.A.; Janssen, R.C.; Friedman, J.E. Transgenic increase in n-3/n-6 fatty acid ratio reduces maternal obesity-associated inflammation and limits adverse developmental programming in mice. PLoS ONE 2013, 8, e67791. [Google Scholar] [CrossRef] [PubMed]
- Clarke, S.D. Nonalcoholic steatosis and steatohepatitis. I. Molecular mechanism for polyunsaturated fatty acid regulation of gene transcription. Am. J. Physiol. Gastrointest. Liver Physiol. 2001, 281, G865–G869. [Google Scholar] [CrossRef] [Green Version]
- Fox, T.E.; Bewley, M.C.; Unrath, K.A.; Pedersen, M.M.; Anderson, R.E.; Jung, D.Y.; Jefferson, L.S.; Kim, J.K.; Bronson, S.K.; Flanagan, J.M.; et al. Circulating sphingolipid biomarkers in models of type 1 diabetes. J. Lipid Res. 2011, 52, 509–517. [Google Scholar] [CrossRef] [Green Version]
- Oda, E.; Hatada, K.; Kimura, J.; Aizawa, Y.; Thanikachalam, P.V.; Watanabe, K. Relationships between serum unsaturated fatty acids and coronary risk factors: Negative relations between nervonic acid and obesity-related risk factors. Int. Heart J. 2005, 46, 975–985. [Google Scholar] [CrossRef] [Green Version]
- Yamazaki, Y.; Kondo, K.; Maeba, R.; Nishimukai, M.; Nezu, T.; Hara, H. Proportion of nervonic acid in serum lipids is associated with serum plasmalogen levels and metabolic syndrome. J. Oleo Sci. 2014, 63, 527–537. [Google Scholar] [CrossRef] [Green Version]
- Uchida, Y. The role of fatty acid elongation in epidermal structure and function. Dermato-Endocrinology 2011, 3, 65–69. [Google Scholar] [CrossRef] [Green Version]
- Kihara, A. Very long-chain fatty acids: Elongation, physiology and related disorders. J. Biochem. 2012, 152, 387–395. [Google Scholar] [CrossRef]
- Oertel, S.; Scholich, K.; Weigert, A.; Thomas, D.; Schmetzer, J.; Trautmann, S.; Wegner, M.S.; Radeke, H.H.; Filmann, N.; Brüne, B.; et al. Ceramide synthase 2 deficiency aggravates AOM-DSS-induced colitis in mice: Role of colon barrier integrity. Cell Mol. Life Sci. 2017, 74, 3039–3055. [Google Scholar] [CrossRef]
- Chang, Y.; Gao, X.Q.; Shen, N.; He, J.; Fan, X.; Chen, K.; Lin, X.H.; Li, H.M.; Tian, F.-S.; Li, H. A targeted metabolomic profiling of plasma acylcarnitines in nonalcoholic fatty liver disease. Eur Rev. Med. Pharmacol. Sci. 2020, 24, 7433–7441. [Google Scholar] [CrossRef]
- Mihalik, S.J.; Goodpaster, B.H.; Kelley, D.E.; Chace, D.H.; Vockley, J.; Toledo, F.G.; DeLany, J.P. Increased levels of plasma acylcarnitines in obesity and type 2 diabetes and identification of a marker of glucolipotoxicity. Obesity (Silver Spring) 2010, 18, 1695–1700. [Google Scholar] [CrossRef] [Green Version]
- Aguer, C.; McCoin, C.S.; Knotts, T.A.; Thrush, A.B.; Ono-Moore, K.; McPherson, R.; Dent, R.; Hwang, D.H.; Adams, S.H.; Harper, M.E. Acylcarnitines: Potential implications for skeletal muscle insulin resistance. FASEB J. 2015, 29, 336–345. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.; Baker, R.D.; Bhatia, T.; Zhu, L.; Baker, S.S. Pathogenesis of nonalcoholic steatohepatitis. Cell Mol. Life Sci. 2016, 73, 1969–1987. [Google Scholar] [CrossRef]
- Bauerly, K.; Harris, C.; Chowanadisai, W.; Graham, J.; Havel, P.J.; Tchaparian, E.; Satre, M.; Karliner, J.S.; Rucker, R.B. Altering pyrroloquinoline quinone nutritional status modulates mitochondrial, lipid, and energy metabolism in rats. PLoS ONE 2011, 6, e21779. [Google Scholar] [CrossRef]
- Brown, M.S.; Goldstein, J.L. The SREBP pathway: Regulation of cholesterol metabolism by proteolysis of a membrane-bound transcription factor. Cell 1997, 89, 331–340. [Google Scholar] [CrossRef] [Green Version]
- Uyeda, K.; Repa, J.J. Carbohydrate response element binding protein, ChREBP, a transcription factor coupling hepatic glucose utilization and lipid synthesis. Cell Metab. 2006, 4, 107–110. [Google Scholar] [CrossRef] [Green Version]
- Ruderman, N.B.; Xu, X.J.; Nelson, L.; Cacicedo, J.M.; Saha, A.K.; Lan, F.; Ido, Y. AMPK and SIRT1: A long-standing partnership? Am. J. Physiol. Endocrinol. Metab. 2010, 298, E751–E760. [Google Scholar] [CrossRef]
- Zhang, J.; Meruvu, S.; Bedi, Y.S.; Chau, J.; Arguelles, A.; Rucker, R.; Choudhury, M. Pyrroloquinoline quinone increases the expression and activity of Sirt1 and -3 genes in HepG2 cells. Nutr. Res. 2015, 35, 844–849. [Google Scholar] [CrossRef] [Green Version]
- Jonscher, K.R.; Chowanadisai, W.; Rucker, R.B. Pyrroloquinoline-quinone is more than an antioxidant: A vitamin-like accessory factor important in health and disease prevention. Biomolecules 2021, 11, 1441. [Google Scholar] [CrossRef]
- Zhang, H.; Li, J.; Cao, C.; Zhang, B.; Yang, W.; Shi, B.; Shan, A. Pyrroloquinoline quinone inhibits the production of inflammatory cytokines via the SIRT1/NF-κB signal pathway in weaned piglet jejunum. Food Funct. 2020, 11, 2137–2153. [Google Scholar] [CrossRef]
- Devasani, K.; Kaul, R.; Majumdar, A. Supplementation of pyrroloquinoline quinone with atorvastatin augments mitochondrial biogenesis and attenuates low grade inflammation in obese rats. Eur J. Pharmacol. 2020, 881, 173273. [Google Scholar] [CrossRef]
- Ishak, N.S.M.; Ikemoto, K.; Kikuchi, M.; Ogawa, M.; Akutagawa, K.; Akagawa, M. Pyrroloquinoline quinone attenuates fat accumulation in obese mice fed with a high-fat diet, Daphnia magna supplied with a high amount of food, and 3T3-L1 adipocytes. ACS Food Sci. Technol. 2021, 1, 1979–1989. [Google Scholar] [CrossRef]
- Fu, S.; Yang, L.; Li, P.; Hofmann, O.; Dicker, L.; Hide, W.; Lin, X.; Watkins, S.M.; Ivanov, A.R.; Hotamisligil, G.S. Aberrant lipid metabolism disrupts calcium homeostasis causing liver endoplasmic reticulum stress in obesity. Nature 2011, 473, 528–531. [Google Scholar] [CrossRef] [Green Version]
- Uhl, O.; Demmelmair, H.; Segura, M.T.; Florido, J.; Rueda, R.; Campoy, C.; Koletzko, B. Effects of obesity and gestational diabetes mellitus on placental phospholipids. Diabetes Res. Clin. Pract. 2015, 109, 364–371. [Google Scholar] [CrossRef] [PubMed]
- Galiero, R.; Caturano, A.; Vetrano, E.; Cesaro, A.; Rinaldi, L.; Salvatore, T.; Marfella, R.; Sardu, C.; Moscarella, E.; Gragnano, F.; et al. Pathophysiological mechanisms and clinical evidence of relationship between Nonalcoholic fatty liver disease (NAFLD) and cardiovascular disease. Rev. Cardiovasc. Med. 2021, 22, 755–768. [Google Scholar] [CrossRef] [PubMed]
- Dobrinskikh, E.; Al-Juboori, S.I.; Shabeka, U.; Reisz, J.A.; Zheng, C.; Marwan, A.I. Heterogeneous pulmonary response after tracheal occlusion: Clues to fetal lung growth. J. Surg. Res. 2019, 239, 242–252. [Google Scholar] [CrossRef]
- Folz, J.S.; Shalon, D.; Fiehn, O. Metabolomics analysis of time-series human small intestine lumen samples collected in vivo. Food Funct. 2021, 12, 9405–9415. [Google Scholar] [CrossRef]
- Cajka, T.; Fiehn, O. Increasing lipidomic coverage by selecting optimal mobile-phase modifiers in LC–MS of blood plasma. Metabolomics 2016, 12, 34. [Google Scholar] [CrossRef]
- Tsugawa, H.; Ikeda, K.; Takahashi, M.; Satoh, A.; Mori, Y.; Uchino, H.; Okahashi, N.; Yamada, Y.; Tada, I.; Bonini, P.; et al. A lipidome atlas in MS-DIAL 4. Nat. Biotechnol. 2020, 38, 1159–1163. [Google Scholar] [CrossRef]
- DeFelice, B.C.; Mehta, S.S.; Samra, S.; Čajka, T.; Wancewicz, B.; Fahrmann, J.F.; Fiehn, O. Mass Spectral Feature List Optimizer (MS-FLO): A tool to minimize false positive peak reports in untargeted liquid chromatography-mass spectroscopy (LC-MS) data processing. Anal. Chem. 2017, 89, 3250–3255. [Google Scholar] [CrossRef]
- Reisz, J.A.; Zheng, C.; D’Alessandro, A.; Nemkov, T. Untargeted and semi-targeted lipid analysis of biological samples using mass spectrometry-based metabolomics. Methods Mol. Biol. 2019, 1978, 121–135. [Google Scholar] [CrossRef]
- Nemkov, T.; Reisz, J.A.; Gehrke, S.; Hansen, K.C.; D’Alessandro, A. High-throughput metabolomics: Isocratic and gradient mass spectrometry-based methods. Methods Mol. Biol. 2019, 1978, 13–26. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CH | WD | WDPQQ | WDPQQ/WD | p Values | |||
|---|---|---|---|---|---|---|---|
| n = 6 | n = 8 | n = 7 | n = 6 | Interaction | Diet | PQQ | |
| Age (wks) | 23.1 ± 0.6 | 23.3 ± 1.4 | 21.5 ± 0.4 | 20.4 ± 0.03 | 0.113 | 0.176 | 0.952 |
| Body weight (g) | 27.7 ± 1.4 | 43.1 ± 0.8 | 34.3 ± 1.0 | 33.4 ± 0.5 1 | 0.0023 | <0.0001 | <0.0001 |
| Liver mass (% of BW) | 4.5 ± 0.2 | 6.7 ± 0.5 | 4.7 ± 0.1 | 4.4 ± 0.4 1 | 0.0004 | 0.0145 | 0.0952 |
| Fat mass (% of BW) | 17.2 ± 1.8 | 41.3 ± 1.0 | 34.6 ± 2.0 | 32.1 ± 1.4 1 | 0.209 | <0.0001 | <0.0001 |
| Lean mass (% of BW) | 70.2 ± 2.2 | 53.0 ± 1.1 | 59.1 ± 2.0 | 57.0 ± 1.6 2 | 0.345 | <0.0001 | 0.0126 |
| p Value | |||
|---|---|---|---|
| Lipid | CH vs. WD | WDPQQ vs. WD | WDPQQ/WD vs. WD |
| FA (16:1) | 0.0479 | 0.0660 | 0.0593 |
| FA (17:0) | 0.0007 | 0.0394 | 0.2085 |
| FA (20:5) | 0.0093 | 0.0784 | 0.1813 |
| FA (22:4) | 0.0479 | 0.0784 | 0.1499 |
| FA (22:5) | 0.0479 | 0.0920 | 0.1813 |
| PE (18:0/20.4) | <0.0001 | 0.0784 | 0.1264 |
| PE (18:0/22.6) | 0.0093 | 0.2212 | 0.2322 |
| PE (18:1/18.0) | 0.0168 | 0.1334 | 0.2187 |
| PE (18:1/18.1) | 0.0089 | 0.1002 | 0.0866 |
| PE (20:0/18.2) | 0.0479 | 0.2213 | 0.2322 |
| SM (18:0/22.0) | 0.0061 | 0.0089 | 0.0607 |
| SM (18:1/21.0) | 0.0168 | 0.0394 | 0.005 |
| SM (18:1/22.0) | 0.3699 | 0.1181 | 0.3669 |
| SM (18:1/22.1) | 0.0479 | 0.004 | 0.0574 |
| SM (18:1/23.0) | 0.0479 | 0.2388 | 0.988 |
| SM (18:1/24.0) | 0.0005 | 0.1002 | 0.0209 |
| SM (18:2/24.1) | 0.0005 | 0.1002 | 0.0054 |
| WDPQQ vs. WD | Log2(FC) | −Log10(P) |
|---|---|---|
| Fetus | ||
| FA (22:6) | 0.6343 | 1.5199 |
| ethanolamine phosphate | 0.6512 | 2.2746 |
| Weanling | ||
| AC (6:0) | −1.7929 | 1.9059 |
| AC (8:1) | −1.3661 | 1.6884 |
| Adult | ||
| AC (3:0) | −0.8425 | 1.1616 |
| Cer (18:0/18:1) | −1.0216 | 1.8328 |
| DG (18:1/18:1) | −0.6062 | 1.4309 |
| FA (16:1) | 0.9205 | 2.2385 |
| FA (17:0) | 0.6232 | 2.4215 |
| FA (20:5) | −0.7813 | 1.637 |
| FA (22:4) | −0.8306 | 2.0879 |
| FA (22:5) | −0.9707 | 1.8923 |
| PC (16:0/16.0) | 0.6412 | 2.8177 |
| PC (17:0/18.2) | 0.7342 | 1.1069 |
| PE (18:0/20.4) | 0.643 | 2.5834 |
| PE (18:1/18.0) | 0.8959 | 1.8718 |
| PE (18:1/18.1) | 0.9549 | 1.4628 |
| PE (20:4/18.2) | 0.5958 | 3.1917 |
| SM (18:0/22.0) | 0.9733 | 3.6305 |
| SM (18:1/21.0) | 0.6561 | 3.2631 |
| SM (18:1/22.1) | 0.7783 | 3.1681 |
| SM (18:1/24.0) | 1.0566 | 1.8493 |
| SM (18:2/24.1) | 0.6891 | 3.3378 |
| TG (12:0/16:0/16.1) | −0.612 | 1.5556 |
| TG (14:1/18:2/20:0) | 5.0953 | 1.5114 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mandala, A.; Dobrinskikh, E.; Janssen, R.C.; Fiehn, O.; D’Alessandro, A.; Friedman, J.E.; Jonscher, K.R. Maternal Pyrroloquinoline Quinone Supplementation Improves Offspring Liver Bioactive Lipid Profiles throughout the Lifespan and Protects against the Development of Adult NAFLD. Int. J. Mol. Sci. 2022, 23, 6043. https://doi.org/10.3390/ijms23116043
Mandala A, Dobrinskikh E, Janssen RC, Fiehn O, D’Alessandro A, Friedman JE, Jonscher KR. Maternal Pyrroloquinoline Quinone Supplementation Improves Offspring Liver Bioactive Lipid Profiles throughout the Lifespan and Protects against the Development of Adult NAFLD. International Journal of Molecular Sciences. 2022; 23(11):6043. https://doi.org/10.3390/ijms23116043
Chicago/Turabian StyleMandala, Ashok, Evgenia Dobrinskikh, Rachel C. Janssen, Oliver Fiehn, Angelo D’Alessandro, Jacob E. Friedman, and Karen R. Jonscher. 2022. "Maternal Pyrroloquinoline Quinone Supplementation Improves Offspring Liver Bioactive Lipid Profiles throughout the Lifespan and Protects against the Development of Adult NAFLD" International Journal of Molecular Sciences 23, no. 11: 6043. https://doi.org/10.3390/ijms23116043
APA StyleMandala, A., Dobrinskikh, E., Janssen, R. C., Fiehn, O., D’Alessandro, A., Friedman, J. E., & Jonscher, K. R. (2022). Maternal Pyrroloquinoline Quinone Supplementation Improves Offspring Liver Bioactive Lipid Profiles throughout the Lifespan and Protects against the Development of Adult NAFLD. International Journal of Molecular Sciences, 23(11), 6043. https://doi.org/10.3390/ijms23116043