1. Introduction
Platelet activation and aggregation upon vascular injury is essential for primary hemostasis, but also has a function in atherogenesis and formation of occlusive thrombi [
1]. The gaseous molecule nitric oxide (NO) is an endogenous inhibitor of platelet aggregation by activating soluble guanylyl cyclase (sGC) and therefore leading to an increase in cyclic guanosine monophosphate (cGMP) production [
2,
3,
4,
5]. cGMP induces a PKGIβ-dependent phosphorylation of the inositol 1,4,5-triphosphate receptor-associated cGMP kinase substrate 1 (IRAG1), resulting in an inhibition of inositol 1,4,5-trisphosphate (IP
3)-induced Ca
2+ release from the endoplasmic reticulum (ER) [
6,
7]. It is shown that the formation of a macro complex consisting of IRAG1, IP
3R1 and PKGIβ mediates the NO/cGMP-dependent inhibition of thrombin-induced Ca
2+ release in platelets and is also an important factor of NO/cGMP-dependent inhibition of platelet aggregation [
8,
9].
Inositol 1,4,5-triphosphate receptor-associated 2 (IRAG2) is a type II membrane protein localized to the cytoplasmic face of the ER and was first described in 1994 by Behrens et al. [
10,
11]. IRAG2 is also known as Jaw1 or lymphoid-restricted membrane protein (LRMP) and shows a homology with IRAG1, especially in its coiled coil domain with a homology of 44% [
12,
13,
14]. The coiled coil domain of IRAG1 is essential for interaction with inositol trisphosphate receptors (IP
3 receptors/IP
3R) and regulation of Ca
2+ release from intracellular stores [
6,
15]. IRAG2 was shown to interact with IP
3R3 via its coiled coil domain in COS7 heterologous expression system and also with IP
3R2 in intestinal tuft cells [
13,
16]. Recently, we observed an interaction of IRAG2 with IP
3R1-3 in the pancreas [
17]. Expression of IRAG2 was found in a variety of tissues and cell lines, e.g., B-cell lines, T-cell lines, spleen, thymus, sinoatrial nodes, sweet, bitter and umami taste-responsive cells or intestinal tuft cells [
10,
12,
13,
16,
18]. Recently, we also detected IRAG2 expression in pancreatic acinar cells [
17].
The physiological and molecular functions of IRAG2 are still widely unknown. Until now, in platelets no data exist regarding the expression and functional role of IRAG2 on platelet aggregation. Due to its homology to IRAG1 [
12,
13,
14], it is possible that IRAG2 has similar interaction partners in platelets and that IRAG2 also has an effect on agonist induction or NO/cGMP-dependent inhibition of platelet aggregation. Furthermore, it is unknown if IRAG2 is phosphorylated by cGMP via PKGI in platelets similar to its homologue IRAG1 and if it thereby modulates NO/cGMP-dependent platelet inhibition.
In our study, we analyzed the expression of IRAG2 in platelets and its interaction with IP3 receptors as well as its possible role in agonist-induced and NO/cGMP-dependent inhibition of platelet aggregation.
3. Discussion
Inositol 1,4,5-triphosphate receptor-associated 2 (IRAG2) is also known as Jaw1 or lymphoid-restricted membrane protein (LRMP). It contains a coiled coil domain and a COOH-terminal hydrophobic anchor domain, which is important for the localization to the endoplasmic reticulum (ER) [
10,
11]. Recently, we detected expression of IRAG2 in pancreatic acinar cells, where it interacts with all types of IP
3 receptors and modulates basal calcium levels and basal secretion of amylase [
17]. IRAG2 has been detected before in a variety of other tissues and cell lines, since its first description in 1994 by Behrens et al. [
10]. Expression was reported, e.g., in thymus, spleen, B- and T-cell lines, sinoatrial nodes or sweet, bitter and umami taste-responsive cells [
10,
12,
13,
18]. IRAG2 shares a homology with inositol 1,4,5-triphosphate receptor-associated cGMP kinase substrate 1 (IRAG1) [
12,
13,
14], that is expressed among others in human and in murine platelets [
8]. In our current study, we demonstrate that IRAG2 is, like its homologue, also expressed in murine platelets. Until now, the function of IRAG2 in murine platelets was unknown. However, this expression suggests that IRAG2 might be involved in platelet function, e.g., platelet aggregation. In addition, to our knowledge, this is the first time that the expression of IRAG2 is shown in murine platelets.
It is reported that IRAG2 shares a homology with IRAG1, especially in its coiled coil domain with 44% [
12,
13,
14]. IRAG1 forms a macro complex with the IP
3R1 and PKGIβ in platelets, where the coiled coil domain is essential for the interaction with the IP
3R1 [
6,
8]. Recently, we detected an interaction of IRAG2 with IP
3R1-3 in the murine pancreas [
17]. In addition, an interaction of IRAG2 with different IP
3 receptor subtypes has been detected before in other tissues. In intestinal tuft cells, IRAG2 interacts with IP
3R2 [
16], and in COS7 heterologous expression system interaction has been detected with IP
3R3 [
13]. Therefore, it is possible that this interaction with IP
3 receptors also occurs in platelets. Here, we show that IRAG2 interacts with IP
3R1, IP
3R2 and IP
3R3 in murine platelets. However, in the input platelet lysates IP
3R2 shows a higher molecular weight compared to the co-immunoprecipitated IP
3R2 in IRAG2-WT probes, but we observe no signal in the co-immunoprecipitated probes of IRAG2-KO. Therefore, we assume that this is not an unspecific band for IP
3R2, but that IRAG2 interacts with a shorter fragment of the IP
3R2, which is only detectable after immunoprecipitating the complex of IRAG2 and IP
3R2. In addition, the amount of co-immunoprecipitated IP
3R1-3 is low, and therefore the interaction of IRAG2 and IP
3R1-3 appears to be weak. Furthermore, we investigated if IRAG2 directly interacts with IRAG1 or PKGIβ. In our study, we did not see a direct interaction of IRAG2 neither with PKGIβ nor with IRAG1. Hence, IRAG2 does not form a macro complex in platelets, where PKGIβ or IRAG1 are included. This is not surprising, as the stable interaction site of IRAG1 with the leucine zipper of PKGIβ is located between amino acid 152 and 184 of the N-terminal part of IRAG1, which is lacking in IRAG2 [
7,
19].
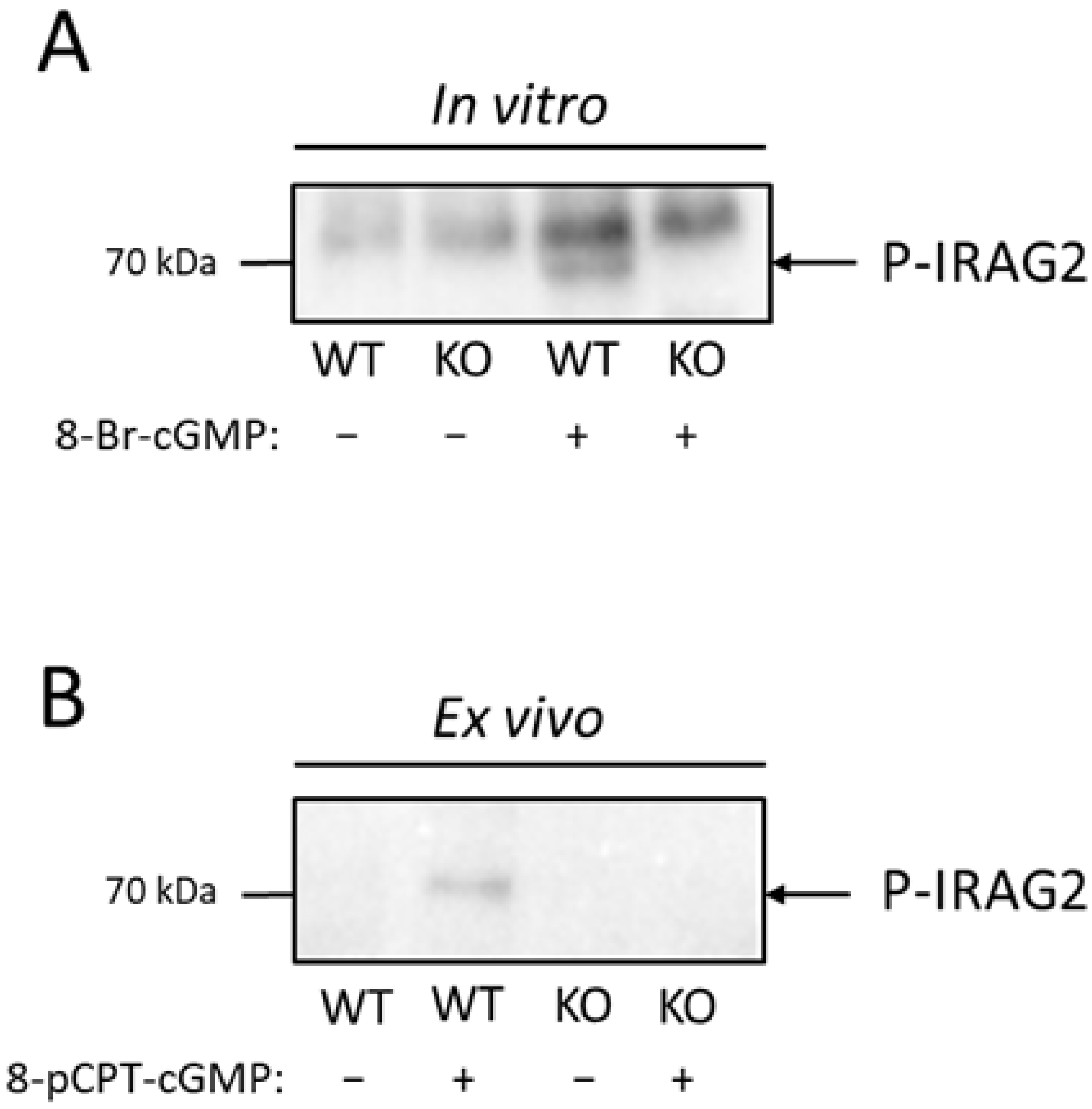
Our finding that IRAG2 reveals no direct interaction with PKGIβ does not exclude a cGMP-dependent phosphorylation of IRAG2 via PKGI. We stimulated lysed platelets as well as intact platelets with the cGMP analogue 8-pCPT-cGMP and thereby detected phosphorylation of IRAG2 in murine platelets. To evaluate this phosphorylation, we used a phospho-(Ser/Thr) PKA substrate antibody that identifies PKA-, PKC- or PKG-dependent phosphorylation at serine or threonine residues of proteins. As cGMP or cGMP analogues such as 8-pCPT-cGMP predominantly activate PKGI [
4,
20], IRAG2 phosphorylation after stimulation with 8-pCPT-cGMP might be mediated mainly through PKGI. To detect which isoform of PKGI, PKGIα or PKGIβ, is responsible for the phosphorylation of IRAG2, further experiments are required. However, in murine platelets it is known that PKGIβ is the predominant isoform and only a small amount of PKGIα is expressed. In human platelets, only PKGIβ is expressed. In addition, IRAG1 phosphorylation is mediated through PKGIβ in murine and in human platelets [
8]. This raises the possibility that IRAG2 phosphorylation in murine platelets is also achieved through PKGIβ. Our result, that IRAG2 is phosphorylated in a cGMP-dependent manner in murine platelets, correlates with the findings of Makhoul et al., who analyzed phosphoproteins in human platelets by a quantitative phosphoproteomics study. They detected phosphorylation of LRMP at amino acids Ser363, Thr375 and Ser418 upon stimulation with NO donors or riociguat in human platelets [
21]. Remarkably, the phosphorylation sites of Ser363 and Thr375 in IRAG2 (consensus sequences: RSAS
363 and RRVT
375) are very homologous to the identified phosphorylation sites of Ser664 and Ser677 in IRAG1 (consensus sequences: RSMS
664 and RRVS
677). For future studies, it is also necessary to detect the exact phosphorylation sites of IRAG2 in murine platelets.
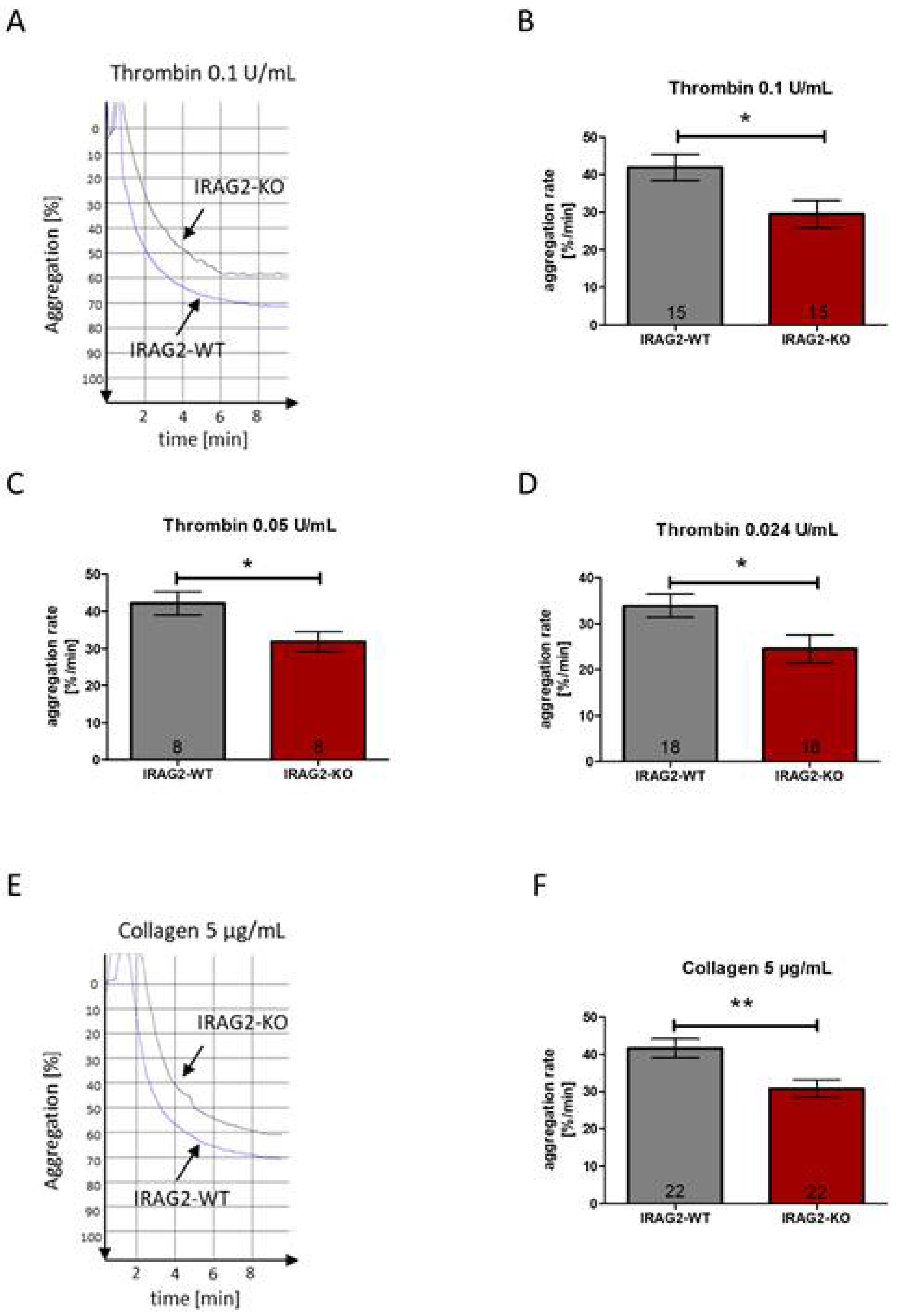
In IRAG1-KO mice, platelet aggregation upon stimulation with agonists such as thrombin, collagen or the thromboxane A2 (TxA2) analogue U46619 is increased compared to IRAG1-WT mice [
9]. These agonists stimulate platelet aggregation mainly by activating the phospholipase C (PLC) and therefore generating inositol trisphosphate (IP
3), which leads to an increase in cytosolic Ca
2+ [
22,
23]. This increase in Ca
2+ results in an activation of GPIIb/IIIa and thus to the aggregation of platelets mediated by fibrinogen [
23,
24]. In our study, we used collagen and thrombin to evoke platelet aggregation to examine the effect of IRAG2 deficiency in platelets. In contrast to IRAG1-KO platelets, IRAG2-KO platelets revealed a lower aggregation rate compared to WT mice. Consequently, IRAG2 augments agonist-induced platelet aggregation and therefore exerts an opposing function on IRAG1. Opposing functions of IRAG1 and IRAG2 have also been observed before by Peters et al., where IRAG1 and IRAG2 both interact with HCN4 channels, but revealed contrary effects on the HCN4 function [
12]. To analyze if the cGMP-dependent phosphorylation of IRAG2 impacts platelet aggregation, we preincubated the platelets with the NO donor natrium nitroprusside (SNP) or the cGMP analogue 8-pCPT-cGMP. NO donors or cGMP analogues cause an inhibition of platelet aggregation [
2,
3,
4,
20]. This effect is mediated by phosphorylation of IRAG1 via PKGI, leading to a reduced secretion of Ca
2+ from the ER and therefore to an inhibition of platelet aggregation [
6,
8,
9]. For IRAG2, we also observed opposing effects compared to IRAG2 on the inhibition of platelet aggregation. IRAG1-KO mice show no inhibition or only a weak inhibition of platelet aggregation after preincubation with SNP or 8-pCPT-cGMP [
9]. In contrast, for IRAG2 we observe a reduced aggregation rate and therefore an enhanced inhibition of platelet aggregation in platelets from IRAG2-KO mice. This leads to the assumption that phosphorylation of IRAG2 by activation of the NO/cGMP pathway enhances platelet aggregation and that IRAG2 might be the counterpart to IRAG1 in murine platelets. Future experiments will require the measurement of Ca
2+ in IRAG2-KO platelets to see if the reduced platelet aggregation in IRAG2-KO platelets occurs due to lower Ca
2+ release after phosphorylation of IRAG2 by activation of the NO/cGMP pathway. In addition, it will be interesting to investigate if IRAG2 might have an impact on thrombus formation after vascular injury. For IRAG1 it is seen that it prevents thrombus formation in a NO/cGMP-dependent manner [
8]. As IRAG2 appears to enhance aggregability of platelets, IRAG2 might have a potential role in the formation of thrombi and therefore in atherogenesis and development of cardiovascular diseases.
In summary, our study reveals new outcomes about the function of IRAG2 in platelets:
IRAG2 is expressed in murine platelets and interacts with IP3R1, IP3R2 and IP3R3; however, no stable interaction of IRAG2 is observed with PKGIβ or IRAG1. Hence, no macro complex consisting of IRAG2, PKGIβ and IP3R1 is formed like it is reported for IRAG1.
IRAG2 is phosphorylated in a cGMP-dependent manner by PKGI.
IRAG2-KO mice reveal a reduced platelet aggregation rate compared to IRAG2-WT mice, indicating that IRAG2 enhances platelet aggregation. When preincubating platelets with SNP or 8-pCPT-cGMP, the aggregation rate upon treatment with thrombin is reduced in platelets of IRAG2-KO mice compared to IRAG2-WT mice. Therefore, the inhibition of platelet aggregation by SNP or cGMP is enhanced in IRAG2-KO.
Taken together, IRAG2 appears to act as a counterpart to IRAG1 in murine platelets with opposing effects on platelet aggregation.
4. Materials and Methods
4.1. Animals
Global
Irag2−/− and
lacZ ×
Irag2−/− mice were generated as previously described [
17]. All experiments were conducted with male and female IRAG2-KO mice (genotype:
Irag2−/−), lacZ × IRAG2-KO mice (genotype:
lacZ ×
Irag2−/−) and their wild type littermates (IRAG2-WT; genotype:
Irag2+/+,
Irag2fl/fl) older than 8 weeks. Animals were bred and maintained in the animal facilities of the University of Regensburg according to the Guidelines of the Federation of European Laboratory Animal Science Associations (Bavaria, Germany; Regierung von Unterfranken: DMS 2532-2-236), following all guidelines according to the German animal protection law, with free access to food and water ad libitum.
4.2. Materials
The following chemicals and antibodies were purchased for this study: 8 pCPT-cGMP, 8-Br-cGMP (BIOLOG Life Science Institute, Bremen, Germany), sodium nitroprusside (SNP; Sigma Aldrich
®, Taufkirchen, Germany), collagen, thrombin (Probe & Go, Osburg, Germany), anti-mouse LRMP (C-terminal) = IRAG2-antibody (Sigma-Aldrich
®, Taufkirchen, Germany; SAB1306900), IP
3R1-antibody (Novusbio, Abingdon, UK; NBP1-21398), IP
3R2-antibody (Santa Cruz, Heidelberg, Germany; sc-7278), IP
3R3-antibody (BD Transduction Laboratories TM, BD Biosciences, San Jose, CA, USA; 610313), β-Galactosidase-antibody (Abcam plc, Cambridge, UK; ab9361), Phospho-(Ser/Thr) PKA substrate-antibody (Cell Signaling Technology, Leiden, The Netherlands; #9621), GAPDH-antibody (Cell Signaling Technology, Leiden, The Netherlands; #2118), secondary antibody goat-anti-mouse (Sigma-Aldrich
®, Taufkirchen, Germany; A4416), secondary antibody goat-anti-rabbit (Dianova GmbH, Hamburg, Germany; 111-035-003), secondary antibody donkey-anti-goat (Dianova GmbH, Hamburg, Germany; 705-035-003), secondary antibody goat-anti-chicken (Dianova GmbH, Hamburg, Germany; DAB87304). The following antibodies are in-house productions: IRAG1-antibody, PKGIβ-antibody [
25].
4.3. Isolation and Preparation of Platelets
Platelets of IRAG2-WT and IRAG2-KO or lacZ × IRAG2-KO mice were isolated as described before [
9]. Briefly, mice were euthanized with CO
2 and blood was drawn by cardiac puncture into 200 µL Alsever’s solution (Sigma Aldrich
®, Taufkirchen, Germany), followed by mixing with 500 µL of buffer B (20 mM HEPES, 138 mM NaCl, 2.9 mM KCl, 1 mM MgCl
2, 0.36 mM NaH
2PO
4, pH 6.2). After centrifugation of the mixture at 70×
g for 15 min (min) at room temperature (RT), supernatant was collected and centrifuged at 600×
g for 5 min at RT. The resulting platelet pellet was resuspended in buffer B (pH 7.4) for platelet aggregation and ex vivo phosphorylation experiments. For Western Blot experiments, (co-)immunoprecipitation and in vitro phosphorylation, platelet pellet was homogenized directly after isolation with 2% Lubrol-buffer (2% nonaethylene glycol monododecyl ether, 150 mM NaCl, 20 mM Tris in ddH
2O, pH 8.0), containing protease inhibitors (1 mM benzamidine, 0.5 µg/mL leupeptin, 300 µM PMSF) and 1× PhosSTOP (Roche, Mannheim, Germany). The homogenate was centrifuged at 18,000×
g for 15 min at 4 °C to remove cell debris and supernatant was collected and stored at −80 °C until further experiments were conducted. Protein concentration was detected using a Lowry kit (Bio-Rad Laboratories, Inc., Munich, Germany).
4.4. In Vitro Phosphorylation of Platelets
For detecting IRAG2 phosphorylation in platelets, 500 µg of lysate was incubated in 200 µL phosphorylation buffer (50 mM MES, 10 mM ATP, pH 7.2, protease and phosphatase inhibitors as described in
Section 4.3) and stimulated with 100 µM 8-Br-cGMP for 20 min at 37 °C or ddH
2O as a control. After phosphorylation, lysates were kept on ice and IRAG2 was immunoprecipitated by IRAG2 antibody as described in
Section 4.6 and analyzed by SDS-Page and Western Blot. Phosphorylation of immunoprecipitated IRAG2 was detected via phospho-(Ser/Thr) PKA substrate antibody.
4.5. Ex Vivo Phosphorylation of Intact Platelets
An amount of 1.75 × 10
8 platelets from IRAG2-WT or IRAG2-KO were preincubated in 500 µL of buffer B (pH 7.4) for 1 h at 37 °C, followed by incubation with 100 µM 8-pCPT-cGMP for 20 min at 37 °C or ddH
2O as a control. After stimulation, platelets were centrifuged at 2000×
g for 5 min at 4 °C and platelet pellet was homogenized with 150 µL of 2% Lubrol buffer (
Section 4.3). The homogenate was centrifuged at 18,000×
g for 15 min at 4 °C and supernatant was collected. An amount of 350 µL of co-immunoprecipitation buffer (
Section 4.6) was added and immunoprecipitation of IRAG2 was carried out as described in
Section 4.6 to detect IRAG2-specific phosphorylation.
4.6. (Co)-Immunoprecipitation
(Co)-immunoprecipitation was carried out as described before [
17]. Briefly, 1000 µg of platelet lysates or 500 µg of phosphorylated platelet lysates were incubated in 500 µL of co-immunoprecipitation buffer (50 mM Tris HCl, 15 mM EGTA, 100 mM NaCl, 0.1% Triton X-100 in ddH
2O, pH 7.5) containing protease inhibitors, 1× PhosSTOP (Roche, Mannheim, Germany), 1 mM DTT and 1 µg of anti-mouse LRMP (C-terminal) antibody = anti-IRAG2 antibody for 90 min on ice. Following incubation, mixture was centrifuged at 18,000×
g for 10 min at 4 °C and supernatant was transferred to 15 µL of washed and blocked (3% BSA in co-immunoprecipitation-buffer, 30 min, 4 °C, Protein-A-Sepharose-Beads (Sigma-Aldrich
®, Taufkirchen, Germany) and incubated overnight at 4 °C. Beads were eluted with 2× Laemmli buffer and supernatant was analyzed by SDS-Page and Western Blot (
Section 4.7).
4.7. Western Blot Analysis
Expression of several proteins was analyzed via Western Blot. Therefore, 35 to 70 µg of protein was used for SDS-Page and blotted on a PVDF membrane (Immobilon-P, Merck KGaA, Darmstadt, Germany) after electrophoresis. Membranes were blocked with 5% non-fatty milk (VWR, Darmstadt, Germany), containing 0.05% Tween® 20 (Sigma-Aldrich®, Taufkirchen, Germany) for 2 h at RT, followed by incubating the membrane with primary antibodies (anti-mouse LRMP (C-terminal) = anti-IRAG2: 1:1000; anti-IP3R1: 1:1000; anti-IP3R2: 1:200; anti-IP3R3: 1:100; anti-β-Galactosidase: 1:1000; anti-IRAG1: 1:200; anti-PKGIβ: 1:200; anti-phospho-(Ser/Thr) PKA substrate: 1:1000) at 4 °C overnight. After incubating the membranes in HRP-conjugated secondary antibodies (goat-anti-mouse: 1:10,000; goat-anti-rabbit: 1:10,000; donkey-anti-goat: 1:10,000; goat-anti-chicken: 1:5000) for 2 h at RT, detection was carried out using ClarityTM Western ECL Substrate (Bio-Rad Laboratories, Inc., Munich, Germany) and ChemiDocTM MP Imaging System (Bio-Rad Laboratories, Inc., Munich, Germany). For statistical analysis, the signal intensity of each band was quantified using Image LabTM Software (Bio-Rad Laboratories, Inc., Munich, Germany) and was normalized to the total protein of the appropriate lane. Therefore, 1.5% trichloroethanol-containing hand-casting SDS-gels were used. Analyzed number of lysates from different animals are indicated by n-numbers in the description of the figure. Quantified samples were applied to one up to three gels (depending on the analyzed n-number) and were evaluated after the same gel-run at the same timepoints.
4.8. Platelet Aggregation
Platelets from IRAG2-WT and IRAG2-KO mice (each 1.0 × 108 platelets/mL) were preincubated in buffer B (20 mM HEPES, 138 mM NaCl, 2.9 mM KCl, 1 mM MgCl2, 0.36 mM NaH2PO4, pH 7.4) for 5 min at 37 °C without stirring and then incubated with or without 8-pCPT-cGMP (150 µM or 200 µM) for 10 min at 37 °C, SNP (2.5 µM) for 2 min at 37 °C or buffer B in equal amounts as a control. Following that, aggregation was initiated by collagen (5 µg/mL) or thrombin (0.024 U/mL, 0.05 U/mL or 0.1 U/mL). Aggregation was measured at 37 °C with stirring (1200 rpm) by an optical aggregometer (Chronolog, Havertown, PA, USA) using the “Aggro/Link Software 5.1”. Results were calculated as maximal slope.
4.9. Statistical Analysis
All data are shown as mean ± SEM. For testing the samples for normality, the Shapiro–Wilk test was used. Normally distributed parameters were analyzed using an unpaired Student’s t-test to calculate significant differences between two means. Non-parametric data were analyzed using the Wilcoxon–Mann–Whitney test. Statistical analysis was performed with “GraphPad Prism version 5.01”. Significant differences in the graphs are shown by asterisks (*) (p < 0.05), (**) (p < 0.01) and (***) (p < 0.001).




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}




