
Glycan-Lectin Interactions as Novel Immunosuppression Drivers in Glioblastoma

 ,
,  , , , , , and
, , , , , and
Abstract
:1. Introduction
2. Glioblastoma and Unique Immunosuppressive Networks
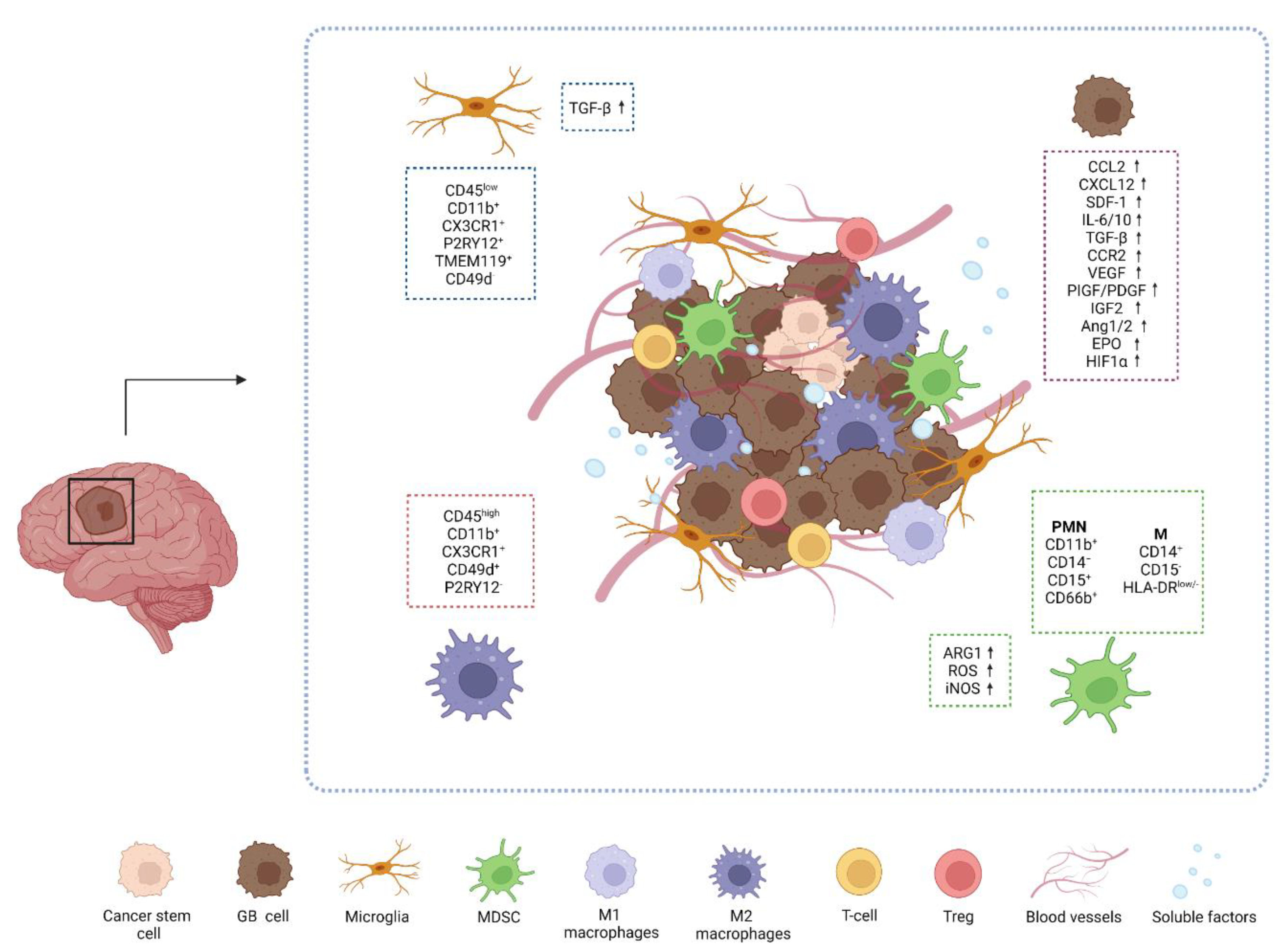
2.1. The GB Microenvironment
2.2. Immune Cells in GB Microenvironment
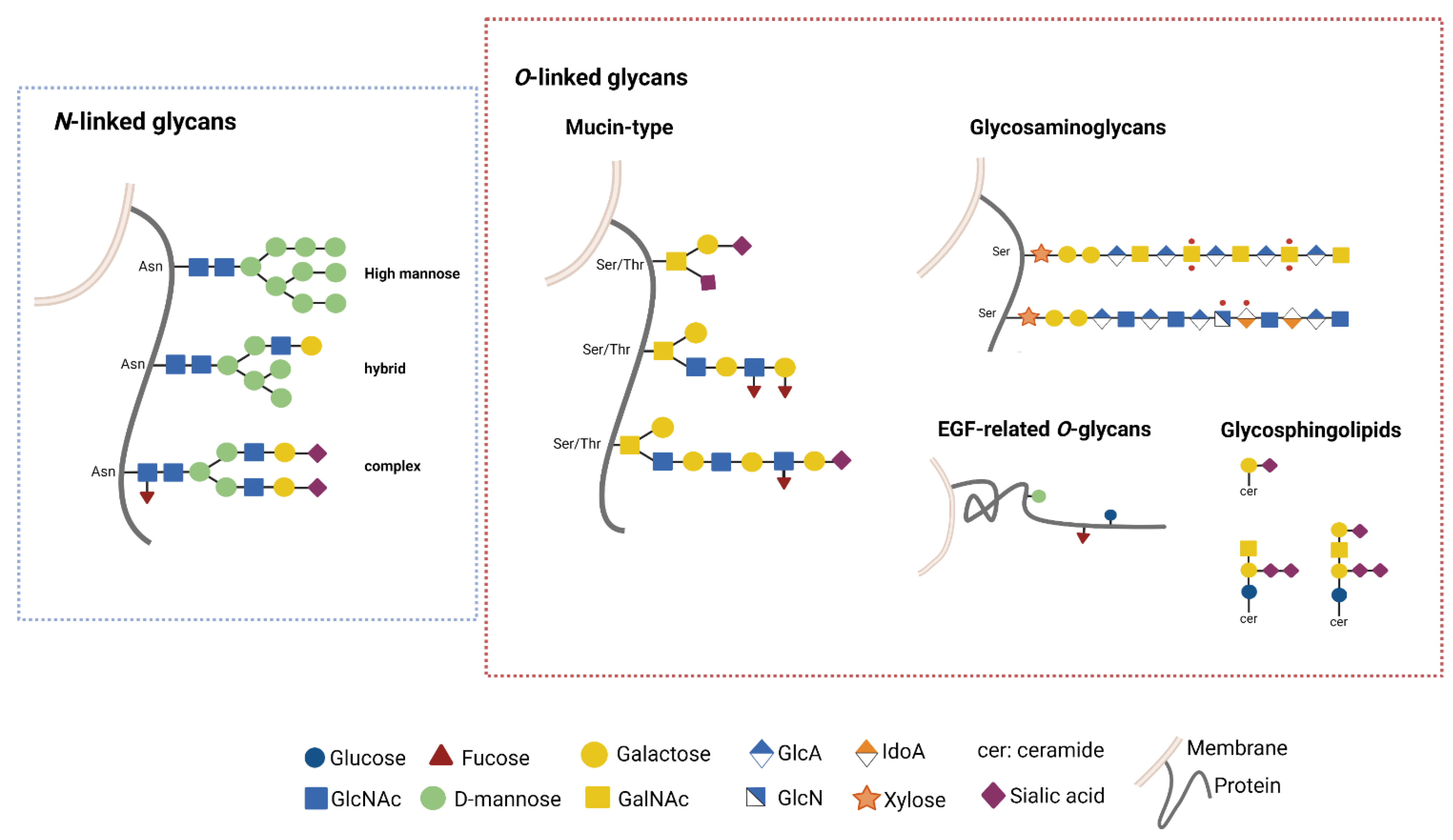
3. GB and Glycosylation Pathways
3.1. Glycosylation Pathways
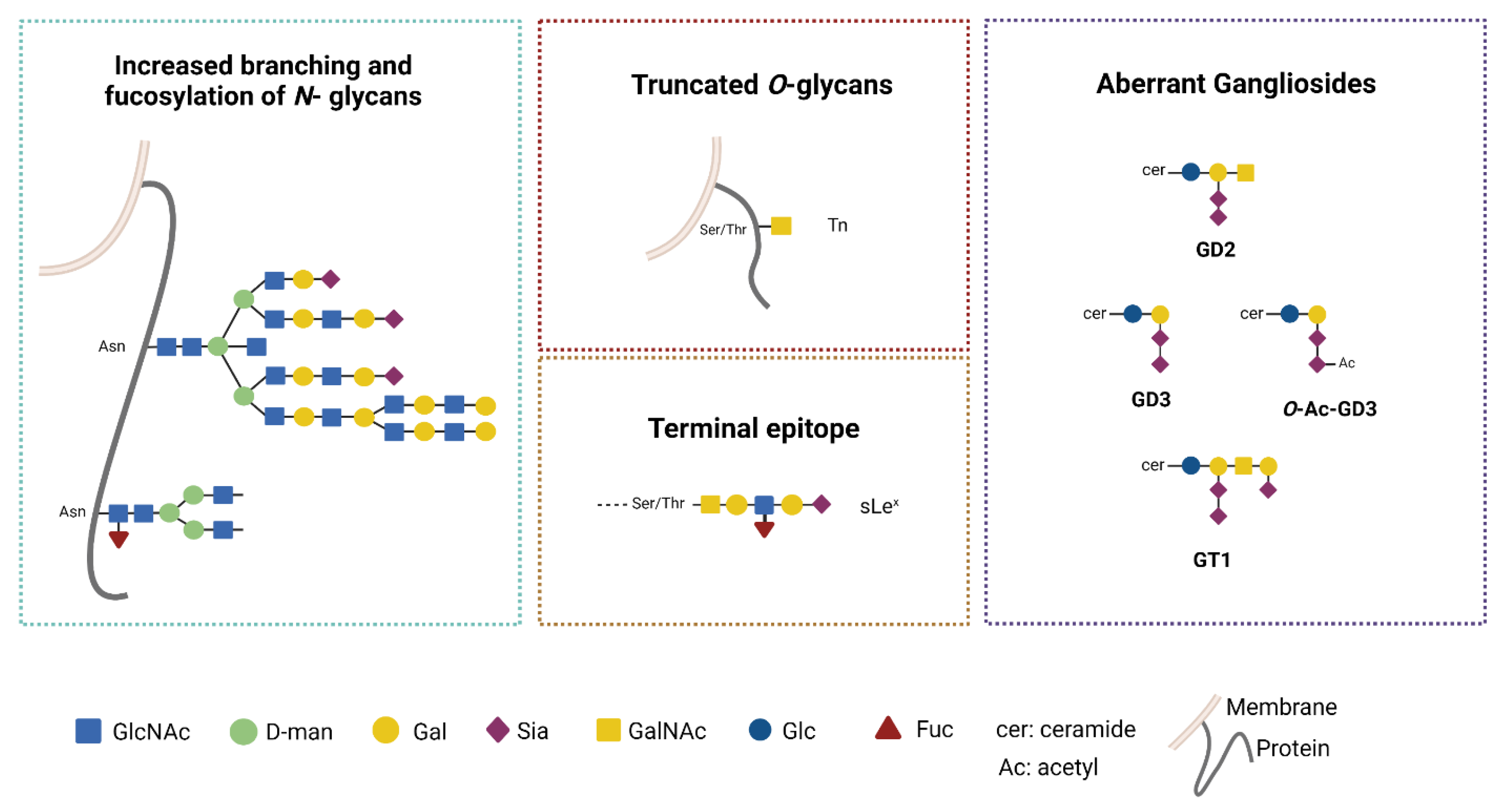
3.2. Aberrant Glycosylation Processes in GB
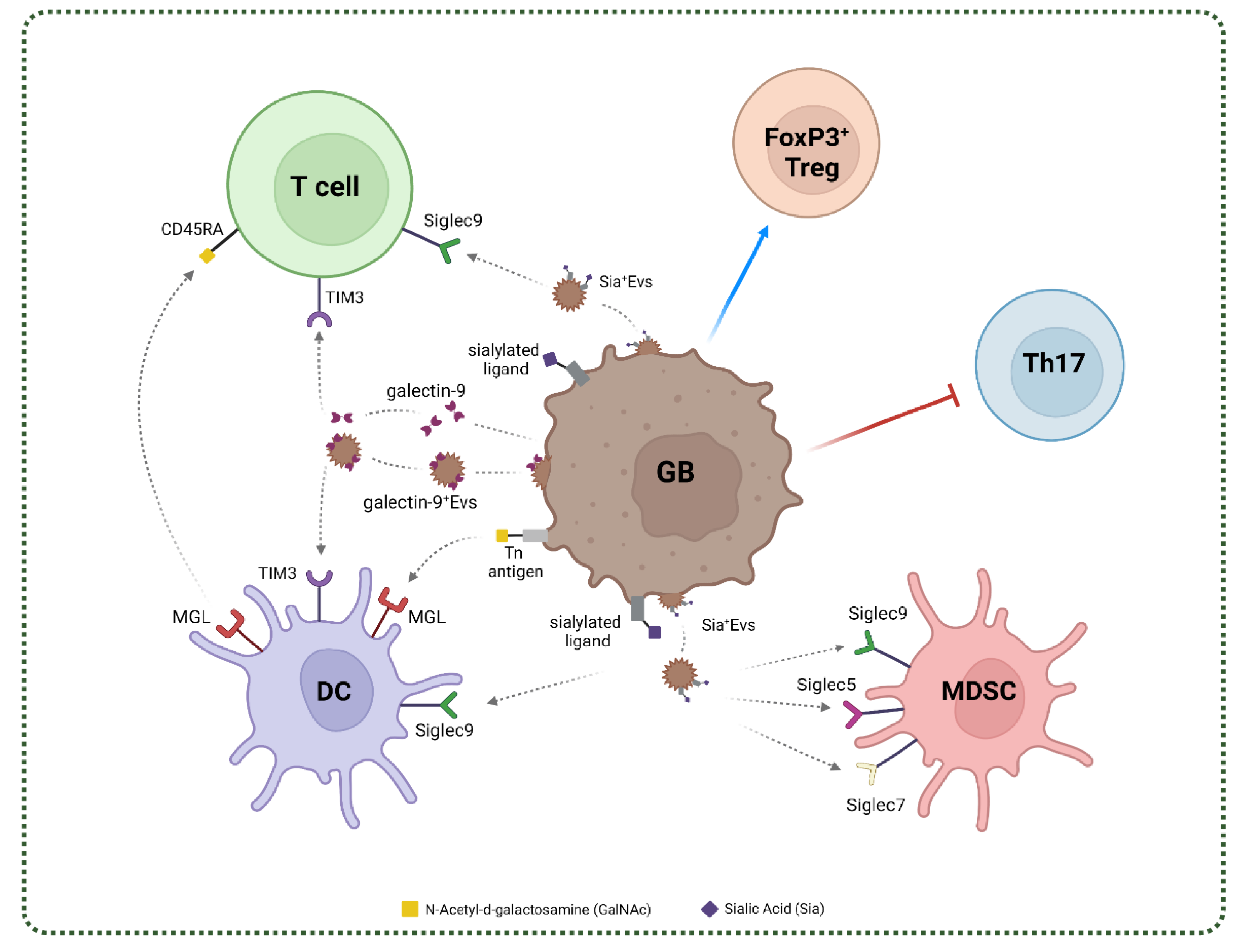
4. Glycan-Lectin Interactions and Immunosuppressive Networks in GB
4.1. C-Type Lectins in GB

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Lectin | Expression | Recognized Carbohydrate Motif | Glycosylated Ligand | Molecular Mechanism | Role in GB | Ref. |
|---|---|---|---|---|---|---|
| MGL | DC and cDC2 Macrophages CD163+ cells Activated MG | GalNAc-α-Ser/Thr | CD45RA Matrix/cell adhesion (VCAN, SDC3, PODXL NID-2, FN1, DAG1, APP, AGRN) Cell metabolism (ERP44,LAMP1/2, QSOX1, SEL1L, LRR8CD) Mucins (MUC1/16/24) | Promotes ERK phosphorylation and NfkB Enhanced secretion of IL10 Activation TLR signaling Promotes DC | TAM/CD163+ cells mediated immunosuppression | [111,112,113,118,119] |
| Siglec5 | Monocytes DC Neutrophils Macrophages | α-(2-3)-Sialic acid α-(2-6)-Sialic acid α-(2-8)-Sialic acid | GBS β-protein | ECM remodelling | MDSCs mediated immunosuppression | [63,73,104,107,120,121] |
| Siglec7 | NK cells Monocytes Macrophages Mast cells DC | α-(2-3)-Sialic acid α-(2-6)-Sialic acid α-(2-8)-Sialic acid disialogangliosides | CD43 GD3 | ECM remodelling | MDSCs mediated immunosuppression | [63,73,104,107,120,121] |
| Siglec9 | NK cells DC T cells Neutrophils Macrophages Monocytes | α-(2-3)-Sialic acid α-(2-6)-Sialic acid α-(2-8)-Sialic acid | Glycophorin Hyaluronic acid MUC1/5 | Modulation of MAPK/ERK Neutrophils inhibition/death M2 polarization ECM remodelling Inhibits macrophages phagocytosis | MDSCs mediated immunosuppression | [63,73,104,107,120,121,122,123] |
4.2. Siglecs in GB
4.3. Galectins in GB
| Lectin | Expression | Recognized Carbohydrate Motif | Glycosylated Ligand | Molecular Mechanism | Role in GB | Ref. |
|---|---|---|---|---|---|---|
| Galectin-1 | Endothelial cells Astrocytes APCs Treg | Lactose poly-N-acetyllactosamine | Immune markers (CD3, CD4, CD7, CD43, CD45, CD69) | Activation of Fas-induced death, mitochondria apoptotic pathway, VEGF-R2/NRP-1; STAT/JAK1-2; c-Jun/AP-1; Lck/ZAP-70 Differentiation of IL-27/IL-10-producing tDCs Increased expression of IL-10 and IL-21 Modulation of the c-Maf/aryl receptor pathway Loss of mitochondrial membrane potential Release of cytocrome c | Tumor progression Angiogenesis Macrophage differentiation MDSC recruitment | [21,132,135,138,139,140,141,142,143,144,145,146] |
| Galectin-3 | Endothelial cells Activated microglia Activated astrocytes Myeloid cells Fibroblasts | Lactose N-acetyllactosamine | Cell adhesion/matrix (Laminin, Vitronectin, Collagen I/IV, MCAM) Immune markers (TCR complex, CD7, CD29, CD45, CD71, LFA-1, TLR-4, LAG-3, CTLA-4) VEGF-R2 | Inhibition of NKp30 signaling pathway Interferes with MICA-NKG2D affinity Increases/impairs cell-matrix adhesion (integrins) Activation of GSK-3β, RAS/PI3K/AKT, MEK/ERK Modulation of β-catenin and RAS/Bcl-2/Myc Increases Akt activity Stabilization of TGF- βR and signaling Supports IL-6 production | Proliferation Motility Resistance to radiotherapy | [21,130,133,134,135,137,156,157,158,160,161,162] |
| Galectin-8 | Endothelial cells | α-(2-3)-Sialic acid Lactose N-acetyllactosamine | Cell adhesion/ matrix (Laminin, Fibronectin, Vitronectin, Collagen IV, CD166, Integrins α1β1/α3β1/ α5β1/α6β1) Immune markers (IL- 2R, TGF-βr, CD44) | Activation of VEGF-R2/NRP-1 and integrin-mediated signaling pathway Modulation of TGF- βR and IL-2R signaling pathway Promotes Treg differentiation and proliferation through STAT5 and Smad3 phosphorylation | Proliferation Prevent apoptosis Migration | [21,85,133,134,135,164,165,166,167,168,169] |
| Galectin-9 | Activated astrocytes Microglia Endothelial cells | Lactose N-acetyllactosamine Forssman pentasaccharide | Immune markers (TIM3, DECTIN-1, CD44, VISTA, CD274, PD-L1, IDO1, LAG3) β3-interini Glut-2 | TGF-β1- induced Treg differentiation Activation of VEGF-R2/NRP-1 pathway Promotes Smad3 phosphorylation Promote expansion of CD11b+Ly-6G+ MDSCs Upregulation of caspase-1, Granzyme B and Bid Downregulation of Lck and Bat3 signaling | Block T helper 17 Expansion of FoxP3+ Treg Apoptosis T cell exhaustion M2 polarization | [21,133,134,135,152,153,154,155] |
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ferris, S.P.; Hofmann, J.W.; Solomon, D.A.; Perry, A. Characterization of Gliomas: From Morphology to Molecules. Virchows Arch. 2017, 471, 257–269. [Google Scholar] [CrossRef] [PubMed]
- Louis, D.N.; Perry, A.; Wesseling, P.; Brat, D.J.; Cree, I.A.; Figarella-Branger, D.; Hawkins, C.; Ng, H.K.; Pfister, S.M.; Reifenberger, G.; et al. The 2021 WHO Classification of Tumors of the Central Nervous System: A Summary. Neuro Oncol. 2021, 23, 1231–1251. [Google Scholar] [CrossRef] [PubMed]
- Tan, A.C.; Ashley, D.M.; López, G.Y.; Malinzak, M.; Friedman, H.S.; Khasraw, M. Management of Glioblastoma: State of the Art and Future Directions. CA Cancer J. Clin. 2020, 70, 299–312. [Google Scholar] [CrossRef] [PubMed]
- Friedman, H.S.; Prados, M.D.; Wen, P.Y.; Mikkelsen, T.; Schiff, D.; Abrey, L.E.; Yung, W.K.A.; Paleologos, N.; Nicholas, M.K.; Jensen, R.; et al. Bevacizumab Alone and in Combination with Irinotecan in Recurrent Glioblastoma. J. Clin. Oncol. 2009, 27, 4733–4740. [Google Scholar] [CrossRef] [Green Version]
- Yang, K.; Wu, Z.; Zhang, H.; Zhang, N.; Wu, W.; Wang, Z.; Dai, Z.; Zhang, X.; Zhang, L.; Peng, Y.; et al. Glioma Targeted Therapy: Insight into Future of Molecular Approaches. Mol. Cancer 2022, 21, 39. [Google Scholar] [CrossRef]
- Liu, S.; Shi, W.; Zhao, Q.; Zheng, Z.; Liu, Z.; Meng, L.; Dong, L.; Jiang, X. Progress and Prospect in Tumor Treating Fields Treatment of Glioblastoma. Biomed. Pharmacother. 2021, 141, 111810. [Google Scholar] [CrossRef]
- Sampson, J.H.; Gunn, M.; Fecci, P.E.; Ashley, D.M. Brain Immunology and Immunotherapy in Brain Tumours John. Nat. Rev. Cancer 2020, 20, 12–25. [Google Scholar] [CrossRef]
- Tomaszewski, W.; Al, E. Brain Tumor Micro-Environment and Host State—Implications for Immunotherapy. Clin. Cancer Res. 2019, 25, 4202–4210. [Google Scholar] [CrossRef] [Green Version]
- Keskin, D.B.; Anandappa, A.J.; Sun, J.; Tirosh, I.; Mathewson, N.D.; Li, S.; Oliveira, G.; Giobbie-Hurder, A.; Felt, K.; Gjini, E.; et al. Neoantigen Vaccine Generates Intratumoral T Cell Responses in Phase Ib Glioblastoma Trial. Nature 2019, 565, 234–239. [Google Scholar] [CrossRef]
- Jackson, C.M.; Choi, J.; Lim, M. Mechanisms of Immunotherapy Resistance: Lessons from Glioblastoma. Nat. Immunol. 2019, 20, 1100–1109. [Google Scholar] [CrossRef]
- Basheer, A.S.; Abas, F.; Othman, I.; Naidu, R. Role of Inflammatory Mediators, Macrophages, and Neutrophils in Glioma Maintenance and Progression: Mechanistic Understanding and Potential Therapeutic Applications. Cancers 2021, 13, 4226. [Google Scholar] [CrossRef] [PubMed]
- Yeo, E.C.F.; Brown, M.P.; Gargett, T.; Ebert, L.M. The Role of Cytokines and Chemokines in Shaping the Immune Microenvironment of Glioblastoma: Implications for Immunotherapy. Cells 2021, 10, 607. [Google Scholar] [CrossRef] [PubMed]
- Brown, N.F.; Carter, T.J.; Ottaviani, D.; Mulholland, P. Harnessing the Immune System in Glioblastoma. Br. J. Cancer 2018, 119, 1171–1181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Leo, A.; Ugolini, A.; Veglia, F. Myeloid Cells in Glioblastoma Microenvironment. Cells 2020, 10, 18. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, J.G.; Balmaña, M.; Macedo, J.A.; Al, E. Glycosylation in Cancer: Selected Roles in Tumour Progression, Immune Modulation and Metastasis. Cell. Immunol. 2018, 333, 46–57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ho, W.L.; Hsu, W.M.; Huang, M.C.; Kadomatsu, K.; Nakagawara, A. Protein Glycosylation in Cancers and Its Potential Therapeutic Applications in Neuroblastoma. J. Hematol. Oncol. 2016, 9, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Fu, C.; Zhao, H.; Wang, Y.; Cai, H.; Xiao, Y.; Zeng, Y.; Chen, H. Tumor-Associated Antigens: Tn Antigen, STn Antigen, and T Antigen. Hla 2016, 88, 275–286. [Google Scholar] [CrossRef]
- Veillon, L.; Fakih, C.; Abou-El-Hassan, H.; Kobeissy, F.; Mechref, Y. Glycosylation Changes in Brain Cancer. ASC Chem. Neurosci. 2018, 9, 51–52. [Google Scholar] [CrossRef]
- Kremsreiter, S.M.; Kroell, A.H.; Weinberger, K.; Boehm, H. Glycan—Lectin Interactions in Cancer and Viral Infections and How to Disrupt Them. Int. J. Mol. Sci. 2021, 22, 10577. [Google Scholar] [CrossRef]
- Pillai, S.; Netravali, I.A.; Cariappa, A.; Mattoo, H. Siglecs and Immune Regulation. Annu. Rev. Immunol. 2012, 30, 357–392. [Google Scholar] [CrossRef] [Green Version]
- Cedeno-Laurent, F. Galectins and Their Ligands: Negative Regulators of Anti-Tumor Immunity. Glycoconj J. 2012, 29, 619–625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drickamer, K.; Taylor, M.E. Recent Insights into Structures and Functions of C-Type Lectins in the Immune System. Curr. Opin. Struct. Biol. 2015, 34, 26–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mayer, S.; Raulf, M.K.; Lepenies, B. C-Type Lectins: Their Network and Roles in Pathogen Recognition and Immunity. Histochem. Cell Biol. 2017, 147, 223–237. [Google Scholar] [CrossRef] [PubMed]
- Spill, F.; Reynolds, D.S.; Kamm, R.D.; Zaman, M.H. Impact of the Physical Microenvironment on Tumor Progression and Metastasis. Curr. Opin. Biotechnol. 2016, 40, 41–48. [Google Scholar] [CrossRef] [Green Version]
- Khalaf, K.; Hana, D.; Chou, J.T.-T.; Singh, C.; Mackiewicz, A.; Kaczmarek, M. Aspects of the Tumor Microenvironment Involved in Immune Resistance and Drug Resistance. Front. Immunol. 2021, 12, 656364. [Google Scholar] [CrossRef]
- Di Cintio, F.; Dal Bo, M.; Baboci, L.; De Mattia, E.; Polano, M.; Toffoli, G. The Molecular and Microenvironmental Landscape of Glioblastomas: Implications for the Novel Treatment Choices. Front. Neurosci. 2020, 14, 603647. [Google Scholar] [CrossRef]
- Colwell, N.; Larion, M.; Giles, A.J.; Seldomridge, A.N.; Sizdahkhani, S.; Gilbert, M.R.; Park, D.M. Hypoxia in the Glioblastoma Microenvironment: Shaping the Phenotype of Cancer Stem-like Cells. Neuro Oncol. 2017, 19, 887–896. [Google Scholar] [CrossRef]
- Soeda, A.; Park, M.; Lee, D.; Mintz, A.; Androutsellis-Theotokis, A.; McKay, R.D.; Engh, J.; Iwama, T.; Kunisada, T.; Kassam, A.B.; et al. Hypoxia Promotes Expansion of the CD133-Positive Glioma Stem Cells through Activation of HIF-1α. Oncogene 2009, 28, 3949–3959. [Google Scholar] [CrossRef] [Green Version]
- Dom, M.; Hern, A.; Plaja, A.; Mart, E. Hypoxia: The Cornerstone of Glioblastoma. Int. J. Mol. Sci. 2021, 22, 12608. [Google Scholar]
- Silva-Filho, A.F.; Sena, W.L.B.; Lima, L.R.A.; Carvalho, L.V.N.; Pereira, M.C.; Santos, L.G.S.; Santos, R.V.C.; Tavares, L.B.; Pitta, M.G.R.; Rêgo, M.J.B.M. Glycobiology Modifications in Intratumoral Hypoxia: The Breathless Side of Glycans Interaction. Cell. Physiol. Biochem. 2017, 41, 1801–1829. [Google Scholar] [CrossRef]
- Kaur, B.; Khwaja, F.W.; Severson, E.A.; Matheny, S.L.; Brat, D.J.; Van Meir, E.G. Hypoxia and the Hypoxia-Inducible-Factor Pathway in Glioma Growth and Angiogenesis. Neuro Oncol. 2005, 7, 134–153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carmeliet, P. VEGF as a Key Mediator of Angiogenesis in Cancer. Oncology 2005, 69, 4–10. [Google Scholar] [CrossRef] [PubMed]
- Duru, G.; van Egmond, M.; Heemskerk, N. A Window of Opportunity: Targeting Cancer Endothelium to Enhance Immunotherapy. Front. Immunol. 2020, 11, 584723. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; He, Z.; Duan, H.; Zhang, D.; Li, J.; Yang, H.; Dorsey, J.F.; Zou, W.; Ali Nabavizadeh, S.; Bagley, S.J.; et al. Synergistic Immunotherapy of Glioblastoma by Dual Targeting of IL-6 and CD40. Nat. Commun. 2021, 12, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Louveau, A.; Smirnov, I.; Keyes, T.J.; Eccles, J.D.; Sherin, J.; Peske, J.D.; Derecki, N.C.; Castle, D.; Mandell, J.W.; Kevin, L.; et al. Structural and Functional Features of Central Nervous System Lymphatics. Nature 2015, 523, 337–341. [Google Scholar] [CrossRef]
- Eder, K.; Kalman, B. The Dynamics of Interactions Among Immune and Glioblastoma Cells. Neuro Mol. Med. 2015, 17, 335–352. [Google Scholar] [CrossRef]
- Zhang, B.; Shen, R.; Cheng, S.; Feng, L. Immune Microenvironments Differ in Immune Characteristics and Outcome of Glioblastoma Multiforme. Cancer Med. 2019, 8, 2897–2907. [Google Scholar] [CrossRef]
- DeCordova, S.; Shastri, A.; Tsolaki, A.G.; Yasmin, H.; Klein, L.; Singh, S.K.; Kishore, U. Molecular Heterogeneity and Immunosuppressive Microenvironment in Glioblastoma. Front. Immunol. 2020, 11, 1402. [Google Scholar] [CrossRef]
- Chen, Z.; Feng, X.; Herting, C.J.; Garcia, V.A.; Nie, K.; Pong, W.W.; Rasmussen, R.; Dwivedi, B.; Seby, S.; Wolf, S.A.; et al. Cellular and Molecular Identity of Tumor-Associated Macrophages in Glioblastoma. Cancer Res. 2017, 77, 2266–2278. [Google Scholar] [CrossRef] [Green Version]
- Pinton, L.; Masetto, E.; Vettore, M.; Solito, S.; Magri, S.; D’Andolfi, M.; Del Bianco, P.; Lollo, G.; Benoit, J.P.; Okada, H.; et al. The Immune Suppressive Microenvironment of Human Gliomas Depends on the Accumulation of Bone Marrow-Derived Macrophages in the Center of the Lesion. J. Immunother. Cancer 2019, 7, 58. [Google Scholar] [CrossRef]
- Brandenburg, S.; Blank, A.; Bungert, A.D.; Vajkoczy, P. Distinction of Microglia and Macrophages in Glioblastoma: Close Relatives, Different Tasks? Int. J. Mol. Sci. 2021, 22, 194. [Google Scholar] [CrossRef] [PubMed]
- Geribaldi-Doldán, N.; Fernández-Ponce, C.; Quiroz, R.N.; Sánchez-Gomar, I.; Escorcia, L.G.; Velásquez, E.P.; Quiroz, E.N. The Role of Microglia in Glioblastoma. Front. Oncol. 2021, 10, 603495. [Google Scholar] [CrossRef] [PubMed]
- Daubon, T.; Hemadou, A.; Romero Garmendia, I.; Saleh, M. Glioblastoma Immune Landscape and the Potential of New Immunotherapies. Front. Immunol. 2020, 11, 585616. [Google Scholar] [CrossRef] [PubMed]
- Satoh, J.; Kino, Y.; Asahina, N.; Takitani, M.; Miyoshi, J.; Ishida, T.; Saito, Y. TMEM119 Marks a Subset of Microglia in the Human Brain. Neuropathology 2016, 36, 39–49. [Google Scholar] [CrossRef] [PubMed]
- Crane, C.A.; Ahn, B.J.; Han, S.J.; Parsa, A.T. Soluble Factors Secreted by Glioblastoma Cell Lines Facilitate Recruitment, Survival, and Expansion of Regulatory T Cells: Implications for Immunotherapy. Neuro. Oncol. 2012, 14, 584–595. [Google Scholar] [CrossRef] [PubMed]
- Buonfiglioli, A.; Hambardzumyan, D. Macrophages and Microglia: The Cerberus of Glioblastoma. Acta Neuropathol. Commun. 2021, 9, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Koshkaki, H.R.; Minasi, S.; Ugolini, A.; Trevisi, G.; Napoletano, C.; Zizzari, I.G.; Gessi, M.; Giangaspero, F.; Mangiola, A.; Nuti, M.; et al. Immunohistochemical Characterization of Immune Infiltrate in Tumor Microenvironment of Glioblastoma. J. Pers. Med. 2020, 10, 112. [Google Scholar] [CrossRef]
- Pyonteck, S.M.; Akkari, L.; Schuhmacher, A.J.; Bowman, R.L.; Sevenich, L.; Quail, D.F.; Olson, O.C.; Quick, M.L.; Huse, J.T.; Teijeiro, V.; et al. CSF-1R Inhibition Alters Macrophage Polarization and Blocks Glioma Progression. Nat. Med. 2013, 19, 1264–1272. [Google Scholar] [CrossRef] [Green Version]
- Lindau, D.; Gielen, P.; Kroesen, M.; Wesseling, P.; Adema, G.J. The Immunosuppressive Tumour Network: Myeloid-Derived Suppressor Cells, Regulatory T Cells and Natural Killer T Cells. Immunology 2013, 138, 105–115. [Google Scholar] [CrossRef]
- Lakshmanachetty, S.; Cruz-Cruz, J.; Hoffmeyer, E.; Cole, A.P.; Mitra, S.S. New Insights into the Multifaceted Role of Myeloid-Derived Suppressor Cells (MDSCs) in High-Grade Gliomas: From Metabolic Reprograming, Immunosuppression, and Therapeutic Resistance to Current Strategies for Targeting MDSCs. Cells 2021, 10, 893. [Google Scholar] [CrossRef]
- Bayik, D.; Zhou, Y.; Park, C.; Hong, C.; Vail, D.; Silver, D.J.; Lauko, A.; Roversi, G.; Watson, D.C.; Lo, A.; et al. Myeloid-Derived Suppressor Cell Subsets Drive Glioblastoma Growth in a Sex.-Specific Manner. Cancer Discov. 2020, 10, 1210–1225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chai, E.; Zhang, L.; Li, C. LOX-1+ PMN-MDSC Enhances Immune Suppression Which Promotes Glioblastoma Multiforme Progression. Cancer Manag. Res. 2019, 11, 7307–7315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dubinski, D.; Wölfer, J.; Hasselblatt, M.; Schneider-Hohendorf, T.; Bogdahn, U.; Stummer, W.; Wiendl, H.; Grauer, O.M. CD4+ T Effector Memory Cell Dysfunction Is Associated with the Accumulation of Granulocytic Myeloid-Derived Suppressor Cells in Glioblastoma Patients. Neuro Oncol. 2016, 18, 807–818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parizi, M.S.; Parizi, F.S.; Abdolhosseini, S.; Vanaei, S.; Manzouri, A.; Ebrahimzadeh, F. Myeloid-Derived Suppressor Cells (MDSCs) in Brain Cancer: Challenges and Therapeutic Strategies. Inflammopharmacology 2021, 29, 1613–1624. [Google Scholar] [CrossRef]
- Raychaudhuri, B.; Rayman, P.; Huang, P.; Grabowski, M.; Hambardzumyan, D.; Finke, J.H.; Vogelbaum, M.A. Myeloid Derived Suppressor Cell Infiltration of Murine and Human Gliomas Is Associated with Reduction of Tumor Infiltrating Lymphocytes. J. Neurooncol. 2015, 122, 293–301. [Google Scholar] [CrossRef]
- Raychaudhuri, B.; Ireland, P.R.J.; Ko, J.; Rini, B.; Borden, E.C.; Garcia, J.; Vogelbaum, M.A.; Finke, J. Myeloid-Derived Suppressor Cell Accumulation and Function in Patients with Newly Diagnosed Glioblastoma. Neuro Oncol. 2011, 13, 591–599. [Google Scholar] [CrossRef] [Green Version]
- Del Bianco, P.; Pinton, L.; Magri, S.; Canè, S.; Masetto, E.; Basso, D.; Padovan, M.; Volpin, F.; d’Avella, D.; Lombardi, G.; et al. Myeloid Diagnostic and Prognostic Markers of Immune Suppression in the Blood of Glioma Patients. Front. Immunol. 2022, 12, 809826. [Google Scholar] [CrossRef]
- Gielen, P.R.; Schulte, B.M.; Kers-Rebel, E.D.; Verrijp, K.; Petersen-Baltussen, H.M.J.M.; Ter Laan, M.; Wesseling, P.; Adema, G.J. Increase in Both CD14-Positive and CD15-Positive Myeloid-Derived Suppressor Cell Subpopulations in the Blood of Patients with Glioma but Predominance of CD15-Positive Myeloid-Derived Suppressor Cells in Glioma Tissue. J. Neuropathol. Exp. Neurol. 2015, 74, 390–400. [Google Scholar] [CrossRef] [Green Version]
- Condamine, T.; Gabrilovich, D.I.; Dominguez, G.A.; Youn, J.-I.; Kossenkov, A.V.; Mony, S.; Alicea-Torres, K.T.; Tcyganov, E.; Hashimoto, A.; Nefedova, Y.; et al. Lectin-Type Oxidized LDL Receptor-1 Distinguishes Population of Human Polymorphonuclear Myeloid-Derived Suppressor Cells in Cancer Patients. Sci. Immunol. 2016, 1, aaf8943. [Google Scholar] [CrossRef] [Green Version]
- Schjoldager, K.T.; Narimatsu, Y.; Joshi, H.J.; Clausen, H. Global View of Human Protein Glycosylation Pathways and Functions. Nat. Rev. Mol. Cell Biol. 2020, 21, 729–749. [Google Scholar] [CrossRef]
- Zhang, X.; Wang, Y. Glycosylation Quality Control by the Golgi Structure. J. Mol. Biol. 2016, 428, 3183–3193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varki, A. Biological Roles of Glycans. Glycobiology 2017, 27, 3–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johannssen, T.; Lepenies, B. Glycan-Based Cell Targeting to Modulate Immune Responses. Trends Biotechnol. 2017, 35, 334–346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, X.; Qian, K. Protein O-GlcNAcylation: Emerging Mechanisms and Functions. Nat. Rev. Mol. Cell Biol. 2017, 18, 452–465. [Google Scholar] [CrossRef]
- Schwarz, F.; Aebi, M. Mechanisms and Principles of N-Linked Protein Glycosylation. Curr. Opin. Struct. Biol. 2011, 21, 576–582. [Google Scholar] [CrossRef]
- Cherepanova, N.; Al, E. N-Linked Glycosylation and Homeostasis of the Endoplasmic Reticulum. Curr. Opin. Cell Biol. 2016, 41, 57–65. [Google Scholar] [CrossRef] [Green Version]
- Angelo, G.D.; Capasso, S.; Sticco, L.; Russo, D. Glycosphingolipids: Synthesis and Functions. FEBS J. 2013, 280, 6338–6353. [Google Scholar] [CrossRef]
- Yu, R.K.; Tsai, Y.T.; Ariga, T.; Yanagisawa, M. Structures, Biosynthesis, and Functions of Gangliosides-an Overview. J. Oleo Sci. 2011, 60, 537–544. [Google Scholar] [CrossRef] [Green Version]
- Sipione, S.; Monyror, J.; Galleguillos, D.; Steinberg, N.; Kadam, V. Gangliosides in the Brain: Physiology, Pathophysiology and Therapeutic Applications. Front. Neurosci. 2020, 14, 572965. [Google Scholar] [CrossRef]
- Varki, A. Glycan-Based Interactions Involving Vertebrate Sialic-Acid-Recognizing Proteins. Nature 2007, 446, 1023–1029. [Google Scholar] [CrossRef]
- Alves, I.; Fernandes, Â.; Santos-Pereira, B.; Azevedo, C.M.; Pinho, S.S. Glycans as a Key Factor in Self and Non-self Discrimination: Impact on the Breach of Immune Tolerance. FEBS Lett. 2022, 1–18. [Google Scholar] [CrossRef]
- Reily, C.; Stewart, T.J.; Renfrow, M.B.; Novak, J. Glycosylation in Health and Disease. Nat. Rev. Nephrol 2019, 15, 346–366. [Google Scholar] [CrossRef] [PubMed]
- Mantuano, N.R.; Natoli, M.; Zippelius, A.; Läubli, H. Tumor-Associated Carbohydrates and Immunomodulatory Lectins as Targets for Cancer Immunotherapy. J. Immunother. Cancer 2020, 8, e001222. [Google Scholar] [CrossRef] [PubMed]
- Tondepu, C.; Karumbaiah, L. Glycomaterials to Investigate the Functional Role of Aberrant Glycosylation in Glioblastoma. Adv. Healthc. Mater. 2022, 11, 2101956. [Google Scholar] [CrossRef]
- Li, Y.; Liu, Y.; Zhu, H.; Chen, X.; Tian, M.; Wei, Y.; Gong, Y.; Jiang, J. N-Acetylglucosaminyltransferase I Promotes Glioma Cell Proliferation and Migration through Increasing the Stability of the Glucose Transporter GLUT1. FEBS Lett. 2020, 594, 358–366. [Google Scholar] [CrossRef]
- Marhuenda, E.; Fabre, C.; Zhang, C.; Martin-Fernandez, M.; Iskratsch, T.; Saleh, A.; Bauchet, L.; Cambedouzou, J.; Hugnot, J.P.; Duffau, H.; et al. Glioma Stem Cells Invasive Phenotype at Optimal Stiffness Is Driven by MGAT5 Dependent Mechanosensing. J. Exp. Clin. Cancer Res. 2021, 40, 1–14. [Google Scholar] [CrossRef]
- Cuello, H.A.; Ferreira, G.M.; Gulino, C.A.; Toledo, A.G.; Segatori, V.I.; Gabri, M.R. Terminally Sialylated and Fucosylated Complex N-Glycans Are Involved in the Malignant Behavior of High-Grade Glioma. Oncotarget 2021, 11, 4822–4835. [Google Scholar] [CrossRef]
- Magalhães, A.; Duarte, H.O.; Reis, C.A. Aberrant Glycosylation in Cancer: A Novel Molecular Mechanism Controlling Metastasis. Cancer Cell 2017, 31, 733–735. [Google Scholar] [CrossRef] [Green Version]
- Olio, F.D.; Pucci, M.; Malagolini, N. The Cancer-Associated Antigens Sialyl Lewis a/x and Sd a: Two Opposite Faces of Terminal Glycosylation. Cancers 2021, 13, 5273. [Google Scholar]
- Wei, K.-C.; Lin, Y.-C.; Chen, C.-H.; Chu, Y.-H.; Huang, C.-Y.; Liao, W.-C.; Liu, C.-H. Fucosyltransferase 8 Modulates Receptor Tyrosine Kinase Activation and Temozolomide Resistance in Glioblastoma Cells. Am. J. Cancer Res. 2021, 11, 5472–5484. [Google Scholar]
- Granta, B.D.; Smithb, C.A.; Castlec, P.E.; Al, E. Oligosaccharyltransferase Inhibition Reduces Receptor Tyrosine Kinase Activation and Enhances Glioma Radiosensitivity. Clin. Cancer Red. 2019, 25, 784–795. [Google Scholar] [CrossRef]
- Chong, Y.K.; Sandanaraj, E.; Koh, L.W.H.; Thangaveloo, M.; Tan, M.S.Y.; Koh, G.R.H.; Toh, T.B.; Lim, G.G.Y.; Holbrook, J.D.; Kon, O.L.; et al. ST3GAL1-Associated Transcriptomic Program in Glioblastoma Tumor Growth, Invasion, and Prognosis. J. Natl. Cancer Inst. 2016, 108, djv326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imamaki, R.; Ogawa, K.; Kizuka, Y.; Komi, Y.; Kojima, S.; Kotani, N.; Honke, K.; Honda, T.; Taniguchi, N.; Kitazume, S. Glycosylation Controls Cooperative PECAM-VEGFR2-Β3 Integrin Functions at the Endothelial Surface for Tumor Angiogenesis. Oncogene 2018, 37, 4287–4299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kroes, R.A.; He, H.; Emmett, M.R.; Nilsson, C.L.; Leach, F.E.; Amster, I.J.; Marshall, A.G.; Moskal, J.R. Overexpression of ST6GalNAcV, a Ganglioside-Specific A2,6- Sialyltransferase, Inhibits Glioma Growth in Vivo. Proc. Natl. Acad. Sci. USA 2010, 107, 12646–12651. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, H.; Oviedo, A.; Sweeley, C.; Saito, T.; Moskal, J.R. A2, 6-Sialylation of Cell-Surface N-Glycans Inhibits Glioma Formation in Vivo. Cancer Res. 2001, 61, 6822–6829. [Google Scholar]
- Burchell, J.M.; Beatson, R.; Graham, R.; Taylor-Papadimitriou, J.; Tajadura-Ortega, V. O-Linked Mucin-Type Glycosylation in Breast Cancer. Biochem. Soc. Trans. 2018, 46, 779–788. [Google Scholar] [CrossRef]
- Lou, Y.W.; Wang, P.Y.; Yeh, S.C.; Chuang, P.K.; Li, S.T.; Wu, C.Y.; Khoo, K.H.; Hsiao, M.; Hsu, T.L.; Wong, C.H. Stage-Specific Embryonic Antigen-4 as a Potential Therapeutic Target in Glioblastoma Multiforme and Other Cancers. Proc. Natl. Acad. Sci. USA 2014, 111, 2482–2487. [Google Scholar] [CrossRef] [Green Version]
- Dusoswa, S.A.; Verhoeff, J.; Abels, E.; Méndez-Huergo, S.P.; Croci, D.O.; Kuijper, L.H.; de Miguel, E.; Wouters, V.M.C.J.; Best, M.G.; Rodriguez, E.; et al. Glioblastomas Exploit Truncated O-Linked Glycans for Local and Distant Immune Modulation via the Macrophage Galactose-Type Lectin. Proc. Natl. Acad. Sci. USA 2020, 117, 3693–3703. [Google Scholar] [CrossRef]
- Mennel, H.D.; Wiegandt, H.; Bauer, B.L.; Jennemann, R.; Rodden, A.F.; Schachenmayr, W. Tissue Architecture and Glycosphingolipid Content in Human Gliomas II–IV. Pathol. Res. Pract. 1991, 187, 157–165. [Google Scholar] [CrossRef]
- Nakamura, O.; Iwamori, M.; Matsutani, M.; Takakura, K. Ganglioside GD3 Shedding by Human Gliomas. Acta Neurochir. 1991, 109, 34–36. [Google Scholar] [CrossRef]
- Iwasawa, T.; Zhang, P.; Ohkawa, Y.; Momota, H.; Wakabayashi, T.; Ohmi, Y.; Bhuiyan, R.H.; Furukawa, K.; Furukawa, K. Enhancement of Malignant Properties of Human Glioma Cells by Ganglioside GD3/GD2. Int. J. Oncol. 2018, 52, 1255–1266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spaeth, E.L.; Dembinski, J.L.; Sasser, A.K.; Watson, K.; Klopp, A.; Hall, B.; Andreeff, M.; Marini, F. Mesenchymal Stem Cell Transition to Tumor-Associated Fibroblasts Contributes to Fibrovascular Network Expansion and Tumor Progression. PLoS ONE 2009, 4, e4992. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Head, B.P.; Al, E. Interaction of Membrane/Lipid Rafts with the Cytoskeleton: Impact on Signaling and Function: Membrane/Lipid Rafts, Mediators of Cytoskeletal Arrangement and Cell Signaling. Biochim. Biophys. Acta 2014, 1838. [Google Scholar] [CrossRef]
- Yeh, S.C.; Wang, P.Y.; Lou, Y.W.; Khoo, K.H.; Hsiao, M.; Hsu, T.L.; Wong, C.H. Glycolipid GD3 and GD3 Synthase Are Key Drivers for Glioblastoma Stem Cells and Tumorigenicity. Proc. Natl. Acad. Sci. USA 2016, 113, 5592–5597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fabris, D.; Rožman, M.; Sajko, T.; Vukelić, Ž. Aberrant Ganglioside Composition in Glioblastoma Multiforme and Peritumoral Tissue: A Mass Spectrometry Characterization. Biochimie 2017, 137, 56–68. [Google Scholar] [CrossRef] [PubMed]
- Sarbu, M.; Petrica, L.; Clemmer, D.E.; Vukelić, Ž.; Zamfir, A.D. Gangliosides of Human Glioblastoma Multiforme: A Comprehensive Mapping and Structural Analysis by Ion Mobility Tandem Mass Spectrometry. J. Am. Soc. Mass Spectrom. 2021, 32, 1249–1257. [Google Scholar] [CrossRef] [PubMed]
- Groux-Degroote, S.; Delannoy, P. Cancer-Associated Glycosphingolipids as Tumor Markers and Targets for Cancer Immunotherapy. Int. J. Mol. Sci. 2021, 22, 6145. [Google Scholar] [CrossRef]
- Belousov, A.; Titov, S.; Shved, N.; Garbuz, M.; Malykin, G.; Gulaia, V.; Kagansky, A.; Kumeiko, V. The Extracellular Matrix and Biocompatible Materials in Glioblastoma Treatment. Front. Bioeng. Biotechnol. 2019, 7, 341. [Google Scholar] [CrossRef] [Green Version]
- Barnes, J.M.; Kaushik, S.; Bainer, R.O.; Sa, J.K.; Woods, E.C.; Kai, F.B.; Przybyla, L.; Lee, M.; Lee, H.W.; Tung, J.C.; et al. A Tension-Mediated Glycocalyx–Integrin Feedback Loop Promotes Mesenchymal-like Glioblastoma. Nat. Cell Biol. 2018, 20, 1203–1214. [Google Scholar] [CrossRef]
- Ma, D.; Liu, S.; Lal, B.; Al, E. Extracellular Matrix Protein Tenascin C Increases Phagocytosis Mediated by CD47 Loss of Function in Glioblastoma. Cancer Res. 2019, 79, 2697–2708. [Google Scholar] [CrossRef] [Green Version]
- Sethi, M.K.; Downs, M.; Shao, C.; Hackett, W.E.; Phillips, J.J.; Zaia, J. In-Depth Matrisome and Glycoproteomic Analysis of Human Brain Glioblastoma Versus Control Tissue. Mol. Cell. Proteom. 2022, 21, 100216. [Google Scholar] [CrossRef] [PubMed]
- Berois, N.; Pittini, A.; Osinaga, E. Targeting Tumor Glycans for Cancer Therapy: Successes, Limitations, and Perspectives. Cancers 2022, 14, 645. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, E.; Schetters, S.T.T.; van Kooyk, Y. The Tumour Glyco-Code as a Novel Immune Checkpoint for Immunotherapy. Nat. Rev. Immunol. 2018, 18, 204–211. [Google Scholar] [CrossRef] [PubMed]
- Raposo, C.D.; Canelas, A.B.; Barros, M.T. Human Lectins, Their Carbohydrate Affinities and Where to Find Them. Biomolecules 2021, 11, 188. [Google Scholar] [CrossRef] [PubMed]
- Le Mercier, M.; Fortin, S.; Mathieu, V.; Kiss, R.; Lefranc, F. Galectins and Gliomas. Brain Pathol. 2010, 20, 17–27. [Google Scholar] [CrossRef]
- Fan, H.W.; Ni, Q.; Fan, Y.N.; Ma, Z.X.; Li, Y. Bin C-Type Lectin Domain Family 5, Member A (CLEC5A, MDL-1) Promotes Brain Glioblastoma Tumorigenesis by Regulating PI3K/Akt Signalling. Cell Prolif. 2019, 52, e12584. [Google Scholar] [CrossRef]
- Santegoets, K.C.M.; Gielen, P.R.; Büll, C.; Schulte, B.M.; Kers-Rebel, E.D.; Küsters, B.; Bossman, S.A.J.F.H.; ter Laan, M.; Wesseling, P.; Adema, G.J. Expression Profiling of Immune Inhibitory Siglecs and Their Ligands in Patients with Glioma. Cancer Immunol. Immunother. 2019, 68, 937–949. [Google Scholar] [CrossRef] [Green Version]
- Dambuza, I.M.; Brown, G.D. C-Type Lectins in Immunity: Recent Developments. Curr. Opin. Immunol. 2015, 32, 21–27. [Google Scholar] [CrossRef] [Green Version]
- Sancho, D.; Sousa, C.R. Signaling by Myeloid C-Type Lectin Receptors in Immunity and Homeostasis. Annu. Rev. Immunol. 2012, 30, 491–529. [Google Scholar] [CrossRef] [Green Version]
- Brown, G.D.; Willment, J.A.; Whitehead, L. C-Type Lectins in Immunity and Homeostasis. Nat. Rev. Immunol. 2018, 18, 374–389. [Google Scholar] [CrossRef]
- Wu, X.; Saito, T.; Saido, T.C.; Barron, A.M.; Ruedl, C. Microglia and CD206+ Border-Associated Mouse Macrophages Maintain Their Embryonic Origin during Alzheimer’s Disease. eLife 2021, 10, e71879. [Google Scholar] [CrossRef] [PubMed]
- Ilarregui, J.M.; Kooij, G.; Rodríguez, E.; Van Der Pol, S.M.A.; Koning, N.; Kalay, H.; Van Der Horst, J.C.; Van Vliet, S.J.; García-Vallejo, J.J.; De Vries, H.E.; et al. Macrophage Galactose-Type Lectin (MGL) Is Induced on M2 Microglia and Participates in the Resolution Phase of Autoimmune Neuroinflammation. J. Neuroinflammation 2019, 16, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Zizzari, I.G.; Napoletano, C.; Battisti, F.; Rahimi, H.; Caponnetto, S.; Pierelli, L.; Nuti, M.; Rughetti, A. MGL Receptor and Immunity: When the Ligand Can Make the Difference. J. Immunol. Res. 2015, 2015, 450695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Napoletano, C.; Rughetti, A.; Agervig Tarp, M.P.; Coleman, J.; Bennett, E.P.; Picco, G.; Sale, P.; Denda-Nagai, K.; Irimura, T.; Mandel, U.; et al. Tumor-Associated Tn-MUC1 Glycoform Is Internalized through the Macrophage Galactose-Type C-Type Lectin and Delivered to the HLA Class I and II Compartments in Dendritic Cells. Cancer Res. 2007, 67, 8358–8367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zizzari, I.G.; Martufi, P.; Battisti, F.; Rahimi, H.; Caponnetto, S.; Bellati, F.; Nuti, M.; Rughetti, A.; Napoletano, C. The Macrophage Galactose-Type C-Type Lectin (MGL) Modulates Regulatory T Cell Functions. PLoS ONE 2015, 10, e0132617. [Google Scholar] [CrossRef] [Green Version]
- Eggink, L.L.; Roby, K.F.; Cote, R.; Kenneth Hoober, J. An Innovative Immunotherapeutic Strategy for Ovarian Cancer: CLEC10A and Glycomimetic Peptides. J. Immunother. Cancer 2018, 6, 28. [Google Scholar] [CrossRef] [Green Version]
- Napoletano, C.; Steentoff, C.; Battisti, F.; Ye, Z.; Rahimi, H.; Zizzari, I.G.; Dionisi, M.; Cerbelli, B.; Tomao, F.; French, D.; et al. Investigating Patterns of Immune Interaction in Ovarian Cancer: Probing the O-glycoproteome by the Macrophage Galactose-like C-type Lectin (Mgl). Cancers 2020, 12, 2841. [Google Scholar] [CrossRef]
- Heger, L.; Balk, S.; Lühr, J.J.; Heidkamp, G.F.; Lehmann, C.H.K.; Hatscher, L.; Purbojo, A.; Hartmann, A.; Garcia-Martin, F.; Nishimura, S.; et al. CLEC10A Is a specific marker for human CD1c+ dendritic cells and enhances their toll-like receptor 7/8-Induced cytokine secretion. Front. Immunol. 2018, 9, 744. [Google Scholar] [CrossRef]
- Van Vliet, S.J.; Bay, S.; Vuist, I.M.; Kalay, H.; García-Vallejo, J.J.; Leclerc, C.; van Kooyk, Y. MGL signaling augments TLR2-mediated responses for enhanced IL-10 and TNF-α secretion. J. Leukoc. Biol. 2013, 94, 315–323. [Google Scholar] [CrossRef]
- Pearce, O.M.T.; Läubli, H. Sialic acids in cancer biology and immunity. Glycobiology 2015, 26, 111–128. [Google Scholar] [CrossRef] [Green Version]
- Gianchecchi, E.; Arena, A.; Fierabracci, A. Sialic acid-siglec axis in human immune regulation, involvement in autoimmunity and cancer and potential therapeutic treatments. Int. J. Mol. Sci. 2021, 22, 5774. [Google Scholar] [CrossRef] [PubMed]
- Wielgat, P.; Rogowski, K.; Niemirowicz-Laskowska, K. Sialic Acid-Siglec Axis as Molecular Checkpoints Targeting of Immune System: Smart Players in Pathology and Conventional Therapy. Int. J. Mol. Sci. 2020, 21, 4361. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Gil, A.; Schnaar, R.L. Siglec Ligands. Cells 2021, 10, 1260. [Google Scholar] [CrossRef]
- Bochner, B.S.; Zimmermann, N. Role of Siglecs and Related Glycan-Binding Proteins in Immune Responses and Immunoregulation. J. Allergy Clin. Immunol. 2015, 135, 598–608. [Google Scholar] [CrossRef] [Green Version]
- Quarles, R.H. Myelin-Associated Glycoprotein (MAG): Past, Present and Beyond. J. Neurochem. 2007, 100, 1431–1448. [Google Scholar] [CrossRef]
- Liao, H.; Duka, T.; Teng, F.Y.H.; Sun, L.; Bu, W.Y.; Ahmed, S.; Tang, B.L.; Xiao, Z.C. Nogo-66 and Myelin-Associated Glycoprotein (MAG) Inhibit the Adhesion and Migration of Nogo-66 Receptor Expressing Human Glioma Cells. J. Neurochem. 2004, 90, 1156–1162. [Google Scholar] [CrossRef] [PubMed]
- Van de Wall, S.; Santegoets, K.C.M.; van Houtum, E.J.H.; Büll, C.; Adema, G.J. Sialoglycans and Siglecs Can Shape the Tumor Immune Microenvironment. Trends Immunol. 2020, 41, 274–285. [Google Scholar] [CrossRef] [PubMed]
- Dusoswa, S.A.; Horrevorts, S.K.; Ambrosini, M.; Kalay, H.; Paauw, N.J.; Nieuwland, R.; Pegtel, M.D.; Würdinger, T.; Van Kooyk, Y.; Garcia-Vallejo, J.J. Glycan Modification of Glioblastoma-Derived Extracellular Vesicles Enhances Receptor-Mediated Targeting of Dendritic Cells. J. Extracell. Vesicles 2019, 8, 1648995. [Google Scholar] [CrossRef] [Green Version]
- Battisti, F.; Napoletano, C.; Koshkaki, H.R.; Belleudi, F.; Zizzari, I.G.; Ruscito, I.; Palchetti, S.; Bellati, F.; Panici, P.B.; Torrisi, M.R.; et al. Tumor-Derived Microvesicles Modulate Antigen Cross-Processing via Reactive Oxygen Species-Mediated Alkalinization of Phagosomal Compartment in Dendritic Cells. Front. Immunol. 2017, 8, 1179. [Google Scholar] [CrossRef]
- Li, C.-H.; Chang, Y.-C.; Chan, M.-H.; Yang, Y.-F.; Liang, S.-M.; Hsiao, M. Galectins in Cancer and the Microenvironment: Functional Roles, Therapeutic Developments, and Perspectives. Biomedicines 2021, 9, 1159. [Google Scholar] [CrossRef]
- Thiemann, S.; Baum, L.G. Galectins and Immune Responses-Just How Do They Do Those Things They Do? Annu. Rev. Immunol. 2016, 34, 243–264. [Google Scholar] [CrossRef] [PubMed]
- Elola, M.T.; Ferragut, F.; Méndez-Huergo, S.P.; Croci, D.O.; Bracalente, C.; Rabinovich, G.A. Galectins: Multitask Signaling Molecules Linking Fibroblast, Endothelial and Immune Cell Programs in the Tumor Microenvironment. Cell. Immunol. 2018, 333, 34–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johannes, L.; Jacob, R.; Leffler, H. Galectins at a Glance. J. Cell Sci. 2018, 131, jcs208884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilson, R.C.; Gunasinghe, S.D.; Johannes, L.; Gaus, K. Galectin-3 Modulation of T-Cell Activation: Mechanisms of Membrane Remodelling. Prog. Lipid Res. 2019, 76, 101010. [Google Scholar] [CrossRef] [PubMed]
- Méndez-Huergo, S.P.; Blidner, A.G.; Rabinovich, G.A. Galectins: Emerging Regulatory Checkpoints Linking Tumor Immunity and Angiogenesis. Curr. Opin. Immunol. 2017, 45, 8–15. [Google Scholar] [CrossRef] [PubMed]
- Laderach, D.J.; Compagno, D. Unraveling How Tumor-Derived Galectins Contribute to Anti-Cancer Immunity Failure. Cancers 2021, 13, 4529. [Google Scholar] [CrossRef] [PubMed]
- Nio-Kobayashi, J.; Itabashi, T. Galectins and Their Ligand Glycoconjugates in the Central Nervous System Under Physiological and Pathological Conditions. Front. Neuroanat. 2021, 15, 767330. [Google Scholar] [CrossRef]
- Huang, Y.; Wang, H.C.; Zhao, J.; Wu, M.H.; Shih, T.C. Immunosuppressive Roles of Galectin-1 in the Tumor Microenvironment. Biomolecules 2021, 11, 1398. [Google Scholar] [CrossRef]
- Jung, T.Y.; Jung, S.; Ryu, H.H.; Jeong, Y.; Jin, Y.H.; Jin, S.G.; Kim, I.Y.; Kang, S.S.; Kim, H.S. Role of Galectin-1 in Migration and Invasion of Human Glioblastoma Multiforme Cell Lines: Laboratory Investigation. J. Neurosurg. 2008, 109, 273–284. [Google Scholar] [CrossRef] [Green Version]
- Fortin, S.; Le Mercier, M.; Camby, I.; Spiegl-Kreinecker, S.; Berger, W.; Lefranc, F.; Kiss, R. Galectin-1 Is Implicated in the Protein Kinase c ε/Vimentin-Controlled Trafficking of Integrin-Β1 in Glioblastoma Cells. Brain Pathol. 2010, 20, 39–49. [Google Scholar] [CrossRef] [Green Version]
- Camby, I.; Belot, N.; Lefranc, F.; Sadeghi, N.; De Launoit, Y.; Kaltner, H.; Musette, S.; Darro, F.; Danguy, A.; Salmon, I.; et al. Galectin-1 Modulates Human Glioblastoma Cell Migration into the Brain through Modifications to the Actin Cytoskeleton and Levels of Expression of Small GTPases. J. Neuropathol. Exp. Neurol. 2002, 61, 585–596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toussaint, L.G.; Nilson, A.E.; Goble, J.M.; Ballman, K.V.; James, C.D.; Lefranc, F.; Kiss, R.; Uhm, J.H. Galectin-1, a Gene Preferentially Expressed at the Tumor Margin, Promotes Glioblastoma Cell Invasion. Mol. Cancer 2012, 11, 32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verschuere, T.; Toelen, J.; Maes, W.; Poirier, F.; Boon, L.; Tousseyn, T.; Mathivet, T.; Gerhardt, H.; Mathieu, V.; Kiss, R.; et al. Glioma-Derived Galectin-1 Regulates Innate and Adaptive Antitumor Immunity. Int. J. Cancer 2014, 134, 873–884. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.R.; Wu, M.Y.; Dai, L.J.; Huang, Y.; Shan, M.Y.; Ma, S.N.; Wang, J.; Peng, H.; Ding, Y.; Zhang, Q.F.; et al. Nuclear FAM289-Galectin-1 Interaction Controls FAM289-Mediated Tumor Promotion in Malignant Glioma. J. Exp. Clin. Cancer Res. 2019, 38, 394. [Google Scholar] [CrossRef] [Green Version]
- Chen, Q.; Han, B.; Meng, X.; Duan, C.; Yang, C.; Wu, Z.; Magafurov, D.; Zhao, S.; Safin, S.; Jiang, C.; et al. Immunogenomic Analysis Reveals LGALS1 Contributes to the Immune Heterogeneity and Immunosuppression in Glioma. Int. J. Cancer 2019, 145, 517–530. [Google Scholar] [CrossRef] [PubMed]
- Chou, S.Y.; Yen, S.L.; Huang, C.C.; Huang, E.Y. Galectin-1 Is a Poor Prognostic Factor in Patients with Glioblastoma Multiforme after Radiotherapy. BMC Cancer 2018, 18, 105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Woensel, M.; Wauthoz, N.; Rosière, R.; Mathieu, V.; Kiss, R.; Lefranc, F.; Steelant, B.; Dilissen, E.; Van Gool, S.W.; Mathivet, T.; et al. Development of SiRNA-Loaded Chitosan Nanoparticles Targeting Galectin-1 for the Treatment of Glioblastoma Multiforme via Intranasal Administration. J. Control. Release 2016, 227, 71–81. [Google Scholar] [CrossRef]
- Van Woensel, M.; Mathivet, T.; Wauthoz, N.; Rosière, R.; Garg, A.D.; Agostinis, P.; Mathieu, V.; Kiss, R.; Lefranc, F.; Boon, L.; et al. Sensitization of Glioblastoma Tumor Micro-Environment to Chemo- and Immunotherapy by Galectin-1 Intranasal Knock-down Strategy. Sci. Rep. 2017, 7, 1217. [Google Scholar] [CrossRef]
- Danhier, F.; Messaoudi, K.; Lemaire, L.; Benoit, J.P.; Lagarce, F. Combined Anti-Galectin-1 and Anti-EGFR SiRNA-Loaded Chitosan-Lipid Nanocapsules Decrease Temozolomide Resistance in Glioblastoma: In Vivo Evaluation. Int. J. Pharm. 2015, 481, 154–161. [Google Scholar] [CrossRef]
- Yuan, F.; Ming, H.; Wang, Y.; Yang, Y.; Yi, L.; Li, T.; Ma, H.; Tong, L.; Zhang, L.; Liu, P.; et al. Molecular and Clinical Characterization of Galectin-9 in Glioma through 1027 Samples. J. Cell. Physiol. 2020, 235, 4326–4334. [Google Scholar] [CrossRef] [Green Version]
- Knudsen, A.M.; Rudkjøbing, S.J.; Sørensen, M.D.; Dahlrot, R.H.; Kristensen, B.W. Expression and Prognostic Value of the Immune Checkpoints Galectin-9 and PD-L1 in Glioblastomas. J. Neuropathol. Exp. Neurol. 2021. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Cai, Y.; Peng, Y.; Xu, B.; Hui, W.; Jiang, Y. Exosomal LGALS9 in the Cerebrospinal Fluid of Glioblastoma Patients Suppressed Dendritic Cell Antigen Presentation and Cytotoxic T-Cell Immunity. Cell Death Dis. 2020, 11, 896. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Han, H.; He, X.; Li, S.; Wu, C.; Yu, C.; Wang, S. Expression of the Galectin-9-Tim-3 Pathway in Glioma Tissues Is Associated with the Clinical Manifestations of Glioma. Oncol. Lett. 2016, 11, 1829–1834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, Q.Y.; Li, Y.S.; Qiao, X.H.; Yang, J.W.; Guo, X.L. Targeting Galectins in T Cell-Based Immunotherapy within Tumor Microenvironment. Life Sci. 2021, 277, 119426. [Google Scholar] [CrossRef]
- Videla-Richardson, G.A.; Morris-Hanon, O.; Torres, N.I.; Esquivel, M.I.; Vera, M.B.; Ripari, L.B.; Croci, D.O.; Sevlever, G.E.; Rabinovich, G.A. Galectins as Emerging Glyco-Checkpoints and Therapeutic Targets in Glioblastoma. Int. J. Mol. Sci. 2022, 23, 316. [Google Scholar] [CrossRef]
- Colomb, F.; Wang, W.; Simpson, D.; Zafar, M.; Beynon, R.; Rhodes, J.M.; Yu, L.G. Galectin-3 Interacts with the Cell-Surface Glycoprotein CD146 (MCAM, MUC18) and Induces Secretion of Metastasispromoting Cytokines from Vascular Endothelial Cells. J. Biol. Chem. 2017, 292, 8381–8389. [Google Scholar] [CrossRef] [Green Version]
- Nangia-Makker, P.; Hogan, V.; Raz, A. Galectin-3 and Cancer Stemness. Glycobiology 2018, 28, 172–181. [Google Scholar] [CrossRef] [Green Version]
- Ikemori, R.Y.; Machado, C.M.L.; Furuzawa, K.M.; Nonogaki, S.; Osinaga, E.; Umezawa, K.; De Carvalho, M.A.; Verinaud, L.; Chammas, R. Galectin-3 up-Regulation in Hypoxic and Nutrient Deprived Microenvironments Promotes Cell Survival. PLoS ONE 2014, 9, e111592. [Google Scholar] [CrossRef] [Green Version]
- Park, S.H.; Min, H.S.; Kim, B.; Myung, J.; Paek, S.H. Galectin-3: A Useful Biomarker for Differential Diagnosis of Brain Tumors. Neuropathology 2008, 28, 497–506. [Google Scholar] [CrossRef]
- Wang, H.; Song, X.; Huang, Q.; Xu, T.; Yun, D.; Wang, Y.; Hu, L.; Yan, Y.; Chen, H.; Lu, D.; et al. LGALS3 Promotes Treatment Resistance in Glioblastoma and Is Associated with Tumor Risk and Prognosis. Cancer Epidemiol. Biomark. Prev. 2019, 28, 760–769. [Google Scholar] [CrossRef] [Green Version]
- Seguin, L.; Odouard, S.; Corlazzoli, F.; Haddad, S.; Moindrot, L.; Calvo Tardón, M.; Yebra, M.; Koval, A.; Marinari, E.; Bes, V.; et al. Macropinocytosis Requires Gal-3 in a Subset of Patient-Derived Glioblastoma Stem Cells. Commun. Biol. 2021, 4, 718. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Zhang, S.; Chen, J.; Li, D. Increased LGALS3 Expression Independently Predicts Shorter Overall Survival in Patients with the Proneural Subtype of Glioblastoma. Cancer Med. 2019, 8, 2031–2040. [Google Scholar] [CrossRef] [PubMed]
- Ideo, H.; Matsuzaka, T.; Nonaka, T.; Seko, A.; Yamashita, K. Galectin-8-N-Domain Recognition Mechanism for Sialylated and Sulfated Glycans. J. Biol. Chem. 2011, 286, 11346–11355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Metz, C.; Döger, R.; Riquelme, E.; Cortés, P.; Holmes, C.; Shaughnessy, R.; Oyanadel, C.; Grabowski, C.; González, A.; Soza, A. Galectin-8 Promotes Migration and Proliferation and Prevents Apoptosis in U87 Glioblastoma Cells. Biol. Res. 2016, 49, 33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zick, Y.; Eisenstein, M.; Goren, R.A.; Hadari, Y.R.; Levy, Y.; Ronen, D. Role of Galectin-8 as a Modulator of Cell Adhesion and Cell Growth. Glycoconj. J. 2002, 19, 517–526. [Google Scholar] [CrossRef] [PubMed]
- Gentilini, L.D.; Jaworski, F.M.; Tiraboschi, C.; Pérez, I.G.; Kotler, M.L.; Chauchereau, A.; Laderach, D.J.; Compagno, D. Stable and High Expression of Galectin-8 Tightly Controls Metastatic Progression of Prostate Cancer. Oncotarget 2017, 8, 44654–44668. [Google Scholar] [CrossRef] [Green Version]
- Friedel, M.; André, S.; Goldschmidt, H.; Gabius, H.J.; Schwartz-Albiez, R. Galectin-8 Enhances Adhesion of Multiple Myeloma Cells to Vascular Endothelium and Is an Adverse Prognostic Factor. Glycobiology 2016, 26, 1048–1058. [Google Scholar] [CrossRef]
- Cludts, S.; Decaestecker, C.; Mahillon, V.; Chevalier, D.; Kaltner, H.; André, S.; Remmelink, M.; Leroy, X.; Gabius, H.J.; Saussez, S. Galectin-8 up-Regulation during Hypopharyngeal and Laryngeal Tumor Progression and Comparison with Galectin-1, -3 and -7. Anticancer Res. 2009, 29, 4933–4940. [Google Scholar]
- Wu, S.; Liu, H.; Zhang, H.; Lin, C.; Li, R.; Cao, Y.; He, H.; Li, H.; Shen, Z.; Qin, J.; et al. Galectin-8 Is Associated with Recurrence and Survival of Patients with Non-Metastatic Gastric Cancer after Surgery. Tumor Biol. 2016, 37, 12635–12642. [Google Scholar] [CrossRef]
- Wu, S.; Yang, W.; Zhang, H.; Ren, Y.; Fang, Z.; Yuan, C.; Yao, Z. The Prognostic Landscape of Tumor-Infiltrating Immune Cells and Immune Checkpoints in Glioblastoma. Technol. Cancer Res. Treat. 2019, 18, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Erhart, F.; Buchroithner, J.; Reitermaier, R.; Fischhuber, K.; Klingenbrunner, S.; Sloma, I.; Hibsh, D.; Kozol, R.; Efroni, S.; Ricken, G.; et al. Immunological Analysis of Phase II Glioblastoma Dendritic Cell Vaccine (Audencel) Trial: Immune System Characteristics Influence Outcome and Audencel up-Regulates Th1-Related Immunovariables. Acta Neuropathol. Commun. 2018, 6, 135. [Google Scholar] [CrossRef] [PubMed]
- Fucikova, J.; Kepp, O.; Kasikova, L.; Petroni, G.; Yamazaki, T.; Liu, P.; Zhao, L.; Spisek, R.; Kroemer, G.; Galluzzi, L. Detection of Immunogenic Cell Death and Its Relevance for Cancer Therapy. Cell Death Dis. 2020, 11, 1013. [Google Scholar] [CrossRef] [PubMed]
- Scirocchi, F.; Napoletano, C.; Pace, A.; Rahimi Koshkaki, H.; Di Filippo, A.; Zizzari, I.G.; Nuti, M.; Rughetti, A. Immunogenic Cell Death and Immunomodulatory Effects of Cabozantinib. Front. Oncol. 2021, 11, 755433. [Google Scholar] [CrossRef]
- Wargo, J.A.; Reuben, A.; Cooper, Z.A.; Oh, K.S.; Sullivan, R.J. Immune Effects of Chemoterapy, Radiation, and Targeted Rapy and Opportunities for Combination with Immunorapy. Semin. Oncol. 2015, 42, 601–616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scirocchi, F.; Scagnoli, S.; Botticelli, A.; Filippo, D.; Napoletano, C.; Zizzari, G.; Strigari, L.; Tomao, S.; Cortesi, E.; Rughetti, A.; et al. Immune Effects of CDK4/6 Inhibitors in Patients with HR+/HER2- Metastatic Breast Cancer: Relief from Immunosuppression Is Associated with Clinical Response. eBioMedicine 2022, 79, 104010. [Google Scholar] [CrossRef]
- Ponterio, E.; De Maria, R.; Haas, T.L. Identification of Targets to Redirect CAR T Cells in Glioblastoma and Colorectal Cancer: An Arduous Venture. Front. Immunol. 2020, 11, 565631. [Google Scholar] [CrossRef] [PubMed]
- Kakimi, K.; Karasaki, T.; Matsushita, H.; Sugie, T. Advances in Personalized Cancer Immunotherapy. Breast Cancer 2017, 24, 16–24. [Google Scholar] [CrossRef]
- Wang, H.C.; Chan, L.P.; Cho, S.F. Targeting the Immune Microenvironment in the Treatment of Head and Neck Squamous Cell Carcinoma. Front. Oncol. 2019, 9, 1084. [Google Scholar] [CrossRef] [Green Version]
- Derosiers, N.; Aguilar, W.; DeGaramo, D.A.; Posey, A.D. Sweet Immune Checkpoint Targets to Enhance T Cell Therapy. J. Immunol. 2022, 208, 278–285. [Google Scholar] [CrossRef]
- Chen, Z.; Hambardzumyan, D. Immune Microenvironment in Glioblastoma Subtypes. Front. Immunol. 2018, 9, 1004. [Google Scholar] [CrossRef] [Green Version]
- Sethi, A.; Sanam, S.; Alvala, M. Non-Carbohydrate Strategies to Inhibit Lectin Proteins with Special Emphasis on Galectins. Eur. J. Med. Chem. 2021, 222, 113561. [Google Scholar] [CrossRef] [PubMed]
- Vrbata, D.; Filipová, M.; Tavares, M.R.; Červený, J.; Vlachová, M.; Šírová, M.; Pelantová, H.; Petrásková, L.; Bumba, L.; Konefał, R.; et al. Glycopolymers Decorated with 3-O-Substituted Thiodigalactosides as Potent Multivalent Inhibitors of Galectin-3. J. Med. Chem. 2022, 65, 3866–3878. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.; Ho, M.; Adeniji, O.S.; Giron, L.; Bordoloi, D.; Kulkarni, A.J.; Puchalt, A.P.; Abdel-Mohsen, M.; Muthumani, K. Development of Siglec-9 Blocking Antibody to Enhance Anti-Tumor Immunity. Front. Oncol. 2021, 11, 778989. [Google Scholar] [CrossRef] [PubMed]
- Jiménez, V.A.; Navarrete, K.R.; Duque-Noreña, M.; Marrugo, K.P.; Contreras, M.A.; Campos, C.H.; Alderete, J.B. Rational Design of Novel Glycomimetic Peptides for E-Selectin Targeting. J. Chem. Inf. Model. 2021, 61, 2463–2474. [Google Scholar] [CrossRef] [PubMed]
- Van Vliet, S.J.; Paessens, L.C.; Broks-van den Berg, V.C.M.; Geijtenbeek, T.B.H.; van Kooyk, Y. The C-Type Lectin Macrophage Galactose-Type Lectin Impedes Migration of Immature APCs. J. Immunol. 2008, 181, 3148–3155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, C.; Öhlund, D.; Rickelt, S.; Lidström, T.; Huang, Y.; Hao, L.; Zhao, R.T.; Franklin, O.; Bhatia, S.N.; Tuveson, D.A.; et al. Cancer Cell–Derived Matrisome Proteins Promote Metastasis in Pancreatic Ductal Adenocarcinoma. Cancer Res. 2020, 80, 1461–1474. [Google Scholar] [CrossRef] [Green Version]
- Galon, J.; Bruni, D. Tumor Immunology and Tumor Evolution: Intertwined Histories. Immunity 2020, 52, 55–81. [Google Scholar] [CrossRef]
- Bruni, D.; Angell, H.K.; Galon, J. The Immune Contexture and Immunoscore in Cancer Prognosis and Therapeutic Efficacy. Nat. Rev. Cancer 2020, 20, 662–680. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pace, A.; Scirocchi, F.; Napoletano, C.; Zizzari, I.G.; D’Angelo, L.; Santoro, A.; Nuti, M.; Rahimi, H.; Rughetti, A. Glycan-Lectin Interactions as Novel Immunosuppression Drivers in Glioblastoma. Int. J. Mol. Sci. 2022, 23, 6312. https://doi.org/10.3390/ijms23116312
Pace A, Scirocchi F, Napoletano C, Zizzari IG, D’Angelo L, Santoro A, Nuti M, Rahimi H, Rughetti A. Glycan-Lectin Interactions as Novel Immunosuppression Drivers in Glioblastoma. International Journal of Molecular Sciences. 2022; 23(11):6312. https://doi.org/10.3390/ijms23116312
Chicago/Turabian StylePace, Angelica, Fabio Scirocchi, Chiara Napoletano, Ilaria Grazia Zizzari, Luca D’Angelo, Antonio Santoro, Marianna Nuti, Hassan Rahimi, and Aurelia Rughetti. 2022. "Glycan-Lectin Interactions as Novel Immunosuppression Drivers in Glioblastoma" International Journal of Molecular Sciences 23, no. 11: 6312. https://doi.org/10.3390/ijms23116312
APA StylePace, A., Scirocchi, F., Napoletano, C., Zizzari, I. G., D’Angelo, L., Santoro, A., Nuti, M., Rahimi, H., & Rughetti, A. (2022). Glycan-Lectin Interactions as Novel Immunosuppression Drivers in Glioblastoma. International Journal of Molecular Sciences, 23(11), 6312. https://doi.org/10.3390/ijms23116312