
The Primary Microglial Leukodystrophies: A Review


{kind=link}
{kind=link}
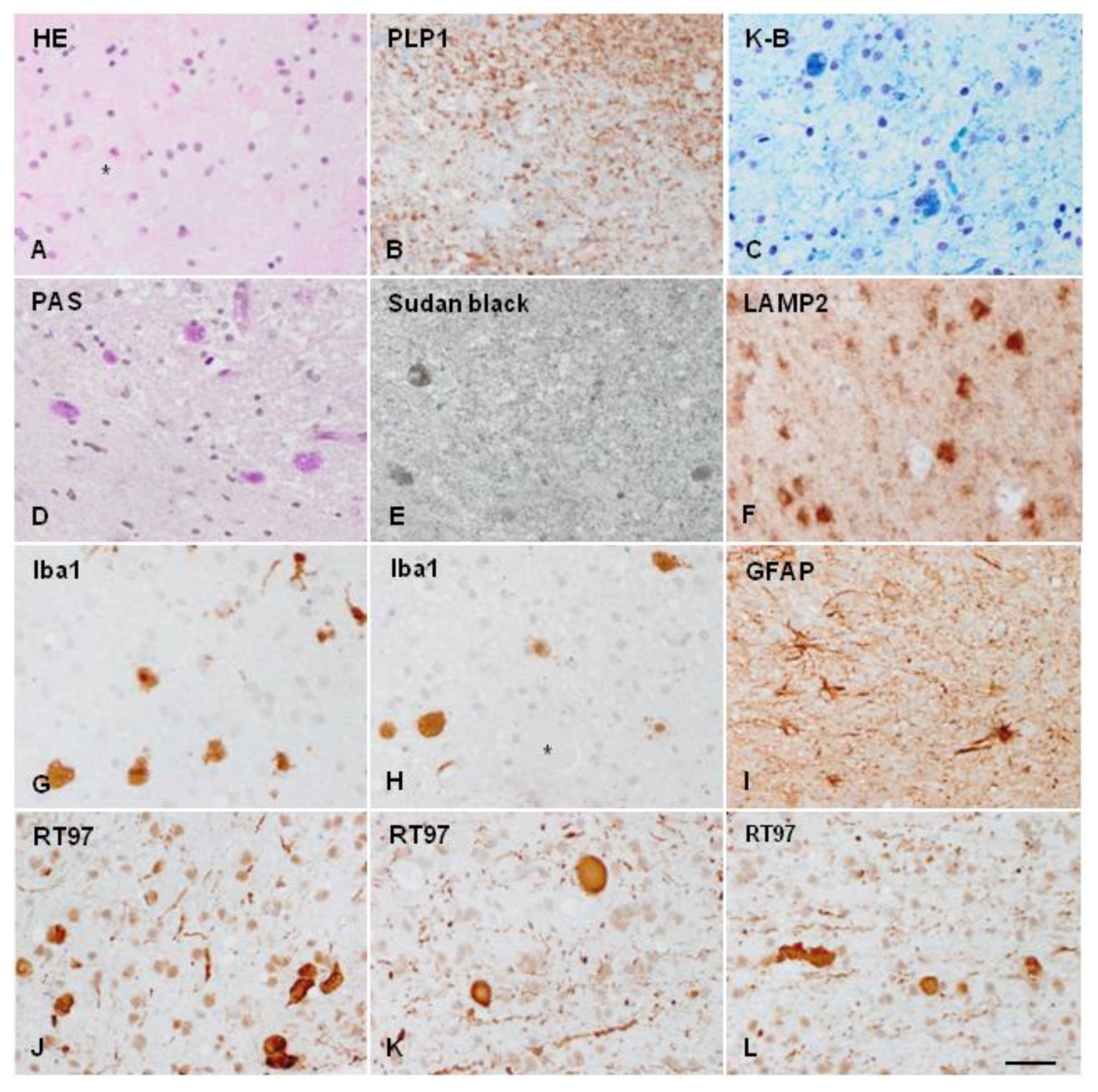{kind=link}
Abstract
:1. Introduction
2. Adult-Onset Leukodystrophy with Axonal Spheroids and Pigmented Glia (POLD, HDLS, ALSP): CRP Linked to Mutations in CSF1R, and LKENP (Leukoencephalopathy, Progressive with Ovarian Failure) Linked to Heterozygous Mutations in AARS2
2.1. Clinical Features
2.1.1. ALSP-CSFR1 or CRL
2.1.2. ALSP-AARS2 or LKENP
2.1.3. Non-ALSP AARS1 Mutations
2.2. Radiological Findings
2.3. Neuropathology
2.4. Genetics
2.4.1. CRL
2.4.2. LKENP
2.4.3. Non-ALSP AARS1 Mutations
2.5. Pathogenesis
2.6. Phenotype Differences between CRP and LKENP

3. Polycystic Membranous Lipomembranous Osteodysplasia with Sclerosing Leukoencephalopathy (PLOSL; Nasu–Hakola Disease)
3.1. Clinical Features
3.2. Radiological Examination
3.3. General Pathology
3.4. Neuropathology
3.5. Genetics
3.6. Pathogenesis
4. Concluding Remarks
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Van Der Knaap, M.S.; Bugiani, M. Leukodystrophies: A proposed classification system based on pathological changes and pathogenetic mechanisms. Acta Neuropathol. 2017, 134, 351–382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Köhler, W.; Curiel, J.; Vanderver, A. Adulthood leukodystrophies. Nat. Rev. Neurol. 2018, 14, 94–105. [Google Scholar] [CrossRef] [PubMed]
- Lynch, D.S.; De Paiva, A.R.B.; Zhang, W.; Bugiardini, E.; Freua, F.; Lucato, L.; de Souza, L.I.M.; Lakshmanan, R.; Kinsella, J.A.; Merwick, A.; et al. Clinical and genetic characterization of leukoencephalopathies in adults. Brain 2017, 140, 1204–1211. [Google Scholar] [CrossRef] [PubMed]
- Van der Knaap, M.S.; Schiffmann, R.; Mochel, F.; Wolf, N. Diagnosis, prognosis, and treatment of leukodystrophies. Lancet Neurol. 2019, 18, 962–972. [Google Scholar] [CrossRef]
- Resende, L.L.; De Paiva, A.R.B.; Kok, F.; Leite, C.D.C.; Lucato, L. Adult Leukodystrophies: A Step-by-Step Diagnostic Approach. RadioGraphics 2019, 39, 153–168. [Google Scholar] [CrossRef] [Green Version]
- Garcia, L.M.; Hacker, J.L.; Sase, S.; Adang, L.; Almad, A. Glial cells in the driver seat of leukodystrophy pathogenesis. Neurobiol. Dis. 2020, 146, 105087. [Google Scholar] [CrossRef]
- Bianchin, M.M.; Martin, K.C.; De Souza, A.C.; De Oliveira, M.A.; Rieder, C.R. Nasu–Hakola disease and primary microglial dysfunction. Nat. Rev. Neurol. 2010, 6, 523. [Google Scholar] [CrossRef]
- Konno, T.; Kasanuki, K.; Ikeuchi, T.; Dickson, D.W.; Wszolek, Z.K. CSF1R-related leukoencephalopathy. A major player in primary microgliopathies. Neurology 2018, 91, 1092–1104. [Google Scholar] [CrossRef]
- Berdowski, W.M.; Sanderson, L.E.; van Ham, T.J. The multicellular interplay of microglia in health and disease: Lessons from leukodystrophy. Dis. Model. Mech. 2021, 14, dmm048925. [Google Scholar] [CrossRef]
- Martin, L.; Martin, J.J. Ludo van Bogaert (1897–1989). Acta Neurol. Belg. 1996, 96, 254–263. [Google Scholar] [CrossRef]
- Gray, F.; Destée, A.; Bourre, J.-M.; Gherardi, R.; Krivosic, I.; Warot, P.; Poirier, J. Pigmentary Type of Orthochromatic Leukodystrophy: A new case with ultrastructural and biochemical study. J. Neuropathol. Exp. Neurol. 1987, 46, 585–596. [Google Scholar] [CrossRef] [PubMed]
- Tuñón, T.; Ferrer, I.; Gállego, J.; Delgado, G.; Villanueva, J.A.; Martinez-Peñuela, J.M. Leucodystrophy with pigmented glial and scavenger cells (pigmentary type of orthochromatic leucodystrophy). Neuropathol. Appl. Neurobiol. 1988, 14, 337–344. [Google Scholar] [PubMed]
- Constantinidis, J.; Wisniewski, T. The dominant form of the pigmentary orthochromatic leukodystrophy. Acta Neuropathol. 1991, 82, 483–487. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Wherrett, J.; Nag, S. A Rare Form of Adult Onset Leukodystrophy: Orthochromatic Leukodystrophy with Pigmented Glia. Can. J. Neurol. Sci. J. Can. Sci. Neurol. 1997, 24, 146–150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taglia, I.; Di Donato, I.; Bianchi, S.; Cerase, A.; Monti, L.; Marconi, R.; Orrico, A.; Rufa, A.; Federico, A.; Dotti, M.T. AARS2-related ovarioleukodystrophy: Clinical and neuroimaging features of three new cases. Acta Neurol. Scand. 2018, 138, 278–283. [Google Scholar] [CrossRef]
- Axelsson, R.; Röyttä, M.; Sourander, P.; Akesson, H.O.; Andersen, O. Hereditary diffuse leucoencephalopathy with spheroids. Acta Psychiatr. Scand. Suppl. 1984, 314, 1–65. [Google Scholar]
- Baba, Y.; Ghetti, B.; Baker, M.C.; Uitti, R.J.; Hutton, M.L.; Yamaguchi, K.; Bird, T.; Lin, W.; DeLucia, M.W.; Dickson, D.W.; et al. Hereditary diffuse leukoencephalopathy with spheroids: Clinical, pathologic and genetic studies of a new kindred. Acta Neuropathol. 2006, 111, 300–311. [Google Scholar] [CrossRef]
- Lin, W.L.; Wszolek, Z.K.; Dickson, D.W. Hereditary diffuse leukoencephalopathy with spheroids: Ultrastructural and immunoe-lectron microscopic studies. Int. J. Clin. Exp. Pathol. 2010, 3, 665–674. [Google Scholar]
- Riku, Y.; Ando, T.; Goto, Y.; Mano, K.; Iwasaki, Y.; Sobue, G.; Yoshida, M. Early Pathologic Changes in Hereditary Diffuse Leukoencephalopathy with Spheroids. J. Neuropathol. Exp. Neurol. 2014, 73, 1183–1190. [Google Scholar] [CrossRef] [Green Version]
- Hoffmann, S.; Murrell, J.; Harms, L.; Miller, K.; Meisel, A.; Brosch, T.; Scheel, M.; Ghetti, B.; Goebel, H.-H.; Stenzel, W. Enlarging the Nosological Spectrum of Hereditary Diffuse Leukoencephalopathy with Axonal Spheroids (HDLS). Brain Pathol. 2014, 24, 452–458. [Google Scholar] [CrossRef]
- Wider, C.; Van Gerpen, J.A.; DeArmond, S.; Shuster, E.A.; Dickson, D.W.; Wszolek, Z.K. Leukoencephalopathy with spheroids (HDLS) and pigmentary leukodystrophy (POLD): A single entity? Neurology 2009, 72, 1953–1959. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Der Knaap, M.S.; Naidu, S.; Kleinschmidt-DeMasters, B.K.; Kamphorst, W.; Weinstein, H.C. Autosomal dominant diffuse leukoencephalopathy with neuroaxonal spheroids. Neurology 2000, 54, 463. [Google Scholar] [CrossRef] [PubMed]
- Marotti, J.D.; Tobias, S.; Fratkin, J.D.; Powers, J.M.; Rhodes, C.H. Adult onset leukodystrophy with neuroaxonal spheroids and pigmented glia: Report of a family, historical perspective, and review of the literature. Acta Neuropathol. 2004, 107, 481–488. [Google Scholar] [CrossRef] [PubMed]
- Itoh, K.; Shiga, K.; Shimizu, K.; Muranishi, M.; Nakagawa, M.; Fushiki, S. Autosomal dominant leukodystrophy with axonal spheroids and pigmented glia: Clinical and neuropathological characteristics. Acta Neuropathol. 2005, 111, 39–45. [Google Scholar] [CrossRef] [PubMed]
- Ali, Z.S.; Van Der Voorn, J.P.; Powers, J.M. A Comparative Morphologic Analysis of Adult Onset Leukodystrophy with Neuroaxonal Spheroids and Pigmented Glia-A Role for Oxidative Damage. J. Neuropathol. Exp. Neurol. 2007, 66, 660–672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Freeman, S.H.; Hyman, B.T.; Sims, K.B.; Hedley-Whyte, E.T.; Vossough, A.; Frosch, M.P.; Schmahmann, J.D. Adult Onset Leukodystrophy with Neuroaxonal Spheroids: Clinical, Neuroimaging and Neuropathologic Observations. Brain Pathol. 2008, 19, 39–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lynch, D.S.; Jaunmuktane, Z.; Sheerin, U.-M.; Phadke, R.; Brandner, S.; Milonas, I.; Dean, A.; Bajaj, N.; McNicholas, N.; Costello, D.; et al. Hereditary leukoencephalopathy with axonal spheroids: A spectrum of phenotypes from CNS vasculitis to parkinsonism in an adult onset leukodystrophy series. J. Neurol. Neurosurg. Psychiatry 2015, 87, 512–519. [Google Scholar] [CrossRef]
- Sundal, C.; Lash, J.; Aasly, J.; Øygarden, S.; Roeber, S.; Kretzschman, H.; Garbern, J.Y.; Tselis, A.; Rademakers, R.; Dickson, D.W.; et al. Hereditary diffuse leukoencephalopathy with axonal spheroids (HDLS): A misdiagnosed disease entity. J. Neurol. Sci. 2012, 314, 130–137. [Google Scholar] [CrossRef] [Green Version]
- Nicholson, A.M.; Baker, M.C.; Finch, N.A.; Rutherford, N.J.; Wider, C.; Graff-Radford, N.R.; Nelson, P.T.; Clark, H.B.; Wszolek, Z.K.; Dickson, D.W.; et al. CSF1R mutations link POLD and HDLS as a single disease entity. Neurology 2013, 80, 1033–1040. [Google Scholar] [CrossRef]
- Wider, C.; Wszolek, Z.K. Hereditary diffuse leukoencephalopathy with axonal spheroids: More than just a rare disease. Neurology 2013, 82, 102–103. [Google Scholar] [CrossRef] [Green Version]
- Sundal, C.; Wszolek, Z.K. CSFR1-related adult-onset leukoencephalopathy with axonal spheroids and pigmented glia. In GeneReviews® [Internet]; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Stephens, K., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 2017. [Google Scholar]
- Rademakers, R.; Baker, M.; Nicholson, A.M.; Rutherford, N.J.; Finch, N.; Soto-Ortolaza, A.; Lash, J.; Wider, C.; Wojtas, A.; DeJesus-Hernandez, M.; et al. Mutations in the colony stimulating factor 1 receptor (CSF1R) gene cause hereditary diffuse leukoencephalopathy with spheroids. Nat. Genet. 2011, 44, 200–205. [Google Scholar] [CrossRef] [PubMed]
- Dallabona, C.; Diodato, D.; Kevelam, S.H.; Haack, T.B.; Wong, L.-J.; Salomons, G.S.; Baruffini, E.; Melchionda, L.; Mariotti, C.; Strom, T.M.; et al. Novel (ovario) leukodystrophy related to AARS2 mutations. Neurology 2014, 82, 2063–2071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simons, C.; Griffin, L.B.; Helman, G.; Golas, G.; Pizzino, A.; Bloom, M.; Murphy, J.L.P.; Crawford, J.; Evans, S.H.; Topper, S.; et al. Loss-of-Function Alanyl-tRNA Synthetase Mutations Cause an Autosomal-Recessive Early-Onset Epileptic Encephalopathy with Persistent Myelination Defect. Am. J. Hum. Genet. 2015, 96, 675–681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Helman, G.; Mendes, M.I.; Nicita, F.; Darbelli, L.; Sherbini, O.; Moore, T.; Derksen, A.; Pizzino, A.; Carrozzo, R.; Torraco, A.; et al. Expanded phenotype of AARS1-related white matter disease. Genet. Med. 2021, 23, 2352–2359. [Google Scholar] [CrossRef]
- Konno, T.; Yoshida, K.; Mizuno, T.; Kawarai, T.; Tada, M.; Nozaki, H.; Ikeda, S.-I.; Nishizawa, M.; Onodera, O.; Wszolek, Z.K.; et al. Clinical and genetic characterization of adult-onset leukoencephalopathy with axonal spheroids and pigmented glia associated with CSF1R mutation. Eur. J. Neurol. 2016, 24, 37–45. [Google Scholar] [CrossRef]
- Sundal, C.; Fujioka, S.; Van Gerpen, J.A.; Wider, C.; Nicholson, A.M.; Baker, M.; Shuster, E.A.; Aasly, J.; Spina, S.; Ghetti, B.; et al. Parkinsonian features in hereditary diffuse leukoencephalopathy with spheroids (HDLS) and CSF1R mutations. Park. Relat. Disord. 2013, 19, 869–877. [Google Scholar] [CrossRef] [Green Version]
- Kleinfeld, K.; Mobley, B.; Hedera, P.; Wegner, A.; Sriram, S.; Pawate, S. Adult-onset leukoencephalopathy with neuroaxonal spheroids and pigmented glia: Report of five cases and a new mutation. J. Neurol. 2012, 260, 558–571. [Google Scholar] [CrossRef]
- Kimura, T.; Ishizawa, K.; Mitsufuji, T.; Abe, T.; Nakazato, Y.; Yoshida, K.; Sasaki, A.; Araki, N. A clinicopathological and genetic study of sporadic diffuse leukoencephalopathy with spheroids: A report of two cases. Neuropathol. Appl. Neurobiol. 2013, 39, 837–843. [Google Scholar] [CrossRef] [Green Version]
- Guerreiro, R.; Kara, E.; Le Ber, I.; Bras, J.; Rohrer, J.D.; Taipa, R.; Lashley, T.; Dupuits, C.; Gurunlian, N.; Mochel, F.; et al. Genetic Analysis of Inherited Leukodystrophies. JAMA Neurol. 2013, 70, 875–882. [Google Scholar] [CrossRef] [Green Version]
- Konno, T.; Tada, M.; Koyama, A.; Nozaki, H.; Harigaya, Y.; Nishimiya, J.; Matsunaga, A.; Yoshikura, N.; Ishihara, K.; Arakawa, M.; et al. Haploinsufficiency of CSF-1R and clinicopathologic characterization in patients with HDLS. Neurology 2013, 82, 139–148. [Google Scholar] [CrossRef] [Green Version]
- Kawakami, I.; Iseki, E.; Kasanuki, K.; Minegishi, M.; Sato, K.; Hino, H.; Shibuya, K.; Fujisawa, K.; Higashi, S.; Akiyama, H.; et al. A family with hereditary diffuse leukoencephalopathy with spheroids caused by a novel c.2442+2T>C mutation in the CSF1R gene. J. Neurol. Sci. 2016, 367, 349–355. [Google Scholar] [CrossRef] [PubMed]
- Ikeuchi, T.; Mezaki, N.; Miura, T. Cognitive dysfunction and symptoms of movement disorders in adult-onset leukoencephalopathy with axonal spheroids and pigmented glia. Park. Relat. Disord. 2018, 46, S39–S41. [Google Scholar] [CrossRef] [PubMed]
- Leng, C.; Lu, L.; Wang, G.; Zhang, Y.; Xu, Y.; Lin, X.; Shen, N.; Xu, X.; Qun, S.; Sun, M.; et al. A novel dominant-negative mutation of the CSF1R gene causes adult-onset leukoencephalopathy with axonal spheroids and pigmented glia. Am. J. Transl. Res. 2019, 11, 6093–6101. [Google Scholar] [PubMed]
- Calatayud, T.; Turkalp, Z.T.; Gonzales, A.A.; Muñoz, D.G. Adult-onset leukoencephalopathy with axonal spheroids and pigmented glia: Report on a case with morphometric studies. Clin. Neuropathol. 2013, 32, 492–501. [Google Scholar] [CrossRef]
- Tian, W.-T.; Zhan, F.-X.; Liu, Q.; Luan, X.-H.; Zhang, C.; Shang, L.; Zhang, B.-Y.; Pan, S.-J.; Miao, F.; Hu, J.; et al. Clinicopathologic characterization and abnormal autophagy of CSF1R-related leukoencephalopathy. Transl. Neurodegener. 2019, 8, 32. [Google Scholar] [CrossRef] [Green Version]
- Funayama, M.; Sugihara, M.; Takata, T.; Mimura, M.; Ikeuchi, T. Remarkable behavioural signs and progressive non-fluent aphasia in a patient with adult-onset leucoencephalopathy with axonal spheroids and pigmented glia. Psychogeriatrics 2018, 19, 282–285. [Google Scholar] [CrossRef]
- Di Donato, I.; Stabile, C.; Bianchi, S.; Taglia, I.; Mignarri, A.; Salvatore, S.; Giorgio, E.; Brusco, A.; Simone, I.; Dotti, M.T.; et al. A Novel CSF1R Mutation in a Patient with Clinical and Neuroradiological Features of Hereditary Diffuse Leukoencephalopathy with Axonal Spheroids. J. Alzheimer’s Dis. 2015, 47, 319–322. [Google Scholar] [CrossRef]
- Shu, Y.; Long, L.; Liao, S.; Yang, J.; Li, J.; Qiu, W.; Yang, Y.; Bao, J.; Wu, A.; Hu, X.; et al. Involvement of the optic nerve in mutated CSF1R-induced hereditary diffuse leukoencephalopathy with axonal spheroids. BMC Neurol. 2016, 16, 171. [Google Scholar] [CrossRef] [Green Version]
- Lynch, D.S.; Zhang, W.; Lakshmanan, R.; Kinsella, J.A.; Uzun, G.A.; Karbay, M.; Tufekcioglu, Z.; Hanagasi, H.; Burke, G.; Foulds, N.; et al. Analysis of Mutations in AARS2 in a Series of CSF1R-Negative Patients with Adult-Onset Leukoencephalopathy with Axonal Spheroids and Pigmented Glia. JAMA Neurol. 2016, 73, 1433–1439. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Zhang, X. A novel CSF-1R mutation in a family with hereditary diffuse leukoencephalopathy with axonal spheroids misdiagnosed as hydrocephalus. Neurogenetics 2019, 20, 155–160. [Google Scholar] [CrossRef] [Green Version]
- Mao, C.; Zhou, L.; Zhou, L.; Yang, Y.; Niu, J.; Li, J.; Huang, X.; Ren, H.; Zhao, Y.; Peng, B.; et al. Biopsy histopathology in the diagnosis of adult-onset leukoencephalopathy with axonal spheroids and pigmented glia (ALSP). Neurol. Sci. 2019, 41, 403–409. [Google Scholar] [CrossRef] [PubMed]
- Dong, Q.; Long, L.; Chang, Y.-Y.; Lin, Y.-J.; Liu, M.; Lu, Z.-Q. An adolescence-onset male leukoencephalopathy with remarkable cerebellar atrophy and novel compound heterozygous AARS2 gene mutations: A case report. J. Hum. Genet. 2018, 63, 841–846. [Google Scholar] [CrossRef] [PubMed]
- Van Gerpen, J.A.; Wider, C.; Broderick, D.F.; Dickson, D.W.; Brown, L.A.; Wszolek, Z.K. Insights into the dynamics of hereditary diffuse leukoencephalopathy with axonal spheroids. Neurology 2008, 71, 925–929. [Google Scholar] [CrossRef] [PubMed]
- Sundal, C.; Van Gerpen, J.A.; Nicholson, A.M.; Wider, C.; Shuster, E.A.; Aasly, J.; Spina, S.; Ghetti, B.; Roeber, S.; Garbern, J.; et al. MRI characteristics and scoring in HDLS due to CSF1R gene mutations. Neurology 2012, 79, 566–574. [Google Scholar] [CrossRef] [Green Version]
- Fujioka, S.; Broderick, D.F.; Sundal, C.; Baker, M.C.; Rademakers, R.; Wszolek, Z.K. An adult-onset leukoencephalopathy with axonal spheroids and pigmented glia accompanied by brain calcifications: A case report and a literature review of brain calcifications disorders. J. Neurol. 2013, 260, 2665–2668. [Google Scholar] [CrossRef] [Green Version]
- Bender, B.; Klose, U.; Lindig, T.; Biskup, S.; Nägele, T.; Schöls, L.; Karle, K.N. Imaging features in conventional MRI, spectroscopy and diffusion weighted images of hereditary diffuse leukoencephalopathy with axonal spheroids (HDLS). J. Neurol. 2014, 261, 2351–2359. [Google Scholar] [CrossRef]
- Robinson, J.L.; Suh, E.; Wood, E.M.; Lee, E.B.; Coslett, H.B.; Raible, K.; Lee, V.M.-Y.; Trojanowski, J.Q.; Van Deerlin, V.M. Common neuropathological features underlie distinct clinical presentations in three siblings with hereditary diffuse leukoencephalopathy with spheroids caused by CSF1R p.Arg782His. Acta Neuropathol. Commun. 2015, 3, 42. [Google Scholar] [CrossRef] [Green Version]
- Ayrignac, X.; Nicolas, G.; Carra-Dallière, C.; Hannequin, D.; Labauge, P. Brain Calcifications in Adult-Onset Genetic Leukoencephalopathies. JAMA Neurol. 2017, 74, 1000. [Google Scholar] [CrossRef]
- Codjia, P.; Ayrignac, X.; Mochel, F.; Mouzat, K.; Carra-Dalliere, C.; Castelnovo, G.; Ellie, E.; Etcharry-Bouyx, F.; Verny, C.; Belliard, S.; et al. Adult-Onset Leukoencephalopathy with Axonal Spheroids and Pigmented Glia: An MRI Study of 16 French Cases. Am. J. Neuroradiol. 2018, 39, 1657–1661. [Google Scholar] [CrossRef] [Green Version]
- Konno, T.; Broderick, D.F.; Mezaki, N.; Isami, A.; Kaneda, D.; Tashiro, Y.; Tokutake, T.; Keegan, B.M.; Woodruff, B.K.; Miura, T.; et al. Diagnostic Value of Brain Calcifications in Adult-Onset Leukoencephalopathy with Axonal Spheroids and Pigmented Glia. Am. J. Neuroradiol. 2016, 38, 77–83. [Google Scholar] [CrossRef]
- Abe, T.; Kawarai, T.; Fujita, K.; Sako, W.; Terasawa, Y.; Matsuda, T.; Sakai, W.; Tsukamoto-Miyashiro, A.; Matsui, N.; Izumi, Y.; et al. MR Spectroscopy in Patients with Hereditary Diffuse Leukoencephalopathy with Spheroids and Asymptomatic Carriers of Colony-stimulating Factor 1 Receptor Mutation. Magn. Reson. Med Sci. 2017, 16, 297–303. [Google Scholar] [CrossRef] [Green Version]
- Mendes, A.; Pinto, M.; Vieira, S.; Castro, L.; Carpenter, S. Adult-onset leukodystrophy with axonal spheroids. J. Neurol. Sci. 2010, 297, 40–45. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Saez, E.; Shah, S.; Costa, C.; Fleminger, S.; Connor, S.; Bodi, I. Adult onset leukodystrophy with neuroaxonal spheroids and demyelinating plaque-like lesions. Neuropathology 2011, 32, 285–292. [Google Scholar] [CrossRef] [PubMed]
- Alturkustani, M.; Sharma, M.; Hammond, R.; Ang, L.-C. Adult-Onset Leukodystrophy: Review of 3 Clinicopathologic Phenotypes and a Proposed Classification. J. Neuropathol. Exp. Neurol. 2013, 72, 1090–1103. [Google Scholar] [CrossRef] [PubMed]
- Oyanagi, K.; Kinoshita, M.; Inoue, T.; Nakahara, A.; Tokiwai, M.; Arai, N.; Aoki, N.; Jinnai, K.; Yazawa, I.; Ishihara, K.; et al. Adult onset leukoencephalopathy with axonal spheroids and pigmented glia (ALSP) and Nasu-Hakola disease: Lesion staging and dynamic changes of axons and microglial subsets. Brain Pathol. 2017, 27, 748–769. [Google Scholar] [CrossRef]
- Alturkustani, M.; Keith, J.; Hazrati, L.-N.; Rademakers, R.; Ang, L.-C. Pathologic Staging of White Matter Lesions in Adult-Onset Leukoencephalopathy/Leukodystrophy With Axonal Spheroids. J. Neuropathol. Exp. Neurol. 2015, 74, 233–240. [Google Scholar] [CrossRef] [Green Version]
- Mitsui, J.; Matsukawa, T.; Ishiura, H.; Higasa, K.; Yoshimura, J.; Saito, T.L.; Ahsan, B.; Takahashi, Y.; Goto, J.; Iwata, A.; et al. CSF1R mutations identified in three families with autosomal dominantly inherited leukoencephalopathy. Am. J. Med. Genet. Part B Neuropsychiatr. Genet. 2012, 159B, 951–957. [Google Scholar] [CrossRef]
- Ahmed, R.; Guerreiro, R.; Rohrer, J.D.; Guven, G.; Rossor, M.N.; Hardy, J.; Fox, N.C. A novel A781V mutation in the CSF1R gene causes hereditary diffuse leucoencephalopathy with axonal spheroids. J. Neurol. Sci. 2013, 332, 141–144. [Google Scholar] [CrossRef] [Green Version]
- Karle, K.N.; Biskup, S.; Schüle, R.; Schweitzer, K.J.; Krüger, R.; Bauer, P.; Bender, B.; Nägele, T.; Schöls, L. De novo mutations in hereditary diffuse leukoencephalopathy with axonal spheroids (HDLS). Neurology 2013, 81, 2039–2044. [Google Scholar] [CrossRef]
- Sundal, C.; Baker, M.; Karrenbauer, V.D.K.; Gustavsen, M.; Bedri, S.K.; Glaser, A.; Myhr, K.-M.; Haugarvoll, K.; Zetterberg, H.; Harbo, H.; et al. Hereditary diffuse leukoencephalopathy with spheroids with phenotype of primary progressive multiple sclerosis. Eur. J. Neurol. 2014, 22, 328–333. [Google Scholar] [CrossRef] [Green Version]
- Stabile, C.; Taglia, I.; Battisti, C.; Bianchi, S.; Federico, A. Hereditary diffuse leukoencephalopathy with axonal spheroids (HDLS): Update on molecular genetics. Neurol. Sci. 2016, 37, 1565–1569. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Huang, P.; Tan, Y.; Xiao, Q. A Novel Splicing Mutation in the CSF1R Gene in a Family with Hereditary Diffuse Leukoencephalopathy with Axonal Spheroids. Front. Genet. 2019, 10, 491. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.-J.; Shin, J.-H.; Lee, J.H.; Kim, J.H.; Na, D.L.; Suh, Y.-L.; Hwang, S.J.; Lee, J.-H.; Lee, Y.M.; Shin, M.-J.; et al. Adult-onset leukoencephalopathy with axonal spheroids and pigmented glia linked CSF1R mutation: Report of four Korean cases. J. Neurol. Sci. 2014, 349, 232–238. [Google Scholar] [CrossRef] [PubMed]
- Foulds, N.; Pengelly, R.; Hammans, S.R.; Nicoll, J.; Ellison, D.W.; Ditchfield, A.; Beck, S.; Ennis, S. Adult-Onset Leukoencephalopathy with Axonal Spheroids and Pigmented Glia Caused by a Novel R782G Mutation in CSF1R. Sci. Rep. 2015, 5, 10042. [Google Scholar] [CrossRef] [PubMed]
- Makary, M.S.; Awan, U.; Kisanuki, Y.Y.; Slone, H.W. Adult-onset leukoencephalopathy with axonal spheroids and pigmented glia: Clinical and imaging characteristics. Neuroradiol. J. 2019, 32, 139–142. [Google Scholar] [CrossRef]
- Kraya, T.; Quandt, D.; Pfirrmann, T.; Kindermann, A.; Lampe, L.; Schroeter, M.L.; Kohlhase, J.; Stoevesandt, D.; Hoffmann, K.; Villavicencio-Lorini, P. Functional characterization of a novel CSF1R mutation causing hereditary diffuse leukoencephalopathy with spheroids. Mol. Genet. Genom. Med. 2019, 7, e00595. [Google Scholar] [CrossRef] [Green Version]
- Kondo, Y.; Matsushima, A.; Nagasaki, S.; Nakamura, K.; Sekijima, Y.; Yoshida, K. Factors predictive of the presence of a CSF1R mutation in patients with leukoencephalopathy. Eur. J. Neurol. 2019, 27, 369–375. [Google Scholar] [CrossRef] [Green Version]
- Miura, T.; Mezaki, N.; Konno, T.; Iwasaki, A.; Hara, N.; Miura, M.; Funayama, M.; Unai, Y.; Tashiro, Y.; Okita, K.; et al. Identification and functional characterization of novel mutations including frameshift mutation in exon 4 of CSF1R in patients with adult-onset leukoencephalopathy with axonal spheroids and pigmented glia. J. Neurol. 2018, 265, 2415–2424. [Google Scholar] [CrossRef]
- Chitu, V.; Gökhan, Ş.; Stanley, E.R. Modeling CSF-1 receptor deficiency diseases—How close are we? FEBS J. 2021. [Google Scholar] [CrossRef]
- Oosterhof, N.; Chang, I.J.; Karimiani, E.G.; Kuil, L.E.; Jensen, D.M.; Daza, R.; Young, E.; Astle, L.; van der Linde, H.C.; Shivaram, G.M.; et al. Homozygous Mutations in CSF1R Cause a Pediatric-Onset Leukoencephalopathy and Can Result in Congenital Absence of Microglia. Am. J. Hum. Genet. 2019, 104, 936–947. [Google Scholar] [CrossRef] [Green Version]
- Guo, L.; Bertola, D.R.; Takanohashi, A.; Saito, A.; Segawa, Y.; Yokota, T.; Ishibashi, S.; Nishida, Y.; Yamamoto, G.L.; Franco, J.F.D.S.; et al. Bi-allelic CSF1R Mutations Cause Skeletal Dysplasia of Dysosteosclerosis-Pyle Disease Spectrum and Degenerative Encephalopathy with Brain Malformation. Am. J. Hum. Genet. 2019, 104, 925–935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, D.; Yu, M.; Zhang, W.; Wang, Z.; Yuan, Y. AARS2 Compound Heterozygous Variants in a Case of Adult-Onset Leukoencephalopathy with Axonal Spheroids and Pigmented Glia. J. Neuropathol. Exp. Neurol. 2018, 77, 997–1000. [Google Scholar] [CrossRef] [Green Version]
- Hamatani, M.; Jingami, N.; Tsurusaki, Y.; Shimada, S.; Shimojima, K.; Asada-Utsugi, M.; Yoshinaga, K.; Uemura, N.; Yamashita, H.; Uemura, K.; et al. The first Japanese case of leukodystrophy with ovarian failure arising from novel compound heterozygous AARS2 mutations. J. Hum. Genet. 2016, 61, 899–902. [Google Scholar] [CrossRef]
- Szpisjak, L.; Zsindely, N.; Engelhardt, J.I.; Vecsei, L.; Kovacs, G.G.; Klivenyi, P. Novel AARS2 gene mutation producing leukodystrophy: A case report. J. Hum. Genet. 2016, 62, 329–333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.-M.; Yang, H.-J.; Kwon, J.-H.; Kim, W.-J.; Kim, S.-Y.; Lee, E.-M.; Park, J.-Y.; Weon, Y.C.; Park, S.H.; Gwon, B.-J.; et al. Two Korean siblings with recently described ovarioleukodystrophy related to AARS2 mutations. Eur. J. Neurol. 2017, 24, e21–e22. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, S.; Butala, A.; Mahida, S.; Richter, J.; Mu, W.; Poretti, A.; Vernon, H.; VanGerpen, J.; Atwal, P.S.; Middlebrooks, E.H.; et al. Expansion of the clinical spectrum associated with AARS2-related disorders. Am. J. Med Genet. Part A 2019, 179, 1556–1564. [Google Scholar] [CrossRef] [PubMed]
- Sundal, C.; Carmona, S.; Yhr, M.; Almström, O.; Ljungberg, M.; Hardy, J.; Hedberg-Oldfors, C.; Fred, Å.; Bras, J.; Oldfors, A.; et al. An AARS variant as the likely cause of Swedish type hereditary diffuse leukoencephalopathy with spheroids. Acta Neuropathol. Commun. 2019, 7, 188. [Google Scholar] [CrossRef]
- Peragallo, J.H.; Keller, S.; Van Der Knaap, M.S.; Soares, B.P.; Shankar, S.P. Retinopathy and optic atrophy: Expanding the phenotypic spectrum of pathogenic variants in the AARS2 gene. Ophthalmic Genet. 2017, 39, 99–102. [Google Scholar] [CrossRef]
- Kuo, M.E.; Antonellis, A.; Shakkottai, V.G. Alanyl-tRNA Synthetase 2 (AARS2)-Related Ataxia Without Leukoencephalopathy. Cerebellum 2019, 19, 154–160. [Google Scholar] [CrossRef]
- Tang, Y.; Qin, Q.; Xing, Y.; Guo, D.; Di, L.; Jia, J. AARS 2 leukoencephalopathy: A new variant of mitochondrial encephalomyopathy. Mol. Genet. Genom. Med. 2019, 7, e00582. [Google Scholar] [CrossRef] [Green Version]
- Götz, A.; Tyynismaa, H.; Euro, L.; Ellonen, P.; Hyötyläinen, T.; Ojala, T.; Hämäläinen, R.H.; Tommiska, J.; Raivio, T.; Oresic, M.; et al. Exome Sequencing Identifies Mitochondrial Alanyl-tRNA Synthetase Mutations in Infantile Mitochondrial Cardiomyopathy. Am. J. Hum. Genet. 2011, 88, 635–642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mazurova, S.; Magner, M.; Vidrová, V.K.; Vondráčková, A.; Stranecky, V.; Pristoupilova, A.; Zamecnik, J.; Hansikova, H.; Zeman, J.; Tesarova, M.; et al. Thymidine kinase 2 and alanyl-tRNA synthetase 2 deficiencies cause lethal mitochondrial cardiomyopathy: Case reports and review of the literature. Cardiol. Young 2016, 27, 936–944. [Google Scholar] [CrossRef] [PubMed]
- Sommerville, E.W.; Zhou, X.-L.; Oláhová, M.; Jenkins, J.; Euro, L.; Konovalova, S.; Hilander, T.; Pyle, A.; He, L.; Habeebu, S.; et al. Instability of the mitochondrial alanyl-tRNA synthetase underlies fatal infantile-onset cardiomyopathy. Hum. Mol. Genet. 2018, 28, 258–268. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.J.; Nam, D.E.; Choi, Y.J.; Nam, S.H.; Choi, B.-O.; Chung, K.W. Alanyl-tRNA synthetase 1 (AARS1) gene mutation in a family with intermediate Charcot-Marie-Tooth neuropathy. Genes Genom. 2020, 42, 663–672. [Google Scholar] [CrossRef] [PubMed]
- Marten, L.M.; Brinkert, F.; Smith, D.E.C.; Prokisch, H.; Hempel, M.; Santer, R. Recurrent acute liver failure in alanyl-tRNA synthe-tase-1 (AARS1) deficiency. Mol. Genet. Metab. Rep. 2020, 25, 100681. [Google Scholar] [CrossRef]
- Pixley, F.; Stanley, E.R. CSF-1 regulation of the wandering macrophage: Complexity in action. Trends Cell Biol. 2004, 14, 628–638. [Google Scholar] [CrossRef]
- Erblich, B.; Zhu, L.; Etgen, A.M.; Dobrenis, K.; Pollard, J.W. Absence of Colony Stimulation Factor-1 Receptor Results in Loss of Microglia, Disrupted Brain Development and Olfactory Deficits. PLoS ONE 2011, 6, e26317. [Google Scholar] [CrossRef] [Green Version]
- Chitu, V.; Gokhan, S.; Gulinello, M.; Branch, C.A.; Patil, M.; Basu, R.; Stoddart, C.; Mehler, M.F.; Stanley, E.R. Phenotypic characterization of a Csf1r haploinsufficient mouse model of adult-onset leukodystrophy with axonal spheroids and pigmented glia (ALSP). Neurobiol. Dis. 2014, 74, 219–228. [Google Scholar] [CrossRef] [Green Version]
- Hamatani, M.; Yamashita, H.; Ochi, H.; Ashida, S.; Hashi, Y.; Okada, Y.; Fujii, C.; Kawamura, K.; Kitazawa, R.; Nakagawa, M.; et al. Altered features of monocytes in adult onset leukoencephalopathy with axonal spheroids and pigmented glia: A clue to the pathomechanism of microglial dyshomeostasis. Neurobiol. Dis. 2020, 140, 104867. [Google Scholar] [CrossRef]
- Oosterhof, N.; Kuil, L.E.; van der Linde, H.C.; Burm, S.M.; Berdowski, W.; van Ijcken, W.F.; van Swieten, J.C.; Hol, E.M.; Verheijen, M.H.; van Ham, T.J. Colony-Stimulating Factor 1 Receptor (CSF1R) Regulates Microglia Density and Distribution, but Not Microglia Differentiation In Vivo. Cell Rep. 2018, 24, 1203–1217.e6. [Google Scholar] [CrossRef] [Green Version]
- Perry, V.H.; Nicoll, J.A.R.; Holmes, C. Microglia in neurodegenerative disease. Nat. Rev. Neurol. 2010, 6, 193–201. [Google Scholar] [CrossRef] [PubMed]
- Prinz, M.; Priller, J. Microglia and brain macrophages in the molecular age: From origin to neuropsychiatric disease. Nat. Rev. Neurosci. 2014, 15, 300–312. [Google Scholar] [CrossRef] [PubMed]
- Lakshmanan, R.; Adams, M.E.; Lynch, D.S.; Kinsella, J.A.; Phadke, R.; Schott, J.M.; Murphy, E.; Rohrer, J.D.; Chataway, J.; Houlden, H.; et al. Redefining the phenotype of ALSP and AARS2 mutation–related leukodystrophy. Neurol. Genet. 2017, 3, e135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.-I.; Jeon, B.; Bae, J.; Won, J.K.; Kim, H.-J.; Yim, J.; Kim, Y.J.; Park, S.-H. An Autopsy Proven Case of CSF1R-mutant Adult-onset Leukoencephalopathy with Axonal Spheroids and Pigmented Glia (ALSP) with Premature Ovarian Failure. Exp. Neurobiol. 2019, 28, 119–129. [Google Scholar] [CrossRef]
- Adams, S.J.; Kirk, A.; Auer, R.N. Adult-onset leukoencephalopathy with axonal spheroids and pigmented glia (ALSP): Integrating the literature on hereditary diffuse leukoencephalopathy with spheroids (HDLS) and pigmentary orthochromatic leukodystrophy (POLD). J. Clin. Neurosci. 2018, 48, 42–49. [Google Scholar] [CrossRef] [PubMed]
- Keegan, B.M.; Giannini, C.; Parisi, J.E.; Lucchinetti, C.F.; Boeve, B.F.; Josephs, K.A. Sporadic adult-onset leukoencephalopathy with neuroaxonal spheroids mimicking cerebral MS. Neurology 2008, 70, 1128–1133. [Google Scholar] [CrossRef]
- Levin, J.; Tiedt, S.; Arzberger, T.; Biskup, S.; Schuberth, M.; Stenglein-Krapf, G.; Kreth, F.-W.; Högen, T.; la Fougère, C.; Linn, J.; et al. Diffuse leukoencephalopathy with spheroids: Biopsy findings and a novel mutation. Clin. Neurol. Neurosurg. 2014, 122, 113–115. [Google Scholar] [CrossRef]
- Fernández-Vega, I.; De Heredia-Goñi, K.P.; Santos-Juanes, J.; Imizcoz, M.G.; Zaldumbide, L.; Zarranz, J.J.; Ferrer, I. Sporadic adult-onset leucodystrophy with axonal spheroids and pigmented glia with no mutations in the known targeted genes. Histopathology 2015, 68, 308–312. [Google Scholar] [CrossRef]
- Wang, Y.-L.; Wang, F.-Z.; Li, R.; Jiang, J.; Liu, X.; Xu, J. Recent Advances in Basic Research for CSF1R-Microglial Encephalopathy. Front. Aging Neurosci. 2021, 13, 792840. [Google Scholar] [CrossRef]
- Terayama, K. Two cases of cystic bone disease showing peculiar features. Nippon Seikeigeka Gakkai Zasshi 1961, 35, 626. [Google Scholar]
- Hakola, H.P.A.; Järvi, O.H.; Sourander, P. Osteodysplasia polycystica hereditaria combined with sclerosing leucoencephalopathy. Acta Neurol. Scand. 2009, 46, 79–80. [Google Scholar] [CrossRef]
- Hakola, H.P. Neuropsychiatric and genetic aspects of a new hereditary disease characterized by progressive dementia and lipomembranous polycystic osteodysplasia. Acta Psychiatr. Scand. Suppl. 1972, 232, 1–73. [Google Scholar] [PubMed]
- Hakola, H.P.A.; Iivanainen, M. A new hereditary disease with progressive dementia and polycystic osteodysplasia: Neuroradiological analysis of seven cases. Neuroradiology 1973, 6, 162–168. [Google Scholar] [CrossRef] [PubMed]
- Nasu, T.; Tsukahara, Y.; Terayama, K. A lipid metabolic disease—“Membranous lipodystrophy”—An autopsy case demonstrating numerous peculiar membrane-structures composed of compound lipid in bone and bone marrow and various adipose tissues. Acta Pathol. Jpn. 1973, 23, 539–558. [Google Scholar] [CrossRef] [PubMed]
- Harada, K. A case of “membranous lipodystrophy (Nasu)” with emphasis on psychiatric and neuropathologic aspects. Folia Psychiatr. Neurol. Jpn. 1975, 29, 169–177. [Google Scholar] [PubMed]
- Hakola, P.; Virtama, P. Radiologic bone changes of polycystic lipomembranous osteodysplasia with sclerosing leukoencephalopathy. Skelet. Radiol. 1982, 8, 51–54. [Google Scholar] [CrossRef]
- Minagawa, M.; Maeshiro, H.; Shioda, K.; Hirano, A. Membranous lipodystrophy (Nasu disease): Clinical and neuropathological study of a case. Clin. Neuropathol. 1985, 4, 38–45. [Google Scholar]
- Hakola, H.P. Polycystic lipomembranous osteodysplasia with sclerosing leukoencephalopathy (membranous lipodystrophy): A neuropsychiatric follow-up study. In Monographs of Psychiatria Fennica; Henriksson, M., Huttunen, M., Kuoppasalmi, K., Lindfors, O., Lonnqvist, J., Eds.; Foundation for Psychiatric Research: Helsinki, Finland, 1990; pp. 1–114. [Google Scholar]
- Ishigooka, M.; Hashimoto, T.; Izumiya, K.; Kodama, C.; Nakada, T. Membranous Lipodystrophy (Nasu’s Disease): A Rare Cause of Neuropathic Urinary Incontinence. Urol. Int. 1993, 50, 179–181. [Google Scholar] [CrossRef]
- Verloes, A.; Maquet, P.; Sadzot, B.; Vivario, M.; Thiry, A.; Franck, G. Nasu-Hakola syndrome: Polycystic lipomembranous osteodysplasia with sclerosing leucoencephalopathy and presenile dementia. J. Med. Genet. 1997, 34, 753–757. [Google Scholar] [CrossRef] [Green Version]
- Bm, J.P.; Autti, T.; Raininko, R.; Partanen, J.; Salonen, O.; Puranen, M.; Hakola, P.; Haltia, M. CNS manifestations of Nasu-Hakola disease: A frontal dementia with bone cysts. Neurology 2001, 56, 1552–1558. [Google Scholar] [CrossRef]
- Bianchin, M.M.; Capella, H.M.; Chaves, D.L.; Steindel, M.; Grisard, E.C.; Ganev, G.G.; da Silva Júnior, J.P.; Neto Evaldo, S.; Poffo, M.A.; Walz, R.; et al. Nasu-Hakola disease (polycystic lipomembranous osteodysplasia with sclerosing leukoencephalopathy—PLOSL): A dementia associated with bone cystic lesions. From clinical to genetic and molecular aspects. Cell Mol. Neurobiol. 2004, 24, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Kaneko, M.; Sano, K.; Nakayama, J.; Amano, N. Nasu-Hakola disease: The first case reported by Nasu and review: The 50th An-niversary of Japanese Society of Neuropathology. Neuropathology 2010, 30, 463–470. [Google Scholar] [CrossRef] [PubMed]
- Ilonen, T.; Hakola, P.; Vanhanen, M.; Tiihonen, J. Rorschach assessment of personality functioning in patients with polycystic lipomembranous osteodysplasia with sclerosing leukoencephalopathy. Acta Neuropsychiatr. 2012, 24, 236–244. [Google Scholar] [CrossRef] [PubMed]
- Paloneva, J.; Autti, T.; Hakola, P.; Haltia, M.J. Polycystic lipomembranous osteodysplasia with sclerosing leukoencephalopathy (PLOSL). In GeneReviews® [Internet]; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Stephens, K., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 2020. [Google Scholar]
- Kitajima, I.; Kuriyama, M.; Usuki, F.; Izumo, S.; Osame, M.; Suganuma, T.; Murata, F.; Nagamatsu, K. Nasu-Hakola disease (membranous lipodystrophy): Clinical, histopathological and biochemical studies of three cases. J. Neurol. Sci. 1989, 91, 35–52. [Google Scholar] [CrossRef]
- Montalbetti, L.; Ratti, M.T.; Greco, B.; Aprile, C.; Moglia, A.; Soragna, D. Neuropsychological tests and functional nuclear neuroim-aging provide evidence of subclinical impairment in Nasu-Hakola disease heterozygotes. Funct. Neurol. 2005, 20, 71–75. [Google Scholar]
- Chouery, E.; Delague, V.; Bergougnoux, A.; Koussa, S.; Serre, J.-L.; Mégarbané, A. Mutations in TREM2 lead to pure early-onset dementia without bone cysts. Hum. Mutat. 2008, 29, E194–E204. [Google Scholar] [CrossRef]
- Guerreiro, R.; Bilgic, B.; Guven, G.; Bras, J.; Rohrer, J.; Lohmann, E.; Hanagasi, H.; Gürvit, H.; Emre, M. A novel compound heterozygous mutation in TREM2 found in a Turkish frontotemporal dementia-like family. Neurobiol. Aging 2013, 34, 2890.e1–2890.e5. [Google Scholar] [CrossRef]
- Guerreiro, R.J.; Lohmann, E.; Bras, J.; Gibbs, J.R.; Rohrer, J.; Gurunlian, N.; Dursun, B.; Bilgic, B.; Hanagasi, H.; Gürvit, H.; et al. Using Exome Sequencing to Reveal Mutations in TREM2 Presenting as a Frontotemporal Dementia–like Syndrome Without Bone Involvement. JAMA Neurol. 2013, 70, 78–84. [Google Scholar] [CrossRef] [Green Version]
- Bock, V.; Botturi, A.; Gaviani, P.; Lamperti, E.; Maccagnano, C.; Piccio, L.; Silvani, A.; Salmaggi, A. Polycystic Lipomembranous Osteodysplasia with Sclerosing Leukoencephalopathy (PLOSL): A new report of an Italian woman and review of the literature. J. Neurol. Sci. 2013, 326, 115–119. [Google Scholar] [CrossRef]
- Le Ber, I.; De Septenville, A.; Guerreiro, R.; Bras, J.; Camuzat, A.; Caroppo, P.; Lattante, S.; Couarch, P.; Kabashi, E.; Bouya-Ahmed, K.; et al. Homozygous TREM2 mutation in a family with atypical frontotemporal dementia. Neurobiol. Aging 2014, 35, 2419.e23–2419.e25. [Google Scholar] [CrossRef] [Green Version]
- Matsuo, T.; Suetsugu, M.; Eguchi, M.; Sasaki, M.; Tsuneyoshi, M. Membranous lipodystrophy. A case report. Arch Psychiatr. Nervenkr. 1982, 231, 123–130. [Google Scholar] [CrossRef] [PubMed]
- Nwawka, O.K.; Schneider, R.; Bansal, M.; Mintz, U.N.; Lane, J. Membranous lipodystrophy: Skeletal findings on CT and MRI. Skelet. Radiol. 2014, 43, 1449–1455. [Google Scholar] [CrossRef] [PubMed]
- Hakola, H.P.; Partanen, V.S. Neurophysiological findings in the hereditary presenile dementia characterised by polycystic lipomembranous osteodysplasia and sclerosing leukoencephalopathy. J. Neurol. Neurosurg. Psychiatry 1983, 46, 515–520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bird, T.D.; Koerker, R.M.; Leaird, B.J.; Vlcek, B.W.; Thorning, D.R. Lipomembranous polycystic osteodysplasia (brain, bone, and fat disease): A genetic cause of presenile dementia. Neurology 1983, 33, 81. [Google Scholar] [CrossRef] [PubMed]
- Shibata, K.; Uchiyama, S.; Takeuchi, M.; Kobayashi, I.; Maruyama, S. A case of membranous lipodystrophy (Nasu) with diffuse cerebral, white matter involvement and cerebellar atrophy on brain CT and NM (in Japanese). Rinsho Shinkeigaku 1990, 30, 1232–1237. [Google Scholar]
- Araki, T.; Ohba, H.; Monzawa, S.; Sakuyama, K.; Hachiya, J.; Seki, T.; Takahashi, Y.; Yamaguchi, M. Membranous lipodystrophy: MR imaging appearance of the brain. Radiology 1991, 180, 793–797. [Google Scholar] [CrossRef]
- Coomans, C.; Sieben, A.; Lammens, M.; Groote, C.C.-D.; Vandenbroecke, C.; Goethals, I.; Van Melkebeke, D.; Hemelsoet, D. Early-onset dementia, leukoencephalopathy and brain calcifications: A clinical, imaging and pathological comparison of ALSP and PLOSL/Nasu Hakola disease. Acta Neurol. Belg. 2018, 118, 607–615. [Google Scholar] [CrossRef]
- Takeshita, T.; Kaminaga, T.; Tatsumi, T.; Hatanaka, Y.; Furui, S. Regional cerebral blood flow in a patient with Nasu-Hakola disease. Ann. Nucl. Med. 2005, 19, 309–312. [Google Scholar] [CrossRef]
- Klunemann, H.H.; Ridha, B.H.; Magy, L.; Wherrett, J.R.; Hemelsoet, D.; Keen, R.W.; De Bleecker, J.L.; Rossor, M.; Marienhagen, J.; Klein, H.E.; et al. The genetic causes of basal ganglia calcification, dementia, and bone cysts: DAP12 and TREM2. Neurology 2005, 64, 1502–1507. [Google Scholar] [CrossRef]
- Sageshima, M.; Masuda, H.; Kawamura, K.; Shozawa, T. Membranous lipodystrophy. Light and electron microscopic study of a biopsy case. Acta Pathol. Jpn. 1987, 37, 281–290. [Google Scholar]
- Machinami, R. Degenerative change of adipose tissue; the so-called membranous lipodystrophy. Virchows Arch. 1990, 416, 373–374. [Google Scholar] [CrossRef] [PubMed]
- Mii, Y.; Miyauchi, Y.; Yoshikawa, T.; Honoki, K.; Aoki, M.; Tsutsumi, M.; Maruyama, H.; Funauchi, M.; Konishi, Y.; Tamai, S. Ultrastructural lipid and glycoconjugate cytochemistry of membranous lipodystrophy (Nasu-Hakola disease). Virchows Arch. 1991, 419, 137–142. [Google Scholar] [CrossRef] [PubMed]
- Shboul, M.; Roschger, P.; Ganger, R.; Paschalis, L.; Rokidi, S.; Zandieh, S.; Behunova, J.; Muschitz, C.; Fahrleitner-Pammer, A.; Ng, A.Y.J.; et al. Bone matrix hypermineralization associated with low bone turnover in a case of Nasu-Hakola disease. Bone 2018, 123, 48–55. [Google Scholar] [CrossRef]
- Yagishita, S.; Ito, Y.; Sakai, H.; Amano, N. Membranocystic lesions of the lung in Nasu-Hakola disease. Virchows Arch. 1985, 408, 211–217. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, J. Nasu-Hakola disease: A review of its leukoencephalopathic and membranolipodystrophic features. Neuropathology 2000, 20, 25–29. [Google Scholar] [CrossRef] [PubMed]
- Aoki, N.; Tsuchiya, K.; Togo, T.; Kobayashi, Z.; Uchikado, H.; Katsuse, O.; Suzuki, K.; Fujishiro, H.; Arai, T.; Iseki, E.; et al. Gray matter lesions in Nasu-Hakola disease: A report on three autopsy cases. Neuropathology 2010, 31, 135–143. [Google Scholar] [CrossRef] [PubMed]
- Kalimo, H.; Sourander, P.; Jarvi, O.; Hakola, P. Vascular changes and blood-brain barrier damage in the pathogenesis of polycystic lipomembranous osteodysplasia with sclerosing leukoencephalopathy (membranous lipodystrophy). Acta. Neurol. Scand. 1994, 89, 353–356. [Google Scholar] [CrossRef]
- Matsushita, M.; Oyanagi, S.; Hanawa, S.; Shiraki, H.; Kosaka, K. Nasu-Hakola’s disease (membranous lipodystrophy). Acta Neuropathol. 1981, 54, 89–93. [Google Scholar] [CrossRef]
- Miyazu, K.; Kobayashi, K.; Fukutani, Y.; Nakamura, I.; Hasegawa, H.; Yamaguchi, N.; Saitoh, T. Membranous lipodystrophy (Nasu-Hakola disease) with thalamic degeneration: Report of an autopsied case. Acta Neuropathol. 1991, 82, 414–419. [Google Scholar] [CrossRef]
- Satoh, J.-I.; Kino, Y.; Yanaizu, M.; Saito, Y. Alzheimer’s disease pathology in Nasu-Hakola disease brains. Intractable Rare Dis. Res. 2018, 7, 32–36. [Google Scholar] [CrossRef] [Green Version]
- Maderna, E.; Visonà, S.; Bolcato, V.; Redaelli, V.; Caroppo, P.; Montalbetti, L.; Giaccone, G.; Osculati, A. Neuropathological Alzheimer’s Disease Lesions in Nasu-Hakola Disease with TREM2 Mutation: Atypical Distribution of Neurofibrillary Changes. J. Alzheimer’s Dis. 2021, 79, 25–30. [Google Scholar] [CrossRef] [PubMed]
- Yokoi, S.; Suzuki, K.; Amano, N.; Yagishita, S. Fatty Acid Analysis of Galactolipids and Ganglioside in the Brains of Four Cases of Nasu-Hakola Disease. Psychiatry Clin. Neurosci. 1989, 43, 695–701. [Google Scholar] [CrossRef] [PubMed]
- Iannaccone, S.; Ferini-Strambi, L.; Nemni, R.; Marchettini, P.; Corbo, M.; Pinto, P.; Smirne, S. Pheripheral motor-sensory neuropathy in membranous lipodystrophy (Nasu’s disease): A case report. Clin. Neuropathol. 1992, 11, 49–53. [Google Scholar] [PubMed]
- Paloneva, J.; Kestilä, M.; Wu, J.; Salminen, A.; Böhling, T.; Ruotsalainen, V.; Hakola, P.; Bakker, A.B.; Phillips, J.H.; Pekkarinen, P.; et al. Loss-of-function mutations in TYROBP (DAP12) result in a presenile dementia with bone cysts. Nat. Genet. 2000, 25, 357–361. [Google Scholar] [CrossRef] [PubMed]
- Paloneva, J.; Manninen, T.; Christman, G.; Hovanes, K.; Mandelin, J.; Adolfsson, R.; Bianchin, M.; Bird, T.; Miranda, R.; Salmaggi, A.; et al. Mutations in Two Genes Encoding Different Subunits of a Receptor Signaling Complex Result in an Identical Disease Phenotype. Am. J. Hum. Genet. 2002, 71, 656–662. [Google Scholar] [CrossRef] [Green Version]
- Kondo, T.; Takahashi, K.; Kohara, N.; Hayashi, S.; Matsuo, H.; Yamazaki, M.; Inoue, K.; Miyamoto, K.; Yamamura, T. Heterogeneity of presenile dementia with bone cysts (Nasu-Hakola disease): Three genetic forms. Neurology 2002, 59, 1105–1107. [Google Scholar] [CrossRef]
- Soragna, D.; Tupler, R.; Ratti, M.T.; Montalbetti, L.; Papi, L.; Sestini, R. An Italian family affected by Nasu-Hakola disease with a novel genetic mutation in the TREM2 gene. J. Neurol. Neurosurg. Psychiatry 2003, 74, 825–826. [Google Scholar] [CrossRef] [Green Version]
- Numasawa, Y.; Yamaura, C.; Ishihara, S.; Shintani, S.; Yamazaki, M.; Tabunoki, H.; Satoh, J.-I. Nasu-Hakola disease with a splicing mutation of TREM2 in a Japanese family. Eur. J. Neurol. 2010, 18, 1179–1183. [Google Scholar] [CrossRef]
- Dardiotis, E.; Siokas, V.; Pantazi, E.; Dardioti, M.; Rikos, D.; Xiromerisiou, G.; Markou, A.; Papadimitriou, D.; Speletas, M.; Hadjigeorgiou, G.M. A novel mutation in TREM2 gene causing Nasu-Hakola disease and review of the literature. Neurobiol. Aging 2017, 53, 194.e13–194.e22. [Google Scholar] [CrossRef]
- Kober, D.L.; Brett, T.J. TREM2-Ligand Interactions in Health and Disease. J. Mol. Biol. 2017, 429, 1607–1629. [Google Scholar] [CrossRef]
- Olcese, L.; Cambiaggi, A.; Semenzato, G.; Bottino, C.; Moretta, A.; Vivier, E. Killer-cell activatory receptors for MHC class I mole-cules are included in a multimeric complex expressed by human killer cells. J. Immunol. 1997, 158, 5083–5086. [Google Scholar] [PubMed]
- Lanier, L.L.; Corliss, B.C.; Wu, J.; Leong, C.; Phillips, J.H. Immunoreceptor DAP12 bearing a tyrosine-based activation motif is involved in activating NK cells. Nature 1998, 391, 703–707. [Google Scholar] [CrossRef] [PubMed]
- Tomasello, E.; Olcese, L.; Vély, F.; Geourgeon, C.; Bléry, M.; Moqrich, A.; Gautheret, D.; Djabali, M.; Mattei, M.-G.; Vivier, E. Gene Structure, Expression Pattern, and Biological Activity of Mouse Killer Cell Activating Receptor-associated Protein (KARAP)/DAP-12. J. Biol. Chem. 1998, 273, 34115–34119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campbell, K.S.; Cella, M.; Carretero, M.; Lopez-Botet, M.; Colonna, M. Signaling through human killer cell activating receptors triggers tyrosine phosphorylation of an associated protein complex. Eur. J. Immunol. 1998, 28, 599–609. [Google Scholar] [CrossRef]
- Tomasello, E.; Vivier, E. KARAP/DAP12/TYROBP: Three names and a multiplicity of biological functions. Eur. J. Immunol. 2005, 35, 1670–1677. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, M.B.; Xing, J.; Titus, A.R. The TREM2-DAP12 signaling pathway in Nasu–Hakola disease: A molecular genetics perspective. Res. Rep. Biochem. 2015, 5, 89–100. [Google Scholar] [CrossRef] [Green Version]
- Satoh, J.-I.; Tabunoki, H.; Ishida, T.; Yagishita, S.; Jinnai, K.; Futamura, N.; Kobayashi, M.; Toyoshima, I.; Yoshioka, T.; Enomoto, K.; et al. Phosphorylated Syk expression is enhanced in Nasu-Hakola disease brains. Neuropathology 2012, 32, 149–157. [Google Scholar] [CrossRef]
- Cella, M.; Buonsanti, C.; Strader, C.; Kondo, T.; Salmaggi, A.; Colonna, M. Impaired Differentiation of Osteoclasts in TREM-2–deficient Individuals. J. Exp. Med. 2003, 198, 645–651. [Google Scholar] [CrossRef]
- Paloneva, J.; Mandelin, J.; Kiialainen, A.; Böhling, T.; Prudlo, J.; Hakola, P.; Haltia, M.; Konttinen, Y.T.; Peltonen, L. DAP12/TREM2 Deficiency Results in Impaired Osteoclast Differentiation and Osteoporotic Features. J. Exp. Med. 2003, 198, 669–675. [Google Scholar] [CrossRef] [Green Version]
- Kaifu, T.; Nakahara, J.; Inui, M.; Mishima, K.; Momiyama, T.; Kaji, M.; Sugahara, A.; Koito, H.; Ujike-Asai, A.; Nakamura, A.; et al. Osteopetrosis and thalamic hypomyelinosis with synaptic degeneration in DAP12-deficient mice. J. Clin. Investig. 2003, 111, 323–332. [Google Scholar] [CrossRef] [Green Version]
- Humphrey, M.B.; Daws, M.; Spusta, S.C.; Niemi, E.C.; Torchia, J.A.; Lanier, L.L.; Seaman, W.E.; Nakamura, M.C. TREM2, a DAP12-Associated Receptor, Regulates Osteoclast Differentiation and Function. J. Bone Miner. Res. 2005, 21, 237–245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nataf, S.; Anginot, A.; Vuaillat, C.; Malaval, L.; Fodil, N.; Chereul, E.; Langlois, J.-B.; Dumontel, C.; Cavillon, G.; Confavreux, C.; et al. Brain and Bone Damage in KARAP/DAP12 Loss-of-Function Mice Correlate with Alterations in Microglia and Osteoclast Lineages. Am. J. Pathol. 2005, 166, 275–286. [Google Scholar] [CrossRef] [Green Version]
- Thrash, C.; Torbett, B.E.; Carson, M.J. Developmental Regulation of TREM2 and DAP12 Expression in the Murine CNS: Implications for Nasu-Hakola Disease. Neurochem. Res. 2008, 34, 38–45. [Google Scholar] [CrossRef] [PubMed]
- Colonna, M. TREMs in the immune system and beyond. Nat. Rev. Immunol. 2003, 3, 445–453. [Google Scholar] [CrossRef]
- Ford, J.W.; McVicar, D.W. TREM and TREM-like receptors in inflammation and disease. Curr. Opin. Immunol. 2009, 21, 38–46. [Google Scholar] [CrossRef] [Green Version]
- Jiang, T.; Zhang, Y.-D.; Chen, Q.; Gao, Q.; Zhu, X.-C.; Zhou, J.-S.; Shi, J.-Q.; Lu, H.; Tan, L.; Yu, J.-T. TREM2 modifies microglial phenotype and provides neuroprotection in P301S tau transgenic mice. Neuropharmacology 2016, 105, 196–206. [Google Scholar] [CrossRef]
- Mazaheri, F.; Snaidero, N.; Kleinberger, G.; Madore, C.; Daria, A.; Werner, G.; Krasemann, S.; Capell, A.; Trümbach, D.; Wurst, W.; et al. TREM 2 deficiency impairs chemotaxis and microglial responses to neuronal injury. EMBO Rep. 2017, 18, 1186–1198. [Google Scholar] [CrossRef]
- Brownjohn, P.W.; Smith, J.; Solanki, R.; Lohmann, E.; Houlden, H.; Hardy, J.; Dietmann, S.; Livesey, F.J. Functional Studies of Missense TREM2 Mutations in Human Stem Cell-Derived Microglia. Stem Cell Rep. 2018, 10, 1294–1307. [Google Scholar] [CrossRef] [Green Version]
- Satoh, J.-I.; Tabunoki, H.; Ishida, T.; Yagishita, S.; Jinnai, K.; Futamura, N.; Kobayashi, M.; Toyoshima, I.; Yoshioka, T.; Enomoto, K.; et al. Immunohistochemical characterization of microglia in Nasu-Hakola disease brains. Neuropathology 2011, 31, 363–375. [Google Scholar] [CrossRef]
- Satoh, J.-I.; Kino, Y.; Motohashi, N.; Ishida, T.; Yagishita, S.; Jinnai, K.; Arai, N.; Nakamagoe, K.; Tamaoka, A.; Saito, Y.; et al. Immunohistochemical characterization of CD33 expression on microglia in Nasu-Hakola disease brains. Neuropathology 2015, 35, 529–537. [Google Scholar] [CrossRef]
- Sasaki, A.; Kakita, A.; Yoshida, K.; Konno, T.; Ikeuchi, T.; Hayashi, S.; Matsuo, H.; Shioda, K. Variable expression of microglial DAP12 and TREM2 genes in Nasu-Hakola disease. Neurogenetics 2015, 16, 265–276. [Google Scholar] [CrossRef] [PubMed]
- Satoh, J.-I.; Kino, Y.; Yanaizu, M.; Tosaki, Y.; Sakai, K.; Ishida, T.; Saito, Y. Expression of gp91phox and p22phox, catalytic subunits of NADPH oxidase, on microglia in Nasu-Hakola disease brains. Intractable Rare Dis. Res. 2016, 5, 275–279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohgidani, M.; Kato, T.A.; Setoyama, D.; Sagata, N.; Hashimoto, R.; Shigenobu, K.; Yoshida, T.; Hayakawa, K.; Shimokawa, N.; Miura, D.; et al. Direct induction of ramified microglia-like cells from human monocytes: Dynamic microglial dysfunction in Nasu-Hakola disease. Sci. Rep. 2014, 4, 4957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jadhav, V.S.; Lin, P.B.C.; Pennington, T.; Di Prisco, G.V.; Jannu, A.J.; Xu, G.; Moutinho, M.; Zhang, J.; Atwood, B.K.; Puntambekar, S.S.; et al. Trem2 Y38C mutation and loss of Trem2 impairs neuronal synapses in adult mice. Mol. Neurodegener. 2020, 15, 62. [Google Scholar] [CrossRef]
- Satoh, J.-I.; Kino, Y.; Yanaizu, M.; Tosaki, Y.; Sakai, K.; Ishida, T.; Saito, Y. Expression of GPR17, a regulator of oligodendrocyte differentiation and maturation, in Nasu-Hakola disease brains. Intractable Rare Dis. Res. 2017, 6, 50–54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Satoh, J.-I.; Motohashi, N.; Kino, Y.; Ishida, T.; Yagishita, S.; Jinnai, K.; Arai, N.; Nakamagoe, K.; Tamaoka, A.; Saito, Y.; et al. LC3, an autophagosome marker, is expressed on oligodendrocytes in Nasu-Hakola disease brains. Orphanet J. Rare Dis. 2014, 9, 68. [Google Scholar] [CrossRef] [Green Version]
- Roumier, A.; Béchade, C.; Poncer, J.-C.; Smalla, K.-H.; Tomasello, E.; Vivier, E.; Gundelfinger, E.D.; Triller, A.; Bessis, A. Impaired Synaptic Function in the Microglial KARAP/DAP12-Deficient Mouse. J. Neurosci. 2004, 24, 11421–11428. [Google Scholar] [CrossRef] [Green Version]
- Filipello, F.; Morini, R.; Corradini, I.; Zerbi, V.; Canzi, A.; Michalski, B.; Erreni, M.; Markicevic, M.; Starvaggi-Cucuzza, C.; Otero, K.; et al. The Microglial Innate Immune Receptor TREM2 Is Required for Synapse Elimination and Normal Brain Connectivity. Immunity 2018, 48, 979–991.e8. [Google Scholar] [CrossRef] [Green Version]
- Mecca, C.; Giambanco, I.; Donato, R.; Arcuri, C. Microglia and Aging: The Role of the TREM2–DAP12 and CX3CL1-CX3CR1 Axes. Int. J. Mol. Sci. 2018, 19, 318. [Google Scholar] [CrossRef] [Green Version]
- Turnbull, I.R.; Colonna, M. Activating and inhibitory functions of DAP12. Nat. Rev. Immunol. 2007, 7, 155–161. [Google Scholar] [CrossRef]
- Konishi, H.; Kiyama, H.; Konishi, H.; Kiyama, H. Microglial TREM2/DAP12 Signaling: A Double-Edged Sword in Neural Diseases. Front. Cell. Neurosci. 2018, 12, 206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cignarella, F.; Filipello, F.; Bollman, B.; Cantoni, C.; Locca, A.; Mikesell, R.; Manis, M.; Ibrahim, A.; Deng, L.; Benitez, B.A.; et al. TREM2 activation on microglia promotes myelin debris clearance and remyelination in a model of multiple sclerosis. Acta Neuropathol. 2020, 140, 513–534. [Google Scholar] [CrossRef] [PubMed]
- Kiialainen, A.; Hovanes, K.; Paloneva, J.; Kopra, O.; Peltonen, L. Dap12 and Trem2, molecules involved in innate immunity and neurodegeneration, are co-expressed in the CNS. Neurobiol. Dis. 2005, 18, 314–322. [Google Scholar] [CrossRef] [PubMed]
- Sessa, G.; Podini, P.; Mariani, M.; Meroni, A.; Spreafico, R.; Sinigaglia, F.; Colonna, M.; Panina, P.; Meldolesi, J. Distribution and signaling of TREM2/DAP12, the receptor system mutated in human polycystic lipomembraneous osteodysplasia with sclerosing leukoencephalopathy dementia. Eur. J. Neurosci. 2004, 20, 2617–2628. [Google Scholar] [CrossRef] [PubMed]
- Lynch, D.S.; Wade, C.; De Paiva, A.R.B.; John, N.; Kinsella, J.A.; Merwick, A.; Ahmed, R.M.; Warren, J.; Mummery, C.J.; Schott, J.; et al. Practical approach to the diagnosis of adult-onset leukodystrophies: An updated guide in the genomic era. J. Neurol. Neurosurg. Psychiatry 2018, 90, 543–554. [Google Scholar] [CrossRef] [Green Version]
- Papapetropoulos, S.; Pontius, A.; Finger, E.; Karrenbauer, V.; Lynch, D.S.; Brennan, M.; Zappia, S.; Koehler, W.; Schoels, L.; Hayer, S.N.; et al. Adult-Onset Leukoencephalopathy with Axonal Spheroids and Pigmented Glia: Review of Clinical Manifestations as Foundations for Therapeutic Development. Front. Neurol. 2022, 12, 788168. [Google Scholar] [CrossRef]
- Ayrignac, X.; Carra-Dallière, C.; Codjia, P.; Mouzat, K.; Castelnovo, G.; Ellie, E.; Etcharry-Bouyx, F.; Belliard, S.; Marelli, C.; Portet, F.; et al. Evaluation of CSF1R-related adult onset leukoencephalopathy with axonal spheroids and pigmented glia diagnostic criteria. Eur. J. Neurol. 2021, 29, 329–334. [Google Scholar] [CrossRef]
- Mochel, F.; Delorme, C.; Czernecki, V.; Froger, J.; Cormier, F.; Ellie, E.; Fegueux, N.; Lehéricy, S.; Lumbroso, S.; Schiffmann, R.; et al. Haematopoietic stem cell transplantation in CSF1R-related adult-onset leukoencephalopathy with axonal spheroids and pigmented glia. J. Neurol. Neurosurg. Psychiatry 2019, 90, 1375–1376. [Google Scholar] [CrossRef]
- Gelfand, J.M.; Greenfield, A.L.; Barkovich, M.; Mendelsohn, B.A.; Van Haren, K.; Hess, C.P.; Mannis, G.N. Allogeneic HSCT for adult-onset leukoencephalopathy with spheroids and pigmented glia. Brain 2019, 143, 503–511. [Google Scholar] [CrossRef]
- Tipton, P.W.; Stanley, E.R.; Chitu, V.; Wszolek, Z.K. Is Pre-Symptomatic Immunosuppression Protective in CSF1R-related Leukoencephalopathy? Mov. Disord. 2021, 36, 852–856. [Google Scholar] [CrossRef]
- Tipton, P.W.; Kenney-Jung, D.; Rush, B.K.; Middlebrooks, E.H.; Nascene, D.; Singh, B.; Holtan, S.; Ayala, E.; Broderick, D.F.; Lund, T.; et al. Treatment of CSF1R-Related Leukoencephalopathy: Breaking New Ground. Mov. Disord. 2021, 36, 2901–2909. [Google Scholar] [CrossRef] [PubMed]
- Errichiello, E.; Dardiotis, E.; Mannino, F.; Paloneva, J.; Mattina, T.; Zuffardi, O. Phenotypic Expansion in Nasu-Hakola Disease: Immunological Findings in Three Patients and Proposal of a Unifying Pathogenic Hypothesis. Front. Immunol. 2019, 10, 1685. [Google Scholar] [CrossRef] [PubMed] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ferrer, I. The Primary Microglial Leukodystrophies: A Review. Int. J. Mol. Sci. 2022, 23, 6341. https://doi.org/10.3390/ijms23116341
Ferrer I. The Primary Microglial Leukodystrophies: A Review. International Journal of Molecular Sciences. 2022; 23(11):6341. https://doi.org/10.3390/ijms23116341
Chicago/Turabian StyleFerrer, Isidro. 2022. "The Primary Microglial Leukodystrophies: A Review" International Journal of Molecular Sciences 23, no. 11: 6341. https://doi.org/10.3390/ijms23116341
APA StyleFerrer, I. (2022). The Primary Microglial Leukodystrophies: A Review. International Journal of Molecular Sciences, 23(11), 6341. https://doi.org/10.3390/ijms23116341