
CRISPR/Cas9-Directed Gene Trap Constitutes a Selection System for Corrected BCR/ABL Leukemic Cells in CML


 ,
,  and
and
Abstract
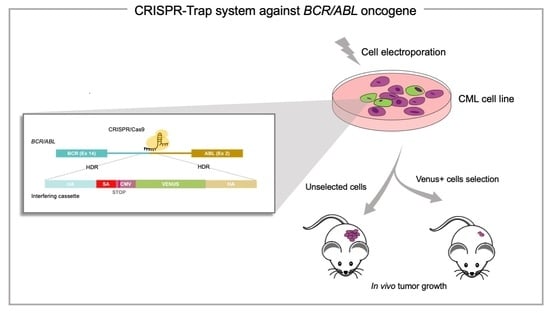
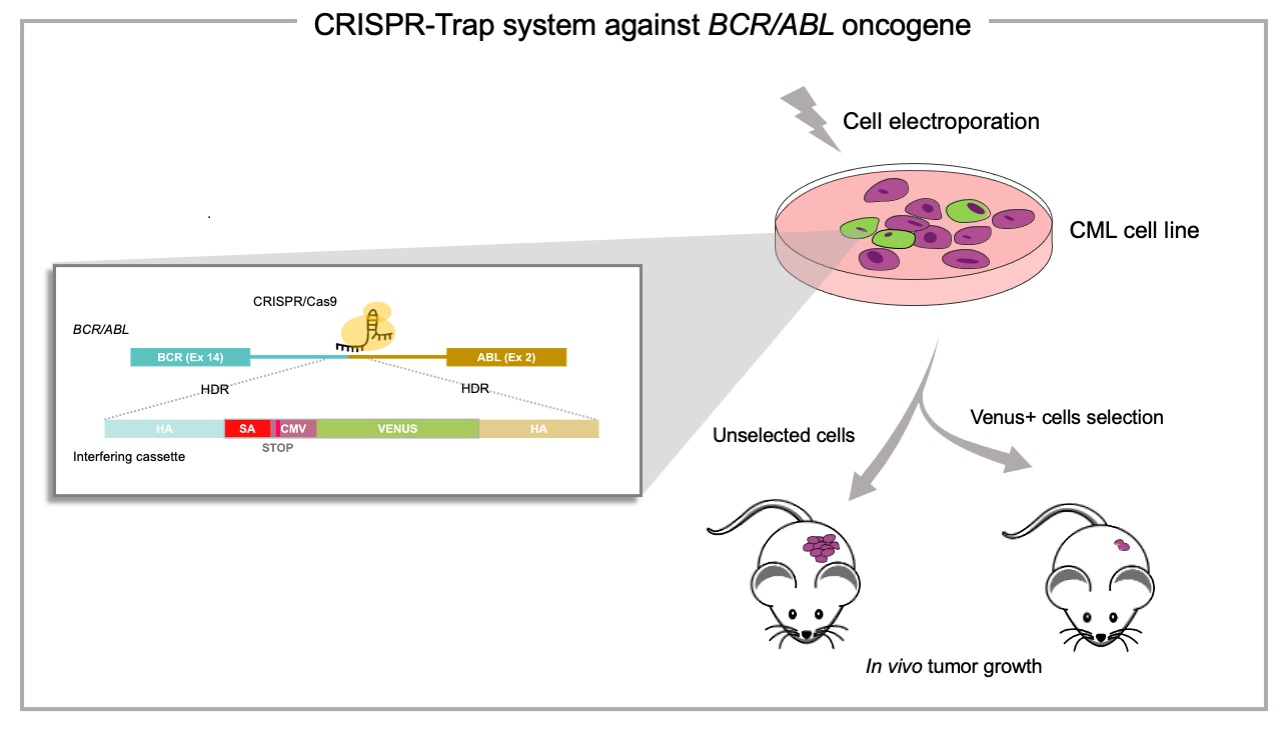
:
1. Introduction
2. Results
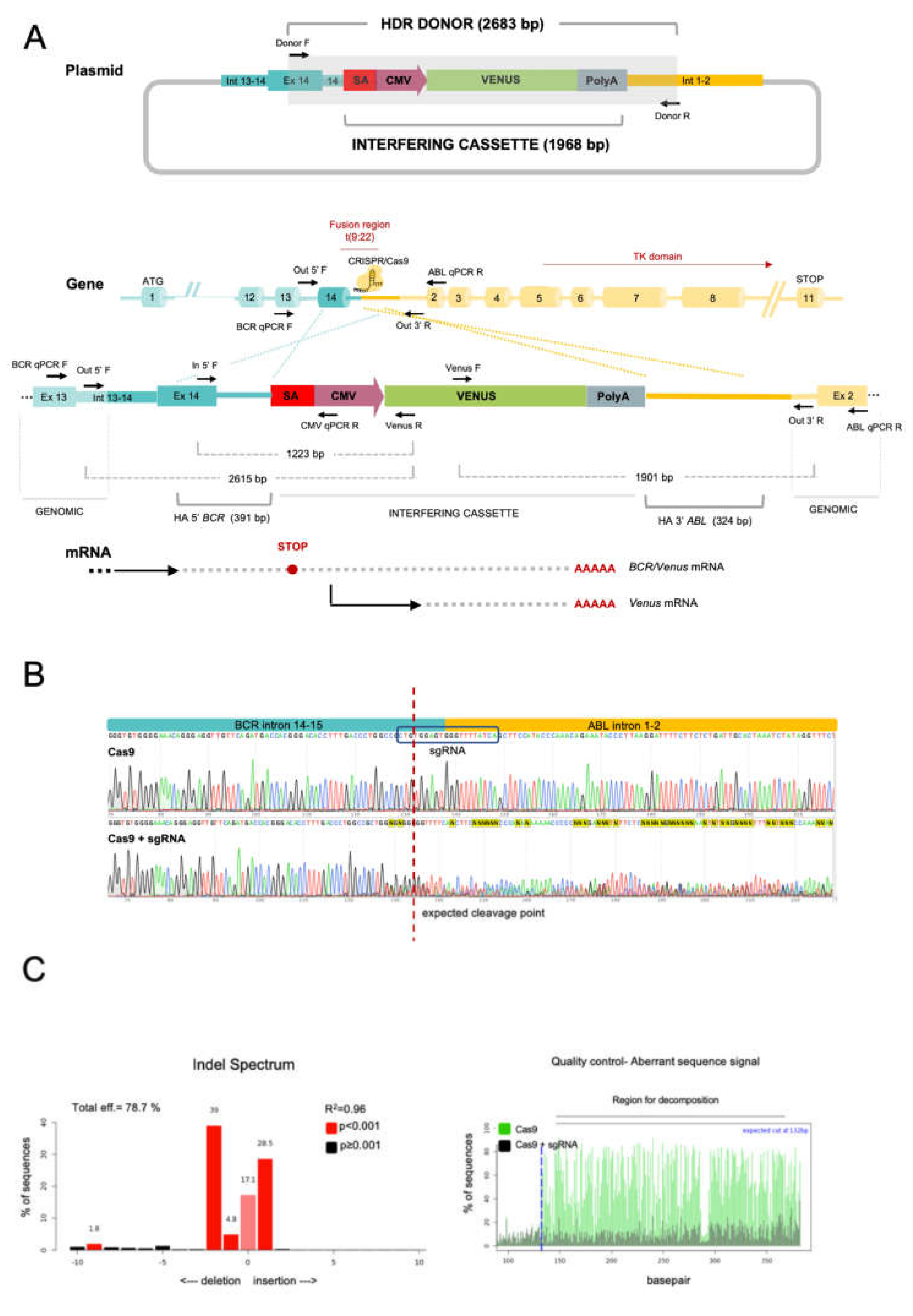
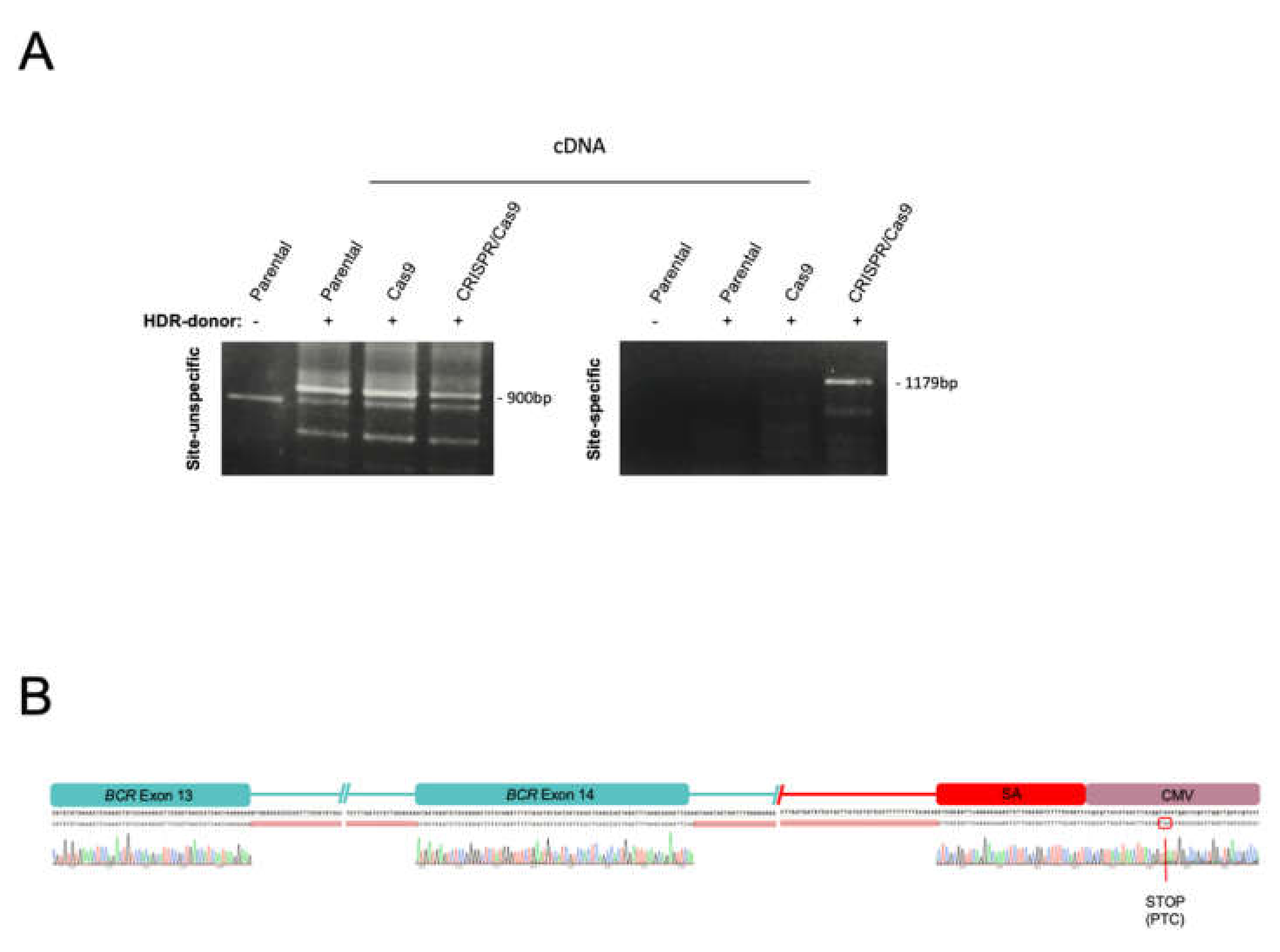
2.1. The CRISPR/Cas9 System Efficiently Directs the Specific Integration of a Gene Trap Donor Cassette at the BCR/ABL Locus
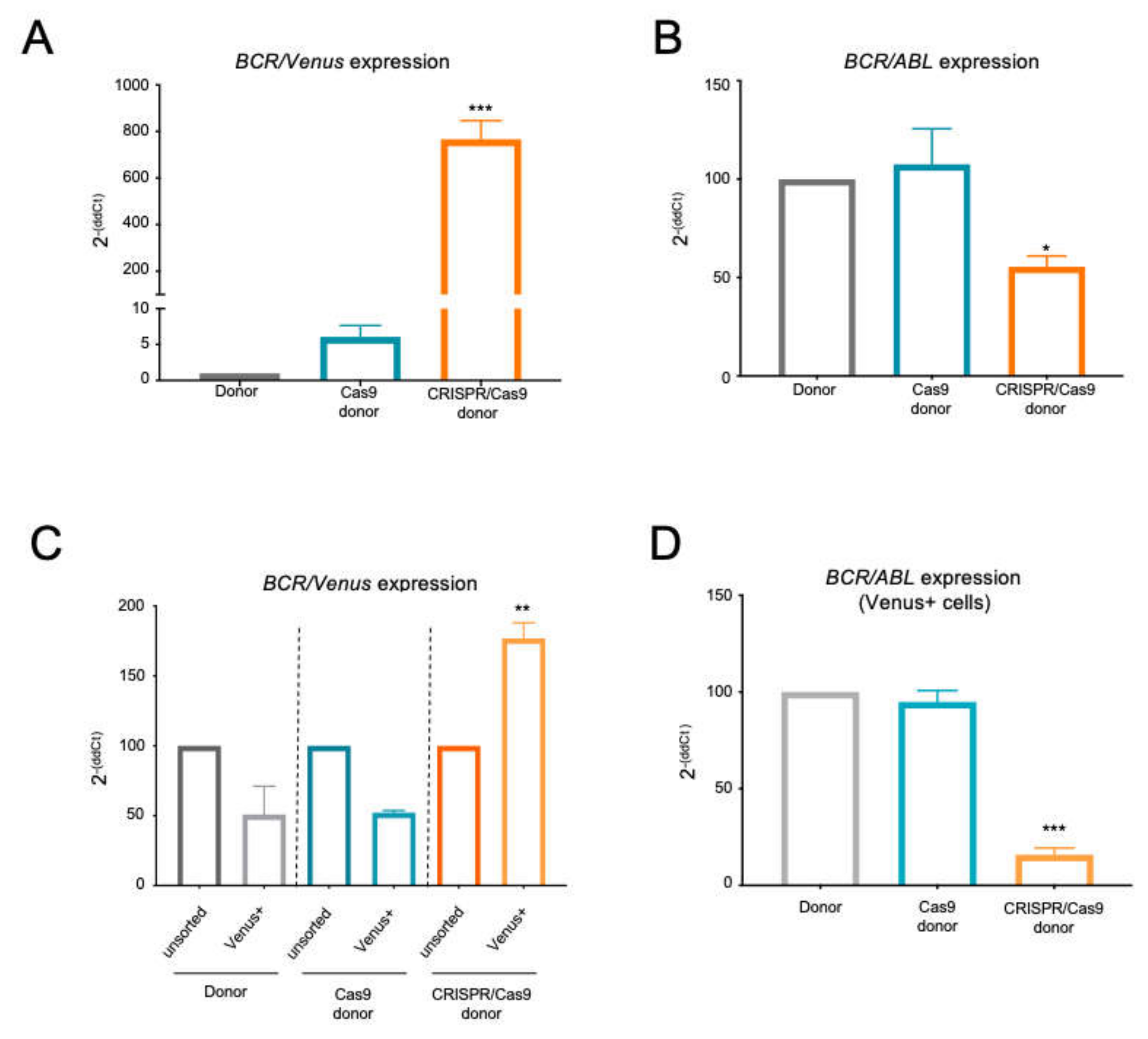
2.2. The BCR/ABL Trapped Allele Is Properly Expressed When the Expression of the Oncogenic Version Is Reduced
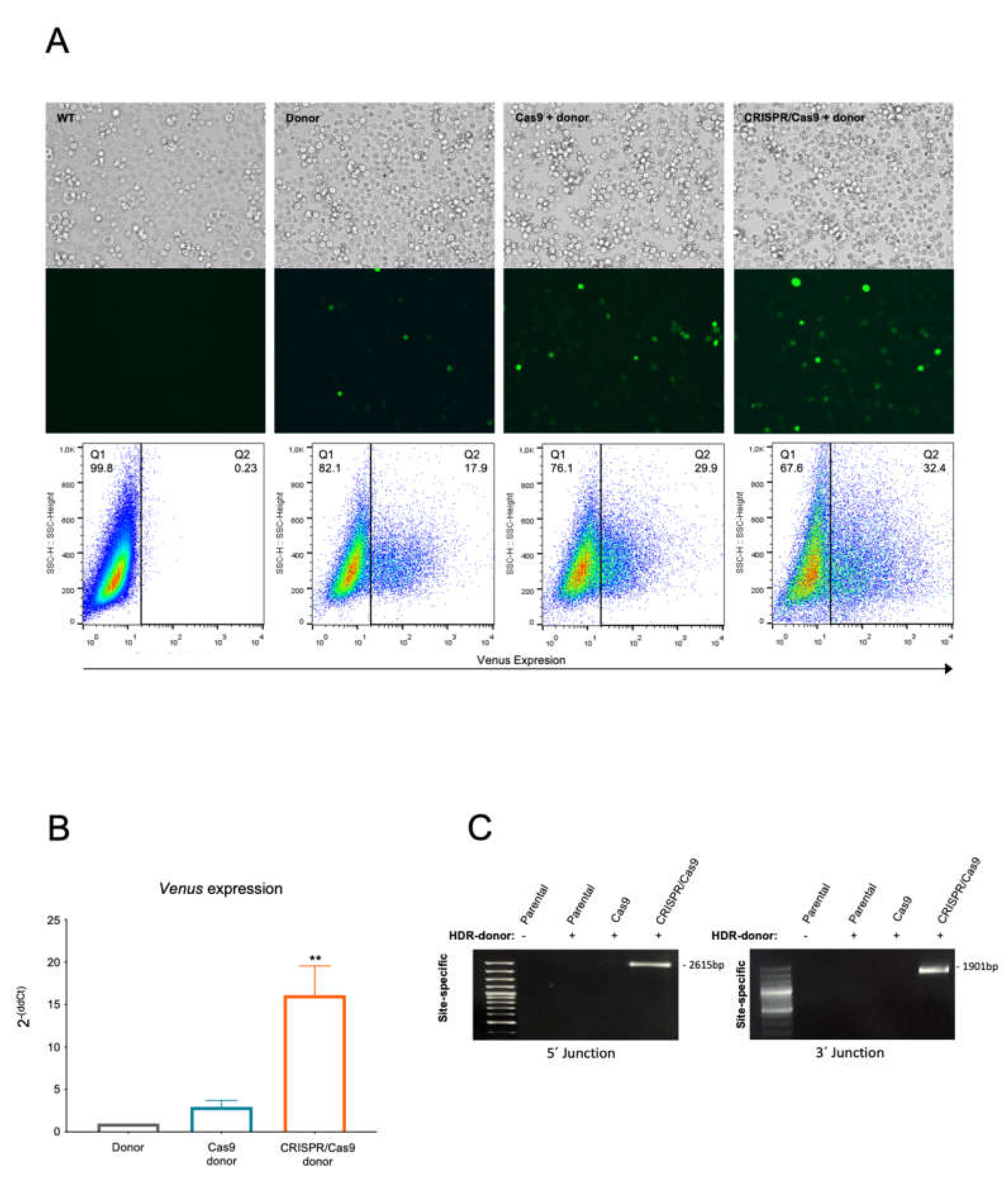
2.3. The BCR/ABL CRISPR-Trap Enables the Selection of Gene-Targeted Cells
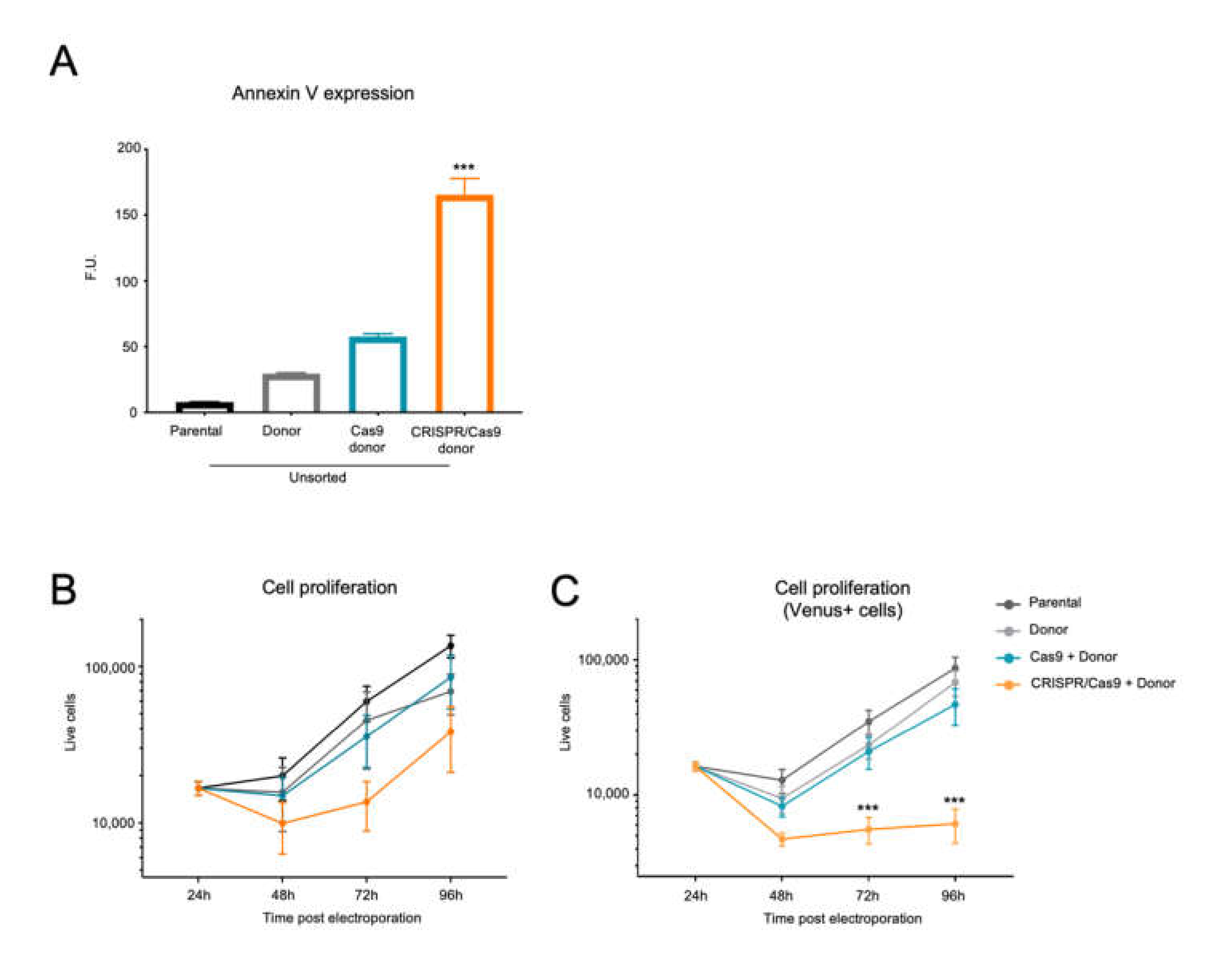
2.4. The BCR/ABL CRISPR-Trap Promotes Apoptosis and Inhibits Proliferation in K562 Leukemic Cells
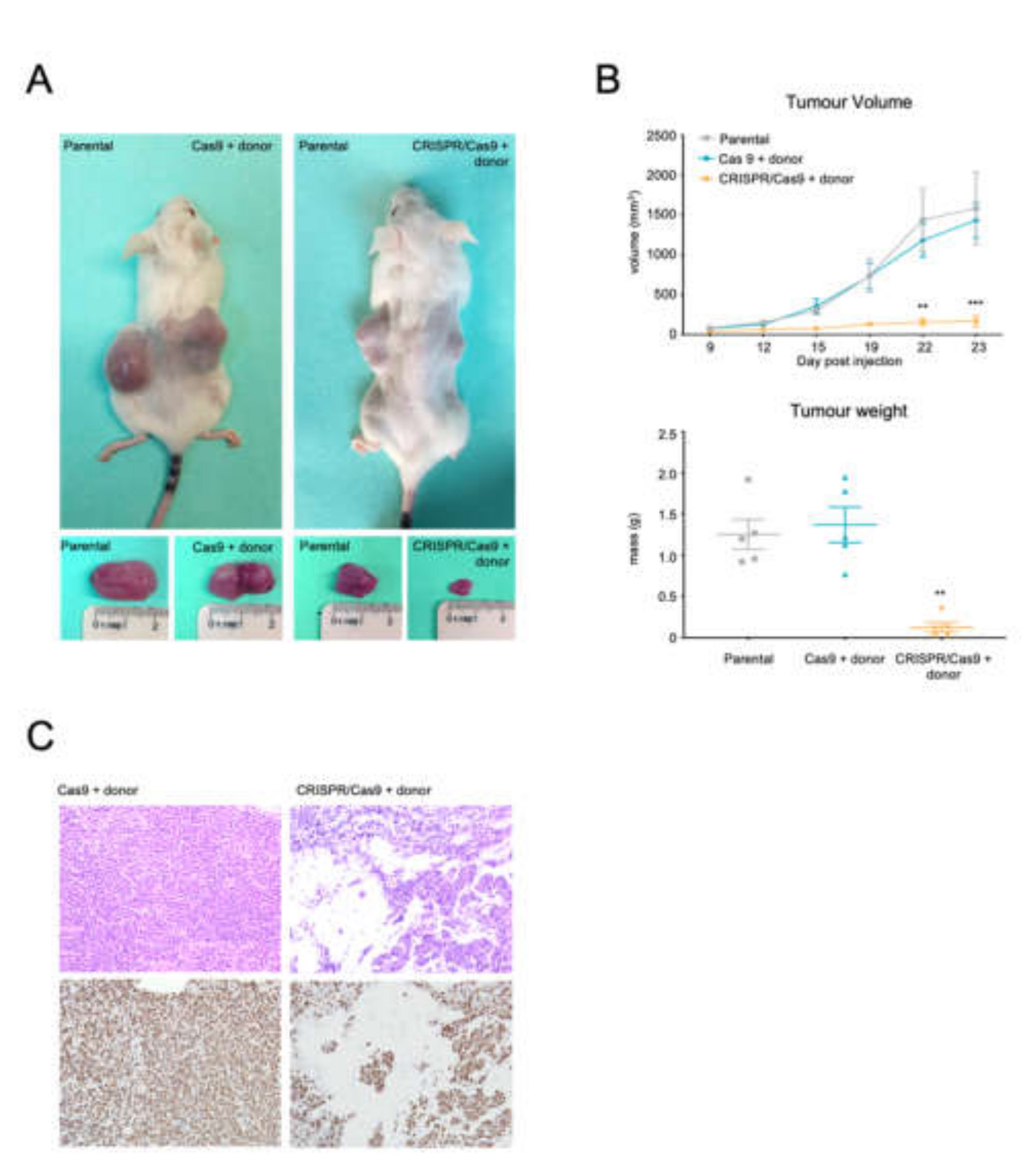
2.5. The CRISPR-Trap System Prevents Tumour Activity of BCR/ABL, Thereby Producing a Therapeutic Effect in a CML Xenograft Model
3. Discussion
4. Materials and Methods
4.1. Cell Lines and Culture Conditions
4.2. Cloning of Targeting Vectors and HDR DNA Donor Obtention
4.3. CRISPR/Cas9 System Design
4.4. CRISPR/Cas9 Ribonucleocomplex Assembly, DNA Donor Delivery and Electroporation
4.5. DNA/RNA Isolation, Retrotranscription and PCR-Based Detection of HDR Events
4.6. Cell Viability and Cell Proliferation Assay
4.7. Flow Cytometry and Cell Sorting
4.8. qPCR
4.9. Mouse Xenograft Tumorigenesis
4.10. Immunohistochemical Studies
4.11. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Acknowledgments
Conflicts of Interest
References
- Maru, Y. Molecular Biology of Chronic Myeloid Leukemia. Int. J. Hematol. 2001, 73, 308–322. [Google Scholar] [CrossRef]
- Quintás-Cardama, A.; Cortes, J. Molecular Biology of Bcr-Abl1-Positive Chronic Myeloid Leukemia. Blood 2009, 113, 1619–1630. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Rowley, J.D. Chronic Myeloid Leukemia: Current Perspectives. Clin. Lab. Med. 2011, 31, 687–698. [Google Scholar] [CrossRef]
- Ross, D.M.; Branford, S.; Seymour, J.F.; Schwarer, A.P.; Arthur, C.; Yeung, D.T.; Dang, P.; Goyne, J.M.; Slader, C.; Filshie, R.J.; et al. Safety and Efficacy of Imatinib Cessation for CML Patients with Stable Undetectable Minimal Residual Disease: Results from the TWISTER Study. Blood 2013, 122, 515–522. [Google Scholar] [CrossRef]
- Bhamidipati, P.K.; Kantarjian, H.; Cortes, J.; Cornelison, A.M.; Jabbour, E. Management of Imatinib-Resistant Patients with Chronic Myeloid Leukemia. Ther. Adv. Hematol. 2013, 4, 103–117. [Google Scholar] [CrossRef]
- Zhang, H.; McCarty, N. CRISPR-Cas9 Technology and Its Application in Haematological Disorders. Br. J. Haematol. 2016, 175, 208. [Google Scholar] [CrossRef]
- Reddy, O.L.; Savani, B.N.; Stroncek, D.F.; Panch, S.R. Advances in Gene Therapy for Hematologic Disease and Considerations for Transfusion Medicine. Semin. Hematol. 2020, 57, 83–91. [Google Scholar] [CrossRef]
- Herzog, R.W.; Hagstrom, J.N. Gene Therapy for Hereditary Hematological Disorders. Am. J. Pharmacogenom. 2001, 1, 137–144. [Google Scholar] [CrossRef]
- Ferrari, S.; Vavassori, V.; Canarutto, D.; Jacob, A.; Castiello, M.C.; Javed, A.O.; Genovese, P. Gene Editing of Hematopoietic Stem Cells: Hopes and Hurdles Toward Clinical Translation. Front. Genome Ed. 2021, 3, 9. [Google Scholar] [CrossRef]
- Lugo, T.G.; Pendergast, A.M.; Muller, A.J.; Witte, O.N. Tyrosine Kinase Activity and Transformation Potency of Bcr-Abl Oncogene Products. Science 1990, 247, 1079–1082. [Google Scholar] [CrossRef]
- Zhang, H.; Li, S. Induction of Chronic Myeloid Leukemia in Mice. Methods Mol. Biol. 2016, 1465, 17–25. [Google Scholar] [CrossRef] [PubMed]
- Warfvinge, R.; Geironson, L.; Sommarin, M.N.E.; Lang, S.; Karlsson, C.; Roschupkina, T.; Stenke, L.; Stentoft, J.; Olsson-Stromberg, U.; Hjorth-Hansen, H.; et al. Single-Cell Molecular Analysis Defines Therapy Response and Immunophenotype of Stem Cell Subpopulations in CML. Blood 2017, 129, 2384–2394. [Google Scholar] [CrossRef]
- Daley, G.Q.; Van Etten, R.A.; Baltimore, D. Blast Crisis in a Murine Model of Chronic Myelogenous Leukemia. Proc. Natl. Acad. Sci. USA 1991, 88, 11335. [Google Scholar] [CrossRef] [Green Version]
- Huang, N.; Huang, Z.; Gao, M.; Luo, Z.; Zhou, F.; Liu, L.; Xiao, Q.; Wang, X.; Feng, W. Induction of Apoptosis in Imatinib Sensitive and Resistant Chronic Myeloid Leukemia Cells by Efficient Disruption of Bcr-Abl Oncogene with Zinc Finger Nucleases. J. Exp. Clin. Cancer Res. 2018, 37, 62. [Google Scholar] [CrossRef]
- Chen, S.-H.; Hsieh, Y.-Y.; Tzeng, H.-E.; Lin, C.-Y.; Hsu, K.-W.; Chiang, Y.-S.; Lin, S.-M.; Su, M.-J.; Hsieh, W.-S.; Lee, C.-H. ABL Genomic Editing Sufficiently Abolishes Oncogenesis of Human Chronic Myeloid Leukemia Cells In Vitro and In Vivo. Cancers 2020, 12, 1399. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Lage, M.; Torres-Ruiz, R.; Puig-Serra, P.; Moreno-Gaona, P.; Martin, M.C.; Moya, F.J.; Quintana-Bustamante, O.; Garcia-Silva, S.; Carcaboso, A.M.; Petazzi, P.; et al. In Vivo CRISPR/Cas9 Targeting of Fusion Oncogenes for Selective Elimination of Cancer Cells. Nat. Commun. 2020, 11, 5060. [Google Scholar] [CrossRef]
- García-Tuñón, I.; Hernández-Sánchez, M.; Ordoñez, J.L.; Alonso-Pérez, V.; Álamo-Quijada, M.; Benito, R.; Guerrero, C.; Hernández-Rivas, J.M.; Sánchez-Martín, M. The CRISPR/Cas9 System Efficiently Reverts the Tumorigenic Ability of BCR/ABL in Vitro and in a Xenograft Model of Chronic Myeloid Leukemia. Oncotarget 2017, 8, 26027–26040. [Google Scholar] [CrossRef] [Green Version]
- Vuelta, E.; García-Tuñón, I.; Hernández-Carabias, P.; Méndez, L.; Sánchez-Martín, M. Future Approaches for Treating Chronic Myeloid Leukemia: CRISPR Therapy. Biology 2021, 10, 118. [Google Scholar] [CrossRef]
- Vuelta, E.; Ordoñez, J.L.; Alonso-Pérez, V.; Méndez, L.; Hernández-Carabias, P.; Saldaña, R.; Sevilla, J.; Sebastián, E.; Muntión, S.; Sánchez-Guijo, F.; et al. CRISPR-Cas9 Technology as a Tool to Target Gene Drivers in Cancer: Proof of Concept and New Opportunities to Treat Chronic Myeloid Leukemia. CRISPR J. 2021, 4, 519–535. [Google Scholar] [CrossRef]
- Kohn, D.B. Update on Gene Therapy for Immunodeficiencies. Clin. Immunol. 2010, 135, 247. [Google Scholar] [CrossRef] [Green Version]
- Reber, S.; Mechtersheimer, J.; Nasif, S.; Benitez, J.A.; Colombo, M.; Domanski, M.; Jutzi, D.; Hedlund, E.; Ruepp, M.-D. CRISPR-Trap: A Clean Approach for the Generation of Gene Knockouts and Gene Replacements in Human Cells. Mol. Biol. Cell 2018, 29, 75. [Google Scholar] [CrossRef]
- Staal, F.J.T.; Aiuti, A.; Cavazzana, M. Autologous Stem-Cell-Based Gene Therapy for Inherited Disorders: State of the Art and Perspectives. Front. Pediatr. 2019, 7, 443. [Google Scholar] [CrossRef]
- Walters, M.C. Update of Hematopoietic Cell Transplantation for Sickle Cell Disease. Curr. Opin. Hematol. 2015, 22, 227–233. [Google Scholar] [CrossRef]
- Boelens, J.J.; Aldenhoven, M.; Purtill, D.; Ruggeri, A.; DeFor, T.; Wynn, R.; Wraith, E.; Cavazzana-Calvo, M.; Rovelli, A.; Fischer, A.; et al. Outcomes of Transplantation Using Various Hematopoietic Cell Sources in Children with Hurler Syndrome after Myeloablative Conditioning. Blood 2013, 121, 3981–3987. [Google Scholar] [CrossRef]
- Van Rhee, F.; Szydlo, R.M.; Hermans, J.; Devergie, A.; Frassoni, F.; Arcese, W.; De Witte, T.; Kolb, H.J.; Niederwiser, D.; Jacobsen, N.; et al. Long-Term Results after Allogeneic Bone Marrow Transplantation for Chronic Myelogenous Leukemia in Chronic Phase: A Report from the Chronic Leukemia Working Party of the European Group for Blood and Marrow Transplantation. Bone Marrow Transplant. 1997, 20, 553–560. [Google Scholar] [CrossRef] [Green Version]
- Morgan, R.A.; Gray, D.; Lomova, A.; Kohn, D.B. Hematopoietic Stem Cell Gene Therapy—Progress and Lessons Learned. Cell Stem Cell 2017, 21, 574. [Google Scholar] [CrossRef] [Green Version]
- Aiuti, A.; Biasco, L.; Scaramuzza, S.; Ferrua, F.; Cicalese, M.P.; Baricordi, C.; Dionisio, F.; Calabria, A.; Giannelli, S.; Castiello, M.C.; et al. Lentiviral Hematopoietic Stem Cell Gene Therapy in Patients with Wiskott-Aldrich Syndrome. Science 2013, 341, 1233151. [Google Scholar] [CrossRef] [Green Version]
- Biffi, A.; Montini, E.; Lorioli, L.; Cesani, M.; Fumagalli, F.; Plati, T.; Baldoli, C.; Martino, S.; Calabria, A.; Canale, S.; et al. Lentiviral Hematopoietic Stem Cell Gene Therapy Benefits Metachromatic Leukodystrophy. Science 2013, 341, 1233158. [Google Scholar] [CrossRef] [Green Version]
- De Ravin, S.S.; Wu, X.; Moir, S.; Anaya-O’Brien, S.; Kwatemaa, N.; Littel, P.; Theobald, N.; Choi, U.; Su, L.; Marquesen, M.; et al. Lentiviral Hematopoietic Stem Cell Gene Therapy for X-Linked Severe Combined Immunodeficiency. Sci. Transl. Med. 2016, 8, 335ra57. [Google Scholar] [CrossRef] [Green Version]
- Cartier, N.; Aubourg, P. Hematopoietic Stem Cell Transplantation and Hematopoietic Stem Cell Gene Therapy in X-Linked Adrenoleukodystrophy. Brain Pathol. 2010, 20, 857–862. [Google Scholar] [CrossRef]
- Diez, B.; Genovese, P.; Roman-Rodriguez, F.J.; Alvarez, L.; Schiroli, G.; Ugalde, L.; Rodriguez-Perales, S.; Sevilla, J.; de Heredia, C.D.; Holmes, M.C.; et al. Therapeutic Gene Editing in CD34+ Hematopoietic Progenitors from Fanconi Anemia Patients. EMBO Mol. Med. 2017, 9, 1574. [Google Scholar] [CrossRef] [PubMed]
- Kabarowski, J.H.S.; Witte, O.N. Consequences of BCR-ABL Expression within the Hematopoietic Stem Cell in Chronic Myeloid Leukemia. Stem Cells 2000, 18, 399–408. [Google Scholar] [CrossRef]
- Tanaka, T.S.; Davey, R.E.; Lan, Q.; Zandstra, P.W.; Stanford, W.L. Development of a Gene-Trap Vector with a Highly Sensitive Fluorescent Protein Reporter System for Expression Profiling. Genesis 2008, 46, 347–356. [Google Scholar] [CrossRef]
- Ishikawa, K.; Kobayashi, Y.; Wakabayashi, Y.; Watanabe, S.; Semba, K. A Highly Sensitive Trap Vector System for Isolating Reporter Cells and Identification of Responsive Genes. Biol. Methods Protoc. 2018, 3, bpy003. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.; Liu, J.; Feng, T.; Guo, Z.; Yin, Y.; Gao, F.; Cao, G.; Du, X.; Wu, S. A HIT-Trapping Strategy for Rapid Generation of Reversible and Conditional Alleles Using a Universal Donor. Genome Res. 2021, 31, 900–909. [Google Scholar] [CrossRef]
- Roth, T.L.; Puig-Saus, C.; Yu, R.; Shifrut, E.; Carnevale, J.; Li, P.J.; Hiatt, J.; Saco, J.; Krystofinski, P.; Li, H.; et al. Reprogramming Human T Cell Function and Specificity with Non-Viral Genome Targeting. Nature 2018, 559, 405. [Google Scholar] [CrossRef]
- McGahon, A.J.; Brown, D.G.; Martin, S.J.; Amarante-Mendes, G.P.; Cotter, T.G.; Cohen, G.M.; Green, D.R. Downregulation of Bcr-Abl in K562 Cells Restores Susceptibility to Apoptosis: Characterization of the Apoptotic Death. Cell Death Differ. 1997, 4, 95–104. [Google Scholar] [CrossRef] [PubMed]
- Rangatia, J.; Bonnet, D. Transient or Long-Term Silencing of BCR-ABL Alone Induces Cell Cycle and Proliferation Arrest, Apoptosis and Differentiation. Leukemia 2006, 20, 68–76. [Google Scholar] [CrossRef]
- Porteus, M.H.; Baltimore, D. Chimeric Nucleases Stimulate Gene Targeting in Human Cells. Science 2003, 300, 763. [Google Scholar] [CrossRef] [Green Version]
- Colicelli, J. ABL Tyrosine Kinases: Evolution of Function, Regulation, and Specificity. Sci. Signal. 2010, 3, re6. [Google Scholar] [CrossRef] [Green Version]
- Melo, J.V.; Deininger, M.W.N. Biology of Chronic Myelogenous Leukemia—Signaling Pathways of Initiation and Transformation. Hematol. Oncol. Clin. N. Am. 2004, 18, 545–568. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.; Monaco, G.; Sun, T.; Ling, X.; Stephens, C.; Xie, S.; Belmont, J.; Arlinghaus, R. Bcr-Abl-Mediated Suppression of Normal Hematopoiesis in Leukemia. Oncogene 2005, 24, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Sánchez-García, I.; Martín-Zanca, D. Regulation of Bcl-2 Gene Expression by BCR-ABL Is Mediated by Ras. J. Mol. Biol. 1997, 267, 225–228. [Google Scholar] [CrossRef] [PubMed]
- Deutsch, E.; Dugray, A.; AbdulKarim, B.; Marangoni, E.; Maggiorella, L.; Vaganay, S.; M’Kacher, R.; Rasy, S.D.; Eschwege, F.; Vainchenker, W.; et al. BCR-ABL down-Regulates the DNA Repair Protein DNA-PKcs. Blood 2001, 97, 2084–2090. [Google Scholar] [CrossRef] [Green Version]
- Koptyra, M.; Cramer, K.; Slupianek, A.; Richardson, C.; Skorski, T. BCR/ABL Promotes Accumulation of Chromosomal Aberrations Induced by Oxidative and Genotoxic Stress. Leukemia 2008, 22, 1969–1972. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaiyachati, B.H.; Kaundal, R.; Zhao, J.; Wu, J.; Flavell, R.; Chi, T. LoxP-FRT Trap (LOFT): A Simple and Flexible System for Conventional and Reversible Gene Targeting. BMC Biol. 2012, 10, 96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Groffen, J.; Stephenson, J.R.; Heisterkamp, N.; de Klein, A.; Bartram, C.R.; Grosveld, G. Philadelphia Chromosomal Breakpoints Are Clustered within a Limited Region, Bcr, on Chromosome 22. Cell 1984, 36, 93–99. [Google Scholar] [CrossRef]
- Heisterkamp, N.; Stam, K.; Groffen, J.; De Klein, A.; Grosveld, G. Structural Organization of the Bcr Gene and Its Role in the Ph’ Translocation. Nature 1985, 315, 758–761. [Google Scholar] [CrossRef]
- Ordóñez, J.L.; Amaral, A.T.; Carcaboso, A.M.; Herrero-Martín, D.; Del Carmen García-Macías, M.; Sevillano, V.; Alonso, D.; Pascual-Pasto, G.; San-Segundo, L.; Vila-Ubach, M.; et al. The PARP Inhibitor Olaparib Enhances the Sensitivity of Ewing Sarcoma to Trabectedin. Oncotarget 2015, 6, 18875. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Sequence |
|---|---|
| Donor F | ACCCACATCCCACATCACCC |
| Donor R | CATGGTCTCCACTATCAAGGG |
| Out 5′ F | ATCAAGGATCTCCGGGCAGC |
| Out 3′ R | CCAAGGCAAATCTGGGAGTTG |
| In 5′ F | TCCACTCAGCCACTGGATTTAAGCA |
| Venus F | TGGTCCTGCTGGAGTTCGTG |
| Venus R | GGACACGCTGAACTTGTGGC |
| BCR qPCR F | AGTTACACGTTCCTGATCTCC |
| ABL qPCR R | TTGGGCTTCACACCATTCCCC |
| CMV qPCR R | GCGGGCCATTTACCGTAAG |
| Venus qPCR R | GCGGGCCATTTACCGTAAG |
| Gapdh qPCR F | TGCACCACCAACTGCTTAGC |
| Gapdh qPCR R | CACCACCTTCTTGATGTCATCA |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vuelta, E.; Ordoñez, J.L.; Sanz, D.J.; Ballesteros, S.; Hernández-Rivas, J.M.; Méndez-Sánchez, L.; Sánchez-Martín, M.; García-Tuñón, I. CRISPR/Cas9-Directed Gene Trap Constitutes a Selection System for Corrected BCR/ABL Leukemic Cells in CML. Int. J. Mol. Sci. 2022, 23, 6386. https://doi.org/10.3390/ijms23126386
Vuelta E, Ordoñez JL, Sanz DJ, Ballesteros S, Hernández-Rivas JM, Méndez-Sánchez L, Sánchez-Martín M, García-Tuñón I. CRISPR/Cas9-Directed Gene Trap Constitutes a Selection System for Corrected BCR/ABL Leukemic Cells in CML. International Journal of Molecular Sciences. 2022; 23(12):6386. https://doi.org/10.3390/ijms23126386
Chicago/Turabian StyleVuelta, Elena, José L. Ordoñez, David J. Sanz, Sandra Ballesteros, Jesús M. Hernández-Rivas, Lucía Méndez-Sánchez, Manuel Sánchez-Martín, and Ignacio García-Tuñón. 2022. "CRISPR/Cas9-Directed Gene Trap Constitutes a Selection System for Corrected BCR/ABL Leukemic Cells in CML" International Journal of Molecular Sciences 23, no. 12: 6386. https://doi.org/10.3390/ijms23126386
APA StyleVuelta, E., Ordoñez, J. L., Sanz, D. J., Ballesteros, S., Hernández-Rivas, J. M., Méndez-Sánchez, L., Sánchez-Martín, M., & García-Tuñón, I. (2022). CRISPR/Cas9-Directed Gene Trap Constitutes a Selection System for Corrected BCR/ABL Leukemic Cells in CML. International Journal of Molecular Sciences, 23(12), 6386. https://doi.org/10.3390/ijms23126386