
Beyond GWAS—Could Genetic Differentiation within the Allograft Rejection Pathway Shape Natural Immunity to COVID-19?

 , , , , ,
, , , , ,  ,
,  , ,
, ,
Abstract
:1. Introduction
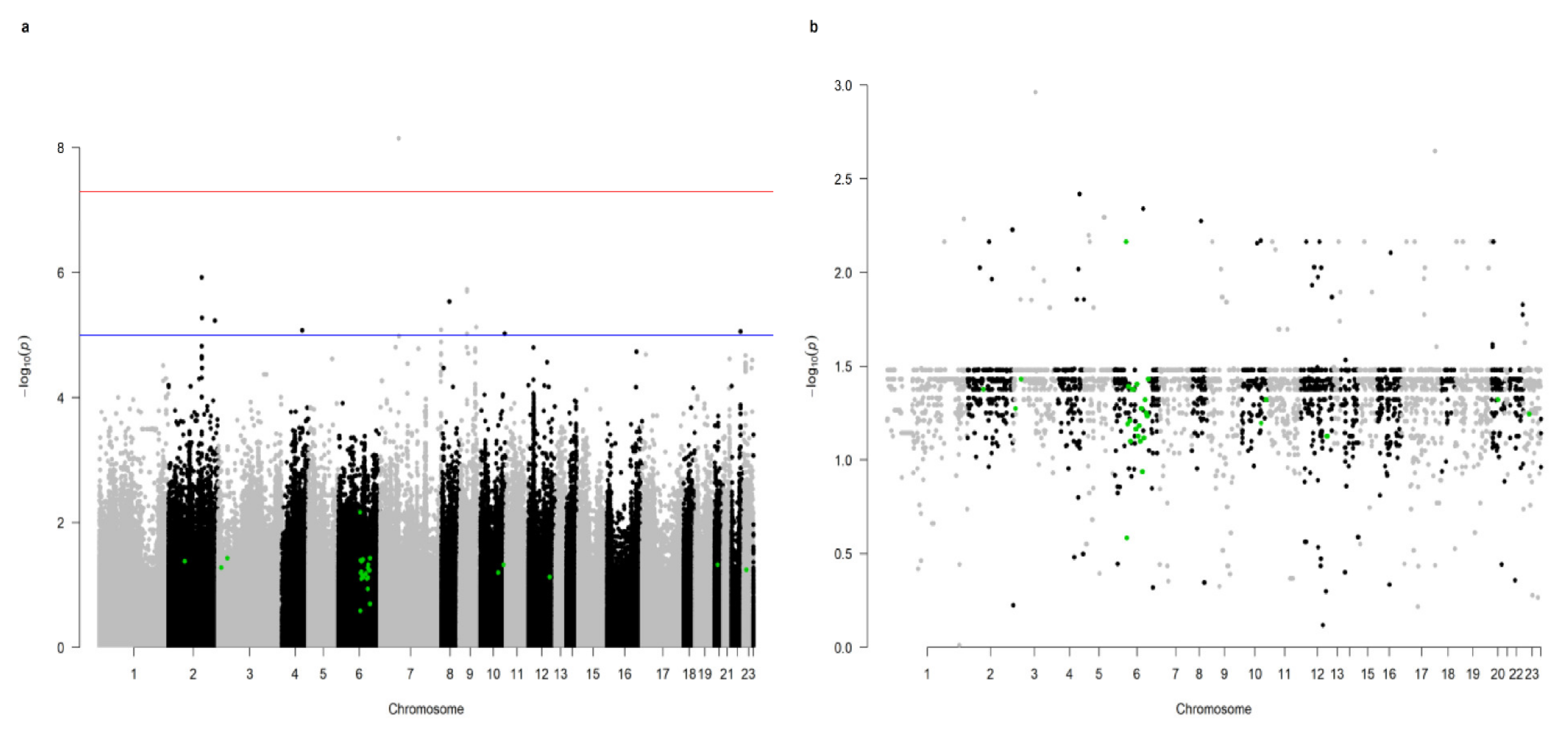
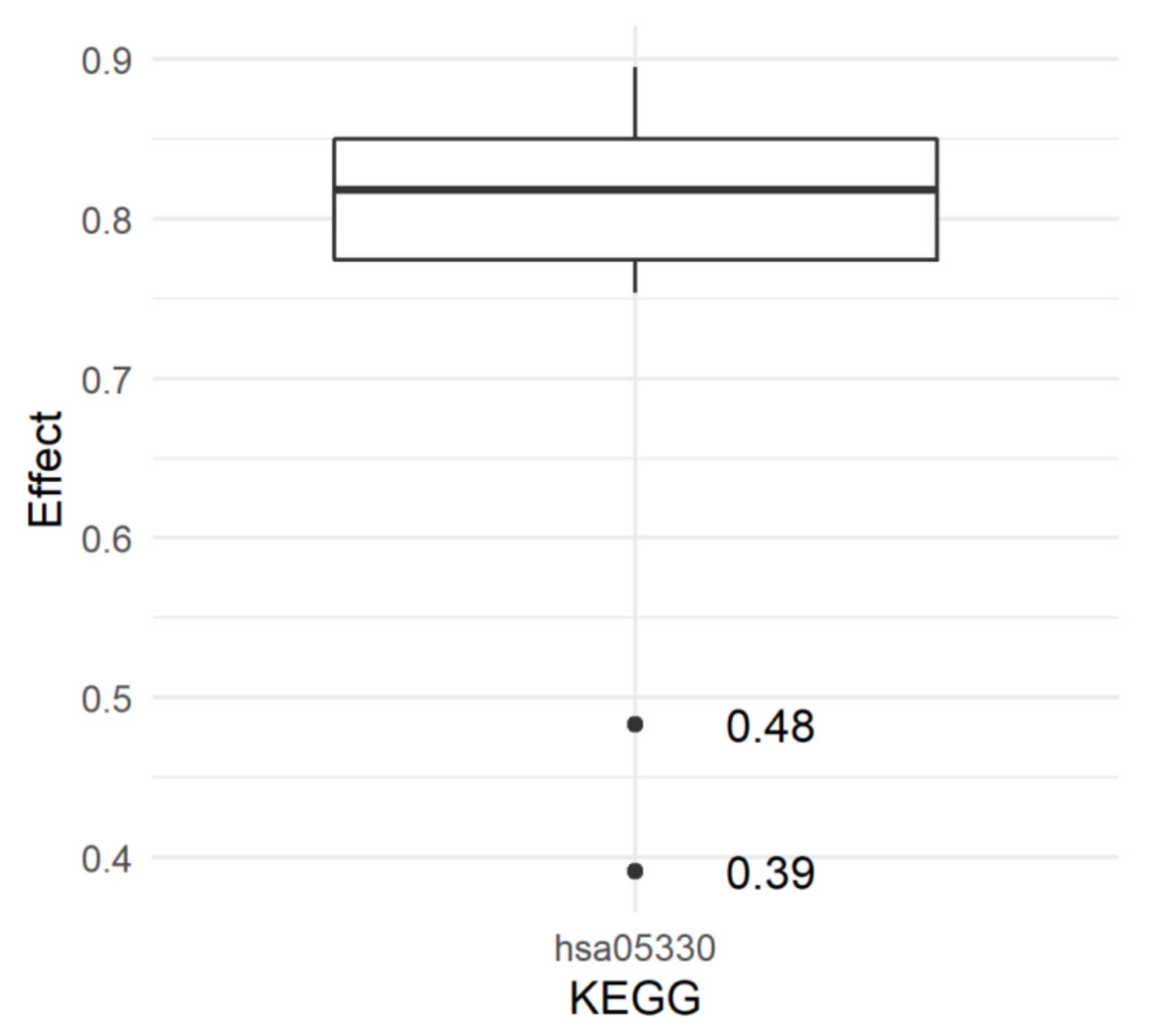
2. Results
3. Discussion
4. Material and Methods
4.1. Sample Collection
4.2. Ethical Policy
4.3. Total Quality Management
4.4. Whole Genome Sequencing
4.5. Pre-Processing of Whole Genome Sequence Data
4.6. Phenotype Encoding
4.7. Genome-Wide Association Study
4.8. Estimation of KEGG Pathway Effects
5. Conclusions
- (1)
- the Allograft rejection pathway (hsa05330) was significant for the resistance to the COVID-19 infection;
- (2)
- 27 SNPs marking genes constituting the Allograft rejection pathway, and the majority of these were located on chromosome 6 (19 SNPs), while the remainder were mapped to chromosomes 2, 3, 10, 12, 20, and X.
- (3)
- the Allograft rejection pathway comprises several immune system components crucial for the self versus non-self recognition, and also the components of antiviral immunity;
- (4)
- no significant metabolic pathway was indicated in the case of susceptibility to COVID-19 and its severe course.
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mosharaf, M.P.; Reza, M.S.; Kibria, M.K.; Ahmed, F.F.; Kabir, M.H.; Hasan, S.; Mollah, M.N.H. Computational identification of host genomic biomarkers highlighting their functions, pathways and regulators that influence SARS-CoV-2 infections and drug repurposing. Sci. Rep. 2022, 12, 4279. [Google Scholar] [CrossRef]
- Zerbo, O.; Lewis, N.; Fireman, B.; Goddard, K.; Skarbinski, J.; Sejvar, J.J.; Azziz-Baumgartner, E.; Klein, N.P. Population-based assessment of risks for severe COVID-19 disease outcomes. Influenza Other Respir. Viruses 2022, 16, 159–165. [Google Scholar] [CrossRef] [PubMed]
- Singh, P.P.; Srivastava, A.; Sultana, G.N.N.; Khanam, N.; Pathak, A.; Suravajhala, P.; Singh, R.; Shrivastava, P.; van Driem, G.; Thangaraj, K.; et al. The major genetic risk factor for severe COVID-19 does not show any association among South Asian populations. Sci. Rep. 2021, 11, 12346. [Google Scholar] [CrossRef] [PubMed]
- Zhou, F.; Yu, T.; Du, R.; Fan, G.; Liu, Y.; Liu, Z.; Xiang, J.; Wang, Y.; Song, B.; Gu, X.; et al. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: A retrospective cohort study. Lancet 2020, 395, 1054–1062. [Google Scholar] [CrossRef]
- Maximiano Sousa, F.; Roelens, M.; Fricker, B.; Thiabaud, A.; Iten, A.; Cusini, A.; Flury, D.; Buettcher, M.; Zukol, F.; Balmelli, C.; et al. CH-Sur Study Group Risk factors for severe outcomes for COVID-19 patients hospitalised in Switzerland during the first pandemic wave, February to August 2020: Prospective observational cohort study. Swiss Med. Wkly. 2021, 151, w20547. [Google Scholar] [CrossRef]
- Sandoval, M.; Nguyen, D.T.; Vahidy, F.S.; Graviss, E.A. Risk factors for severity of COVID-19 in hospital patients age 18–29 years. PLoS ONE 2021, 16, e0255544. [Google Scholar] [CrossRef] [PubMed]
- Satlin, M.J.; Zucker, J.; Baer, B.R.; Rajan, M.; Hupert, N.; Schang, L.M.; Pinheiro, L.C.; Shen, Y.; Sobieszczyk, M.E.; Westblade, L.F.; et al. Changes in SARS-CoV-2 viral load and mortality during the initial wave of the pandemic in New York City. PLoS ONE 2021, 16, e0257979. [Google Scholar] [CrossRef]
- Dadras, O.; Afsahi, A.M.; Pashaei, Z.; Mojdeganlou, H.; Karimi, A.; Habibi, P.; Barzegary, A.; Fakhfouri, A.; Mirzapour, P.; Janfaza, N.; et al. The relationship between COVID-19 viral load and disease severity: A systematic review. Immun. Inflamm. Dis. 2022, 10, e580. [Google Scholar] [CrossRef] [PubMed]
- Duarte-Salles, T.; Vizcaya, D.; Pistillo, A.; Casajust, P.; Sena, A.G.; Lai, L.Y.H.; Prats-Uribe, A.; Ahmed, W.-U.-R.; Alshammari, T.M.; Alghoul, H.; et al. Thirty-Day Outcomes of Children and Adolescents With COVID-19: An International Experience. Pediatrics 2021, 148, e2020042929. [Google Scholar] [CrossRef]
- Garrido, C.; Hurst, J.H.; Lorang, C.G.; Aquino, J.N.; Rodriguez, J.; Pfeiffer, T.S.; Singh, T.; Semmes, E.C.; Lugo, D.J.; Rotta, A.T.; et al. Asymptomatic or mild symptomatic SARS-CoV-2 infection elicits durable neutralizing antibody responses in children and adolescents. JCI Insight 2021, 6, e150909. [Google Scholar] [CrossRef]
- Severe COVID-19 GWAS Group; Ellinghaus, D.; Degenhardt, F.; Bujanda, L.; Buti, M.; Albillos, A.; Invernizzi, P.; Fernández, J.; Prati, D.; Baselli, G.; et al. Genomewide Association Study of Severe Covid-19 with Respiratory Failure. N. Engl. J. Med. 2020, 383, 1522–1534. [Google Scholar] [CrossRef]
- Pairo-Castineira, E.; Clohisey, S.; Klaric, L.; Bretherick, A.D.; Rawlik, K.; Pasko, D.; Walker, S.; Parkinson, N.; Fourman, M.H.; Russell, C.D.; et al. Genetic mechanisms of critical illness in COVID-19. Nature 2021, 591, 92–98. [Google Scholar] [CrossRef] [PubMed]
- Zeberg, H.; Pääbo, S. The major genetic risk factor for severe COVID-19 is inherited from Neanderthals. Nature 2020, 587, 610–612. [Google Scholar] [CrossRef] [PubMed]
- Zeberg, H.; Pääbo, S. A genomic region associated with protection against severe COVID-19 is inherited from Neandertals. Proc. Natl. Acad. Sci. USA 2021, 118, e2026309118. [Google Scholar] [CrossRef]
- Bruchez, A.; Sha, K.; Johnson, J.; Chen, L.; Stefani, C.; McConnell, H.; Gaucherand, L.; Prins, R.; Matreyek, K.A.; Hume, A.J.; et al. MHC class II transactivator CIITA induces cell resistance to Ebola virus and SARS-like coronaviruses. Science 2020, 370, 241–247. [Google Scholar] [CrossRef] [PubMed]
- Schneider, D.S.; Ayres, J.S. Two ways to survive infection: What resistance and tolerance can teach us about treating infectious diseases. Nat. Rev. Immunol. 2008, 8, 889–895. [Google Scholar] [CrossRef] [PubMed]
- Netea, M.G.; Domínguez-Andrés, J.; van de Veerdonk, F.L.; van Crevel, R.; Pulendran, B.; van der Meer, J.W.M. Natural resistance against infections: Focus on COVID-19. Trends Immunol. 2022, 43, 106–116. [Google Scholar] [CrossRef]
- Cardon, L.R.; Palmer, L.J. Population stratification and spurious allelic association. Lancet 2003, 361, 598–604. [Google Scholar] [CrossRef]
- Davidson, S.; Starkey, A.; MacKenzie, A. Evidence of uneven selective pressure on different subsets of the conserved human genome; implications for the significance of intronic and intergenic DNA. BMC Genom. 2009, 10, 614. [Google Scholar] [CrossRef] [Green Version]
- Siu, J.H.Y.; Surendrakumar, V.; Richards, J.A.; Pettigrew, G.J. T cell Allorecognition Pathways in Solid Organ Transplantation. Front. Immunol. 2018, 9, 2548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, A.; David, J.K.; Maden, S.K.; Wood, M.A.; Weeder, B.R.; Nellore, A.; Thompson, R.F. Human Leukocyte Antigen Susceptibility Map for Severe Acute Respiratory Syndrome Coronavirus 2. J. Virol. 2020, 94, 13. [Google Scholar] [CrossRef] [Green Version]
- Weizmann Institute of Science Gene Cards. Available online: https://genecards.weizmann.ac.il/v3/ (accessed on 4 April 2022).
- Mahoney, K.M.; Rennert, P.D.; Freeman, G.J. Combination cancer immunotherapy and new immunomodulatory targets. Nat. Rev. Drug Discov. 2015, 14, 561–584. [Google Scholar] [CrossRef] [PubMed]
- Elgueta, R.; Benson, M.J.; de Vries, V.C.; Wasiuk, A.; Guo, Y.; Noelle, R.J. Molecular mechanism and function of CD40/CD40L engagement in the immune system. Immunol. Rev. 2009, 229, 152–172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McHugh, R.S.; Ahmed, S.N.; Wang, Y.C.; Sell, K.W.; Selvaraj, P. Construction, purification, and functional incorporation on tumor cells of glycolipid-anchored human B7-1 (CD80). Proc. Natl. Acad. Sci. USA 1995, 92, 8059–8063. [Google Scholar] [CrossRef] [Green Version]
- Bromley, S.K.; Iaboni, A.; Davis, S.J.; Whitty, A.; Green, J.M.; Shaw, A.S.; Weiss, A.; Dustin, M.L. The immunological synapse and CD28-CD80 interactions. Nat. Immunol. 2001, 2, 1159–1166. [Google Scholar] [CrossRef]
- Krummel, M.F.; Allison, J.P. CD28 and CTLA-4 have opposing effects on the response of T cells to stimulation. J. Exp. Med. 1995, 182, 459–465. [Google Scholar] [CrossRef] [Green Version]
- Pentcheva-Hoang, T.; Egen, J.G.; Wojnoonski, K.; Allison, J.P. B7-1 and B7-2 Selectively Recruit CTLA-4 and CD28 to the Immunological Synapse. Immunity 2004, 21, 401–413. [Google Scholar] [CrossRef] [Green Version]
- Yokosuka, T.; Saito, T. The Immunological Synapse, TCR Microclusters, and T Cell Activation. In Immunological Synapse; Springer: Berlin/Heidelberg, Germany, 2010; pp. 81–107. [Google Scholar]
- Kong, K.-F.; Yokosuka, T.; Canonigo-Balancio, A.J.; Isakov, N.; Saito, T.; Altman, A. A motif in the V3 domain of the kinase PKC-θ determines its localization in the immunological synapse and functions in T cells via association with CD28. Nat. Immunol. 2011, 12, 1105–1112. [Google Scholar] [CrossRef] [Green Version]
- Galbraith, M.D.; Kinning, K.T.; Sullivan, K.D.; Araya, P.; Smith, K.P.; Granrath, R.E.; Shaw, J.R.; Baxter, R.; Jordan, K.R.; Russell, S.; et al. Specialized interferon action in COVID-19. Proc. Natl. Acad. Sci. USA 2022, 119, e2116730119. [Google Scholar] [CrossRef]
- Ziegler, C.G.K.; Miao, V.N.; Owings, A.H.; Navia, A.W.; Tang, Y.; Bromley, J.D.; Lotfy, P.; Sloan, M.; Laird, H.; Williams, H.B.; et al. Impaired local intrinsic immunity to SARS-CoV-2 infection in severe COVID-19. Cell 2021, 184, 4713–4733.e22. [Google Scholar] [CrossRef]
- Bastard, P.; Rosen, L.B.; Zhang, Q.; Michailidis, E.; Hoffmann, H.-H.; Zhang, Y.; Dorgham, K.; Philippot, Q.; Rosain, J.; Béziat, V.; et al. Autoantibodies against type I IFNs in patients with life-threatening COVID-19. Science 2020, 370, eabd4585. [Google Scholar] [CrossRef] [PubMed]
- Van Laarhoven, A.; Kurver, L.; Overheul, G.J.; Kooistra, E.J.; Abdo, W.F.; van Crevel, R.; Duivenvoorden, R.; Kox, M.; ten Oever, J.; Schouten, J.; et al. Interferon gamma immunotherapy in five critically ill COVID-19 patients with impaired cellular immunity: A case series. Med 2021, 2, 1163–1170.e2. [Google Scholar] [CrossRef]
- Hidalgo, L.G.; Halloran, P.F. Role of IFN-gamma in allograft rejection. Crit. Rev. Immunol. 2002, 22, 317–349. [Google Scholar] [CrossRef]
- Halloran, P.F.; Afrouzian, M.; Ramassar, V.; Urmson, J.; Zhu, L.-F.; Helms, L.M.H.; Solez, K.; Kneteman, N.M. Interferon-γ Acts Directly on Rejecting Renal Allografts to Prevent Graft Necrosis. Am. J. Pathol. 2001, 158, 215–226. [Google Scholar] [CrossRef]
- Roesler, J.; Horwitz, M.E.; Picard, C.; Bordigoni, P.; Davies, G.; Koscielniak, E.; Levin, M.; Veys, P.; Reuter, U.; Schulz, A.; et al. Hematopoietic stem cell transplantation for complete IFN-γ receptor 1 deficiency: A multi-institutional survey. J. Pediatr. 2004, 145, 806–812. [Google Scholar] [CrossRef]
- Merli, P.; Caruana, I.; De Vito, R.; Strocchio, L.; Weber, G.; Del Bufalo, F.; Buatois, V.; Montanari, P.; Cefalo, M.G.; Pitisci, A.; et al. Role of interferon-γ in immune-mediated graft failure after allogeneic hematopoietic stem cell transplantation. Haematologica 2019, 104, 2314–2323. [Google Scholar] [CrossRef] [PubMed]
- Renu, K.; Prasanna, P.L.; Valsala Gopalakrishnan, A. Coronaviruses pathogenesis, comorbidities and multi-organ damage—A review. Life Sci. 2020, 255, 117839. [Google Scholar] [CrossRef] [PubMed]
- Van de Veerdonk, F.L.; Giamarellos-Bourboulis, E.; Pickkers, P.; Derde, L.; Leavis, H.; van Crevel, R.; Engel, J.J.; Wiersinga, W.J.; Vlaar, A.P.J.; Shankar-Hari, M.; et al. A guide to immunotherapy for COVID-19. Nat. Med. 2022, 28, 39–50. [Google Scholar] [CrossRef]
- Garassino, M.C.; Whisenant, J.G.; Huang, L.C.; Trama, A.; Torri, V.; Agustoni, F.; Baena, J.; Banna, G.; Berardi, R.; Bettini, A.C.; et al. COVID-19 in patients with thoracic malignancies (TERAVOLT): First results of an international, registry-based, cohort study. Lancet Oncol. 2020, 21, 914–922. [Google Scholar] [CrossRef]
- Vivarelli, S.; Falzone, L.; Torino, F.; Scandurra, G.; Russo, G.; Bordonaro, R.; Pappalardo, F.; Spandidos, D.A.; Raciti, G.; Libra, M. Immune-checkpoint inhibitors from cancer to COVID-19: A promising avenue for the treatment of patients with COVID-19 (Review). Int. J. Oncol. 2021, 58, 145–157. [Google Scholar] [CrossRef]
- Vivarelli, S.; Falzone, L.; Grillo, C.M.; Scandurra, G.; Torino, F.; Libra, M. Cancer Management during COVID-19 Pandemic: Is Immune Checkpoint Inhibitors-Based Immunotherapy Harmful or Beneficial? Cancers 2020, 12, 2237. [Google Scholar] [CrossRef]
- Qian, W.; Ye, Y.; Zuo, L.; Song, T.; Xu, Q.; Wang, Y.; Qian, J.; Tian, Y. Immune checkpoint inhibitors use and effects on prognosis of COVID-19 infection: A systematic review and meta-analysis. Immunotherapy 2021, 13, 1271–1282. [Google Scholar] [CrossRef]
- Zhang, Q.; Bastard, P.; Liu, Z.; Le Pen, J.; Moncada-Velez, M.; Chen, J.; Ogishi, M.; Sabli, I.; Hodeib, S.; Korol, C.; et al. Inborn errors of type I IFN immunity in patients with life-threatening COVID-19. Science 2020, 370, eabd4570. [Google Scholar] [CrossRef] [PubMed]
- Dustin, M.L. The Immunological Synapse. Cancer Immunol. Res. 2014, 2, 1023–1033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grakoui, A.; Bromley, S.K.; Sumen, C.; Davis, M.M.; Shaw, A.S.; Allen, P.M.; Dustin, M.L. The Immunological Synapse: A Molecular Machine Controlling T Cell Activation. Science 1999, 285, 221–227. [Google Scholar] [CrossRef] [Green Version]
- Schubert, D.A.; Gordo, S.; Sabatino, J.J.; Vardhana, S.; Gagnon, E.; Sethi, D.K.; Seth, N.P.; Choudhuri, K.; Reijonen, H.; Nepom, G.T.; et al. Self-reactive human CD4 T cell clones form unusual immunological synapses. J. Exp. Med. 2012, 209, 335–352. [Google Scholar] [CrossRef]
- Khalil, D.N.; Smith, E.L.; Brentjens, R.J.; Wolchok, J.D. The future of cancer treatment: Immunomodulation, CARs and combination immunotherapy. Nat. Rev. Clin. Oncol. 2016, 13, 273–290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dustin, M.L.; Colman, D.R. Neural and Immunological Synaptic Relations. Science 2002, 298, 785–789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tseng, S.-Y.; Waite, J.C.; Liu, M.; Vardhana, S.; Dustin, M.L. T Cell-Dendritic Cell Immunological Synapses Contain TCR-dependent CD28-CD80 Clusters That Recruit Protein Kinase Cθ. J. Immunol. 2008, 181, 4852–4863. [Google Scholar] [CrossRef] [Green Version]
- Turnis, M.E.; Andrews, L.P.; Vignali, D.A.A. Inhibitory receptors as targets for cancer immunotherapy. Eur. J. Immunol. 2015, 45, 1892–1905. [Google Scholar] [CrossRef]
- Balidis, M.; Mikropoulos, D.; Gatzioufas, Z.; de Politis, P.B.; Sidiropoulos, G.; Vassiliadis, V. Acute corneal graft rejection after anti-severe acute respiratory syndrome-coronavirus-2 vaccination: A report of four cases. Eur. J. Ophthalmol. 2021, 112067212110640. [Google Scholar] [CrossRef] [PubMed]
- Wasser, L.M.; Roditi, E.; Zadok, D.; Berkowitz, L.; Weill, Y. Keratoplasty Rejection After the BNT162b2 messenger RNA Vaccine. Cornea 2021, 40, 1070–1072. [Google Scholar] [CrossRef] [PubMed]
- Phylactou, M.; Li, J.-P.O.; Larkin, D.F.P. Characteristics of endothelial corneal transplant rejection following immunisation with SARS-CoV-2 messenger RNA vaccine. Br. J. Ophthalmol. 2021, 105, 893–896. [Google Scholar] [CrossRef] [PubMed]
- Ravichandran, S.; Natarajan, R. Corneal graft rejection after COVID-19 vaccination. Indian J. Ophthalmol. 2021, 69, 1953. [Google Scholar] [CrossRef] [PubMed]
- Vyhmeister, R.; Enestvedt, C.K.; VanSandt, M.; Schlansky, B. Steroid-Resistant Acute Cellular Rejection of the Liver After Severe Acute Respiratory Syndrome Coronavirus 2 mRNA Vaccination. Liver Transplant. 2021, 27, 1339–1342. [Google Scholar] [CrossRef]
- Ramaswamy, M.; Clel, S.Y.; Cruz, A.C.; Siegel, R.M. Many Checkpoints on the Road to Cell Death:Regulation of Fas–FasL Interactions and Fas Signaling in Peripheral Immune Responses. In Death Receptors and Cognate Ligands in Cancer; Springer: Berlin/Heidelberg, Germany, 2009; pp. 17–47. [Google Scholar]
- Chan, F.K.-M.; Chun, H.J.; Zheng, L.; Siegel, R.M.; Bui, K.L.; Lenardo, M.J. A Domain in TNF Receptors That Mediates Ligand-Independent Receptor Assembly and Signaling. Science 2000, 288, 2351–2354. [Google Scholar] [CrossRef] [PubMed]
- Topham, D.J.; Tripp, R.A.; Doherty, P.C. CD8+ T cells clear influenza virus by perforin or Fas-dependent processes. J. Immunol. 1997, 159, 5197–5200. [Google Scholar] [PubMed]
- Bień, K.; Sokołowska, J.; Bąska, P.; Nowak, Z.; Stankiewicz, W.; Krzyzowska, M. Fas/FasL Pathway Participates in Regulation of Antiviral and Inflammatory Response during Mousepox Infection of Lungs. Mediat. Inflamm. 2015, 2015, 281613. [Google Scholar] [CrossRef]
- Martinez, O.M.; Krams, S.M. Involvement of Fas-Fas Ligand Interactions in Graft Rejection. Int. Rev. Immunol. 1999, 18, 527–546. [Google Scholar] [CrossRef]
- Wang, Y.-L. Correlation of CD95 and soluble CD95 expression with acute rejection status of liver transplantation. World J. Gastroenterol. 2005, 11, 1700. [Google Scholar] [CrossRef] [PubMed]
- Rivero, M.; Crespo, J.; Mayorga, M.; Fabrega, E.; Casafont, F.; Pons-Romero, F. Involvement of the Fas system in liver allograft rejection. Am. J. Gastroenterol. 2002, 97, 1501–1506. [Google Scholar] [CrossRef] [PubMed]
- Ichiba, T.; Hoshi, Y.; Eto, Y.; Tajima, N.; Kuraishi, Y. Characterization of GFR, a novel guanine nucleotide exchange factor for Rap1. FEBS Lett. 1999, 457, 85–89. [Google Scholar] [CrossRef] [Green Version]
- NIH National Early Warning Score. Available online: https://www.mdcalc.com/national-early-warning-score-news#why-use (accessed on 10 March 2022).
- Kaja, E.; Lejman, A.; Sielski, D.; Sypniewski, M.; Gambin, T.; Dawidziuk, M.; Suchocki, T.; Golik, P.; Wojtaszewska, M.; Mroczek, M.; et al. The Thousand Polish Genomes-A Database of Polish Variant Allele Frequencies. Int. J. Mol. Sci. 2022, 23, 4532. [Google Scholar] [CrossRef]
- Chiang, C.; Layer, R.M.; Faust, G.G.; Lindberg, M.R.; Rose, D.B.; Garrison, E.P.; Marth, G.T.; Quinlan, A.R.; Hall, I.M. SpeedSeq: Ultra-fast personal genome analysis and interpretation. Nat. Methods 2015, 12, 966–968. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv 2013, arXiv:1303.3997. [Google Scholar]
- Faust, G.G.; Hall, I.M. SAMBLASTER: Fast duplicate marking and structural variant read extraction. Bioinformatics 2014, 30, 2503–2505. [Google Scholar] [CrossRef] [Green Version]
- Pedersen, B.; Quinlan, A. Mosdepth: Quick coverage calculation for genomes and exomes. Bioinformatics 2018, 34, 867–868. [Google Scholar] [CrossRef]
- Poplin, R.; Chang, P.-C.; Alexander, D.; Schwartz, S.; Colthurst, T.; Ku, A.; Newburger, D.; Dijamco, J.; Nguyen, N.; Afshar, P.T.; et al. A universal SNP and small-indel variant caller using deep neural networks. Nat. Biotechnol. 2018, 36, 983–987. [Google Scholar] [CrossRef]
- Yun, T.; Li, H.; Chang, P.-C.; Lin, M.F.; Carroll, A.; McLean, C.Y. Accurate, scalable cohort variant calls using DeepVariant and GLnexus. Bioinformatics 2020, 36, 5582–5589. [Google Scholar] [CrossRef]
- Danecek, P.; Bonfield, J.K.; Liddle, J.; Marshall, J.; Ohan, V.; Pollard, M.O.; Whitwham, A.; Keane, T.; McCarthy, S.A.; Davies, R.M.; et al. Twelve years of SAMtools and BCFtools. GigaScience 2021, 10, giab008. [Google Scholar] [CrossRef]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.R.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.W.; Daly, M.J.; et al. PLINK: A Tool Set for Whole-Genome Association and Population-Based Linkage Analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Z.; Goddard, M.E.; Hayes, B.J.; Reinhardt, F.; Reents, R. Technical note: Equivalent genomic models with a residual polygenic effect. J. Dairy Sci. 2016, 99, 2016–2025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dennis, G.; Sherman, B.T.; Hosack, D.A.; Yang, J.; Gao, W.; Lane, H.C.; Lempicki, R.A. DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol. 2003, 4, R60. [Google Scholar] [CrossRef] [Green Version]
- Harris, C.R.; Millman, K.J.; van der Walt, S.J.; Gommers, R.; Virtanen, P.; Cournapeau, D.; Wieser, E.; Taylor, J.; Berg, S.; Smith, N.J.; et al. Array programming with NumPy. Nature 2020, 585, 357–362. [Google Scholar] [CrossRef]
- Thiecke, M.J.; Yang, E.J.; Burren, O.S.; Ray-Jones, H.; Spivakov, M. Prioritisation of Candidate Genes Underpinning COVID-19 Host Genetic Traits Based on High-Resolution 3D Chromosomal Topology. Front. Genet. 2021, 12, 745672. [Google Scholar] [CrossRef] [PubMed]
- Falzone, L.; Gattuso, G.; Tsatsakis, A.; Spandidos, D.A.; Libra, M. Current and innovative methods for the diagnosis of COVID-19 infection (Review). Int. J. Mol. Med. 2021, 47, 100. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Chromosome | Position (bp) | Gene ID | Gene Name | Mutation | Genomic Annotation | SNP ID | Median DP | SD |
|---|---|---|---|---|---|---|---|---|
| 2 | 203709432 | ENSG00000178562 | CD28 | G>A | intron | rs984981241 | 28 | 4.145465441 |
| 3 | 119551547 | ENSG00000121594 | CD80 | G>A | intron | novel | 26 | 4.063210675 |
| 3 | 122057921 | ENSG00000114013 | CD86 | G>A | intron | rs186115804 | 30 | 5.143266314 |
| 6 | 29725272 | ENSG00000204642 | HLA-F | C>T | exon | rs374197706 | 33 | 6.119863999 |
| 6 | 29827086 | ENSG00000204632 | HLA-G | T>C | intron | rs538982928 | 33 | 6.636027363 |
| 6 | 29945505 | ENSG00000206503 | HLA-A | C>T | 3′UTR | rs746262450 | 32 | 10.17326727 |
| 6 | 30493031 | ENSG00000204592 | HLA-E | T>C | 3′UTR | rs192326720 | 29 | 5.059734473 |
| 6 | 31271736 | ENSG00000204525 | HLA-C | C>T | exon | rs41548913 | 39 | 8.517438111 |
| 6 | 31356411 | ENSG00000234745 | HLA-B | G>A | exon | rs151341222 | 43 | 9.575215658 |
| 6 | 31576590 | ENSG00000232810 | TNFa | C>T | intron | rs763838774 | 28 | 4.933491701 |
| 6 | 32441500 | ENSG00000204287 | HLA-DRA | T>G | intron | rs1338070938 | 35 | 6.726123187 |
| 6 | 32528966 | ENSG00000198502 | HLA-DRB5 | C>T | intron | rs1168566689 | 39 | 12.87413207 |
| 6 | 32583693 | ENSG00000196126 | HLA-DRB1 | T>TC | intron | novel | 31 | 8.339663173 |
| 6 | 32643698 | ENSG00000196735 | HLA-DQA1 | A>G | Exon of a non-coding transcript | rs1459153928 | 36 | 7.155148333 |
| 6 | 32664633 | ENSG00000179344 | HLA-DQB1 | C>T | intron | rs1184841282 | 36 | 7.686703581 |
| 6 | 32746600 | ENSG00000237541 | HLA-DQA2 | GAGA>G | 3′UTR | novel | 29 | 5.463038801 |
| 6 | 32813318 | ENSG00000241106 | HLA-DOB | CAG>C | intron | novel | 25 | 4.576624558 |
| 6 | 32935042 | ENSG00000242574 | HLA-DMB | G>A | intron | rs779280356 | 25 | 4.040525561 |
| 6 | 32964115 | ENSG00000204257 | HLA-DMA | T>C | intron | novel | 27 | 4.697217326 |
| 6 | 33009100 | ENSG00000204252 | HLA-DOA | G>A | intron | Novel | 33 | 5.700484494 |
| 6 | 33078874 | ENSG00000231389 | HLA-DPA1 | G>A | intron | rs146322130 | 28 | 5.622143954 |
| 6 | 33086775 | ENSG00000223865 | HLA-DPB1 | CTGTT>C | 3′UTR | novel | 31 | 8.032764829 |
| 10 | 70599302 | ENSG00000180644 | PRF1 | G>A | intron | novel | 31 | 4.807858254 |
| 10 | 88956063 | ENSG00000026103 | FAS | T>C | intron | novel | 28 | 4.145442918 |
| 12 | 68156786 | ENSG00000111537 | IFNG | C>T | intron | rs745989394 | 28 | 4.229532615 |
| 20 | 46121566 | ENSG00000101017 | CD40 | G>A | intron | novel | 26 | 3.808906036 |
| X | 136659930 | ENSG00000102245 | CD40L | C>T | 3′UTR | rs879041317 | 12 | 6.017283757 |
| Cluster of Differentiation Protein | Function | References |
|---|---|---|
| CD28 | is one of the proteins expressed on T cells, providing costimulatory signals required for T cell activation and survival; provides a potent signal for the production of various interleukins, especially IL-6; molecules CD80 and CD86 are its ligands; the activity of CD80–CD28 complex stimulates the activation of transcription factors NF-κB, promoting IL-2 production | [25,26,27,28,29] |
| CD40 | is a costimulatory protein, a member of the TNF superfamily, constitutively expressed on B cells and antigen-presenting cells; CD40 binds its ligand CD40L, which is transiently expressed on T cells and other non-immune cells under inflammatory conditions; essential in mediating a broad variety of immune and inflammatory responses including T cell-dependent immunoglobulin class switching, germinal centre formation memory B cell development, to name just a few | [22,23] |
| CD80 | is an immunoglobulin, also a ligand for cytotoxic T-lymphocyte antigen 4 (CTLA-4, also known as CD152), which remains constitutively expressed on most of the T cells; present at APCs and their receptors present on the T cells; present specifically on dendritic cells, activated B cells, and macrophages, but also T cells; malfunctioning CD80 molecules are also involved in some pathological conditions, such as lupus erythematosus | [22,25,26,27,28,29] |
| CD86 | is a costimulatory protein, immunoglobulin, constitutively expressed on dendritic cells, pancreatic Langerhans cells, macrophages, B cells (including memory B cells), and on other antigen-presenting cells; provides costimulatory signals crucial for T cell activation and survival; it is also associated with myocarditis and gallbladder squamous cell carcinoma | [22,25,26,27,28,29] |
| CD152 | also known as CTLA-4 (cytotoxic T-lymphocyte-associated protein); it is a receptor that functions as an immune checkpoint protein and downregulates immune responses; it is constitutively expressed on regulatory T cells but found to be upregulated in conventional T cells after activation, being a phenomenon particularly significant in cancers, thus, being important as a background of immunotherapy utilising checkpoint inhibitors | [25,27,29] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Szyda, J.; Dobosz, P.; Stojak, J.; Sypniewski, M.; Suchocki, T.; Kotlarz, K.; Mroczek, M.; Stępień, M.; Słomian, D.; Butkiewicz, S.; et al. Beyond GWAS—Could Genetic Differentiation within the Allograft Rejection Pathway Shape Natural Immunity to COVID-19? Int. J. Mol. Sci. 2022, 23, 6272. https://doi.org/10.3390/ijms23116272
Szyda J, Dobosz P, Stojak J, Sypniewski M, Suchocki T, Kotlarz K, Mroczek M, Stępień M, Słomian D, Butkiewicz S, et al. Beyond GWAS—Could Genetic Differentiation within the Allograft Rejection Pathway Shape Natural Immunity to COVID-19? International Journal of Molecular Sciences. 2022; 23(11):6272. https://doi.org/10.3390/ijms23116272
Chicago/Turabian StyleSzyda, Joanna, Paula Dobosz, Joanna Stojak, Mateusz Sypniewski, Tomasz Suchocki, Krzysztof Kotlarz, Magdalena Mroczek, Maria Stępień, Dawid Słomian, Sławomir Butkiewicz, and et al. 2022. "Beyond GWAS—Could Genetic Differentiation within the Allograft Rejection Pathway Shape Natural Immunity to COVID-19?" International Journal of Molecular Sciences 23, no. 11: 6272. https://doi.org/10.3390/ijms23116272
APA StyleSzyda, J., Dobosz, P., Stojak, J., Sypniewski, M., Suchocki, T., Kotlarz, K., Mroczek, M., Stępień, M., Słomian, D., Butkiewicz, S., Sztromwasser, P., Liu, J., & Król, Z. J. (2022). Beyond GWAS—Could Genetic Differentiation within the Allograft Rejection Pathway Shape Natural Immunity to COVID-19? International Journal of Molecular Sciences, 23(11), 6272. https://doi.org/10.3390/ijms23116272