
Slaying (Yet Again) the Brain-Eating Zombie Called the “Isochore Theory”: A Segmentation Algorithm Used to “Confirm” the Existence of Isochores Creates “Isochores” Where None Exist

Abstract
:1. Introduction
1.1. Isochores and the Human Genome Sequence
1.2. Segmentation Algorithms
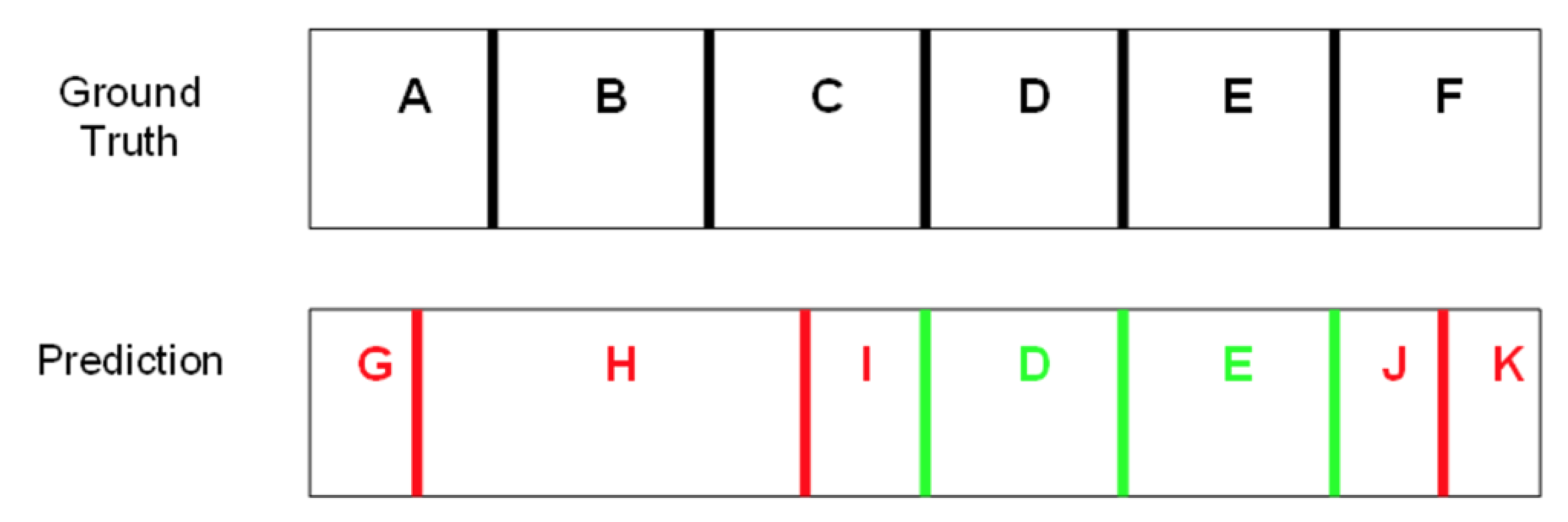
2. Results
3. Discussion
4. Materials and Methods
4.1. Simulated Genomic Sequences
4.2. Scoring of Predicted Domains
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Macaya, G.; Thiery, J.-P.; Bernardi, G. An Approach to the Organization of Eukaryotic Genomes at a Macromolecular Level. J. Mol. Biol. 1976, 108, 237–254. [Google Scholar] [CrossRef]
- Thiery, J.-P.; Macaya, G.; Bernardi, G. An Analysis of Eukaryotic Genomes by Density Gradient Centrifugation. J. Mol. Biol. 1976, 108, 219–235. [Google Scholar] [CrossRef]
- Cuny, G.; Soriano, P.; Macaya, G.; Bernardi, G. The Major Components of the Mouse and Human Genomes. Preparation, Basic Properties and Compositional Heterogeneity. Eur. J. Biochem. 1981, 115, 227–233. [Google Scholar] [CrossRef] [PubMed]
- Elhaik, E.; Graur, D. A Comparative Study and a Phylogenetic Exploration of the Compositional Architectures of Mammalian Nuclear Genomes. PLoS Comput. Biol. 2014, 10, e1003925. [Google Scholar] [CrossRef] [PubMed]
- Elhaik, E.; Landan, G.; Graur, D. Can GC Content at Third-Codon Positions Be Used as a Proxy for Isochore Composition? Mol. Biol. Evol. 2009, 26, 1829–1833. [Google Scholar] [CrossRef] [PubMed]
- International Human Genome Sequencing Consortium; Whitehead Institute for Biomedical Research, Center for Genome Research; Lander, E.S.; Linton, L.M.; Birren, B.; Nusbaum, C.; Zody, M.C.; Baldwin, J.; Devon, K.; Dewar, K.; et al. Initial Sequencing and Analysis of the Human Genome. Nature 2001, 409, 860–921. [Google Scholar] [CrossRef]
- The Bovine Genome Sequencing and Analysis Consortium; Elsik, C.G.; Tellam, R.L.; Worley, K.C.; Gibbs, R.A.; Muzny, D.M.; Weinstock, G.M.; Adelson, D.L.; Eichler, E.E.; Elnitski, L.; et al. The Genome Sequence of Taurine Cattle: A Window to Ruminant Biology and Evolution. Science 2009, 324, 522–528. [Google Scholar] [CrossRef]
- Eyre-Walker, A.; Hurst, L.D. The Evolution of Isochores. Nat. Rev. Genet. 2001, 2, 549–555. [Google Scholar] [CrossRef]
- Häring, D.; Kypr, J. No Isochores in the Human Chromosomes 21 and 22? Biochem. Biophys. Res. Commun. 2001, 280, 567–573. [Google Scholar] [CrossRef]
- Cohen, N.; Dagan, T.; Stone, L.; Graur, D. GC Composition of the Human Genome: In Search of Isochores. Mol. Biol. Evol. 2005, 22, 1260–1272. [Google Scholar] [CrossRef]
- Nekrutenko, A.; Li, W.H. Assessment of Compositional Heterogeneity Within and Between Eukaryotic Genomes. Genome Res. 2000, 10, 1986–1995. [Google Scholar] [CrossRef] [PubMed]
- Costantini, M.; Clay, O.; Auletta, F.; Bernardi, G. An Isochore Map of Human Chromosomes. Genome Res. 2006, 16, 536–541. [Google Scholar] [CrossRef] [PubMed]
- Cozzi, P.; Milanesi, L.; Bernardi, G. Segmenting the Human Genome into Isochores. Evol. Bioinform. Online 2015, 11, 253–261. [Google Scholar] [CrossRef]
- Bernardi, G.; Bernardi, G. Codon usage and genome composition. J. Mol. Evol. 1985, 22, 363–365. [Google Scholar] [CrossRef] [PubMed]
- Belle, E.M.S.; Smith, N.; Eyre-Walker, A. Analysis of the Phylogenetic Distribution of Isochores in Vertebrates and a Test of the Thermal Stability Hypothesis. J. Mol. Evol. 2002, 55, 356–363. [Google Scholar] [CrossRef]
- Nurk, S.; Koren, S.; Rhie, A.; Rautiainen, M.; Bzikadze, A.V.; Mikheenko, A.; Vollger, M.R.; Altemose, N.; Uralsky, L.; Gershman, A.; et al. The Complete Sequence of a Human Genome. Science 2022, 376, 44–53. [Google Scholar] [CrossRef] [PubMed]
- Bernaola-Galván, P.; Román-Roldán, R.; Oliver, J.L. Compositional Segmentation and Long-Range Fractal Correlations in DNA Sequences. Phys. Rev. E 1996, 53, 5181–5189. [Google Scholar] [CrossRef]
- Bernardi, G. Isochores and the Evolutionary Genomics of Vertebrates. Gene 2000, 241, 3–17. [Google Scholar] [CrossRef]
- Grosse, I.; Bernaola-Galván, P.; Carpena, P.; Román-Roldán, R.; Oliver, J.; Stanley, H.E. Analysis of Symbolic Sequences Using the Jensen-Shannon Divergence. Phys. Rev. E 2002, 65, 041905. [Google Scholar] [CrossRef]
- Elhaik, E.; Graur, D.; Josić, K.; Landan, G. Identifying Compositionally Homogeneous and Nonhomogeneous Domains within the Human Genome Using a Novel Segmentation Algorithm. Nucleic Acids Res. 2010, 38, e158. [Google Scholar] [CrossRef]
- Elhaik, E.; Graur, D. IsoPlotter +: A Tool for Studying the Compositional Architecture of Genomes. ISRN Bioinform. 2013, 2013, 725434. [Google Scholar] [CrossRef] [PubMed]
- Afreixo, V.; Rodrigues, J.M.O.S.; Bastos, C.A.C.; Silva, R.M. The Exceptional Genomic Word Symmetry along DNA Sequences. BMC Bioinform. 2016, 17, 59. [Google Scholar] [CrossRef] [PubMed]
- Labena, A.A.; Guo, H.-X.; Dong, C.; Li, L.; Guo, F.-B. The Topologically Associated Domains (TADs) of a Chromatin Correlated with Isochores Organization of a Genome. CBIO 2018, 13, 420–425. [Google Scholar] [CrossRef]
- Arhondakis, S.; Milanesi, M.; Castrignanò, T.; Gioiosa, S.; Valentini, A.; Chillemi, G. Evidence of Distinct Gene Functional Patterns in GC-poor and GC-rich Isochores in Bos taurus. Anim. Genet. 2020, 51, 358–368. [Google Scholar] [CrossRef] [PubMed]
- Ayad, L.A.K.; Dourou, A.-M.; Arhondakis, S.; Pissis, S.P. IsoXpressor: A Tool to Assess Transcriptional Activity within Isochores. Genome Biol. Evol. 2020, 12, 1573–1578. [Google Scholar] [CrossRef]
- Delage, W.J.; Thevenon, J.; Lemaitre, C. Towards a Better Understanding of the Low Recall of Insertion Variants with Short-Read Based Variant Callers. BMC Genom. 2020, 21, 762. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Bernaola-Galván, P.; Carpena, P.; Oliver, J.L. Isochores Merit the Prefix ‘Iso’. Comput. Biol. Chem. 2003, 27, 5–10. [Google Scholar] [CrossRef]
- Mourad, R. Studying 3D Genome Evolution Using Genomic Sequence. Bioinformatics 2019, 36, btz775. [Google Scholar] [CrossRef]
- Nacheva, E.; Mokretar, K.; Soenmez, A.; Pittman, A.M.; Grace, C.; Valli, R.; Ejaz, A.; Vattathil, S.; Maserati, E.; Houlden, H.; et al. DNA Isolation Protocol Effects on Nuclear DNA Analysis by Microarrays, Droplet Digital PCR, and Whole Genome Sequencing, and on Mitochondrial DNA Copy Number Estimation. PLoS ONE 2017, 12, e0180467. [Google Scholar] [CrossRef]
- Arita, M. Writing Information into DNA. In Aspects of Molecular Computing; Jonoska, N., Păun, G., Rozenberg, G., Eds.; Lecture Notes in Computer Science; Springer: Berlin/Heidelberg, Germany, 2003; Volume 2950, pp. 23–35. ISBN 978-3-540-20781-8. [Google Scholar]
- Schmidt, T.; Frishman, D. Assignment of Isochores for All Completely Sequenced Vertebrate Genomes Using a Consensus. Genome Biol. 2008, 9, R104. [Google Scholar] [CrossRef]
- Cock, P.J.A.; Antao, T.; Chang, J.T.; Chapman, B.A.; Cox, C.J.; Dalke, A.; Friedberg, I.; Hamelryck, T.; Kauff, F.; Wilczynski, B.; et al. Biopython: Freely Available Python Tools for Computational Molecular Biology and Bioinformatics. Bioinformatics 2009, 25, 1422–1423. [Google Scholar] [CrossRef] [PubMed]
- Clauset, A.; Shalizi, C.R.; Newman, M.E.J. Power-Law Distributions in Empirical Data. SIAM Rev. 2009, 51, 661–703. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mean for isoPlotter | Median for isoPlottter | Mean for isoSegmenter | Median for isoSegmenter | p-Value * | |
|---|---|---|---|---|---|
| Sensitivity | 0.5362 | 0.52 | 0.2714 | 0.025 | p < 10−16 |
| Precision | 0.3122 | 0.2903 | 0.2854 | 0.0714 | p < 10−16 |
| Jaccard Index | 0.26 | 0.2321 | 0.2394 | 0.0196 | p < 10−16 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Graur, D. Slaying (Yet Again) the Brain-Eating Zombie Called the “Isochore Theory”: A Segmentation Algorithm Used to “Confirm” the Existence of Isochores Creates “Isochores” Where None Exist. Int. J. Mol. Sci. 2022, 23, 6558. https://doi.org/10.3390/ijms23126558
Graur D. Slaying (Yet Again) the Brain-Eating Zombie Called the “Isochore Theory”: A Segmentation Algorithm Used to “Confirm” the Existence of Isochores Creates “Isochores” Where None Exist. International Journal of Molecular Sciences. 2022; 23(12):6558. https://doi.org/10.3390/ijms23126558
Chicago/Turabian StyleGraur, Dan. 2022. "Slaying (Yet Again) the Brain-Eating Zombie Called the “Isochore Theory”: A Segmentation Algorithm Used to “Confirm” the Existence of Isochores Creates “Isochores” Where None Exist" International Journal of Molecular Sciences 23, no. 12: 6558. https://doi.org/10.3390/ijms23126558
APA StyleGraur, D. (2022). Slaying (Yet Again) the Brain-Eating Zombie Called the “Isochore Theory”: A Segmentation Algorithm Used to “Confirm” the Existence of Isochores Creates “Isochores” Where None Exist. International Journal of Molecular Sciences, 23(12), 6558. https://doi.org/10.3390/ijms23126558