
CRISPR/Cas9-Mediated Constitutive Loss of VCP (Valosin-Containing Protein) Impairs Proteostasis and Leads to Defective Striated Muscle Structure and Function In Vivo


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
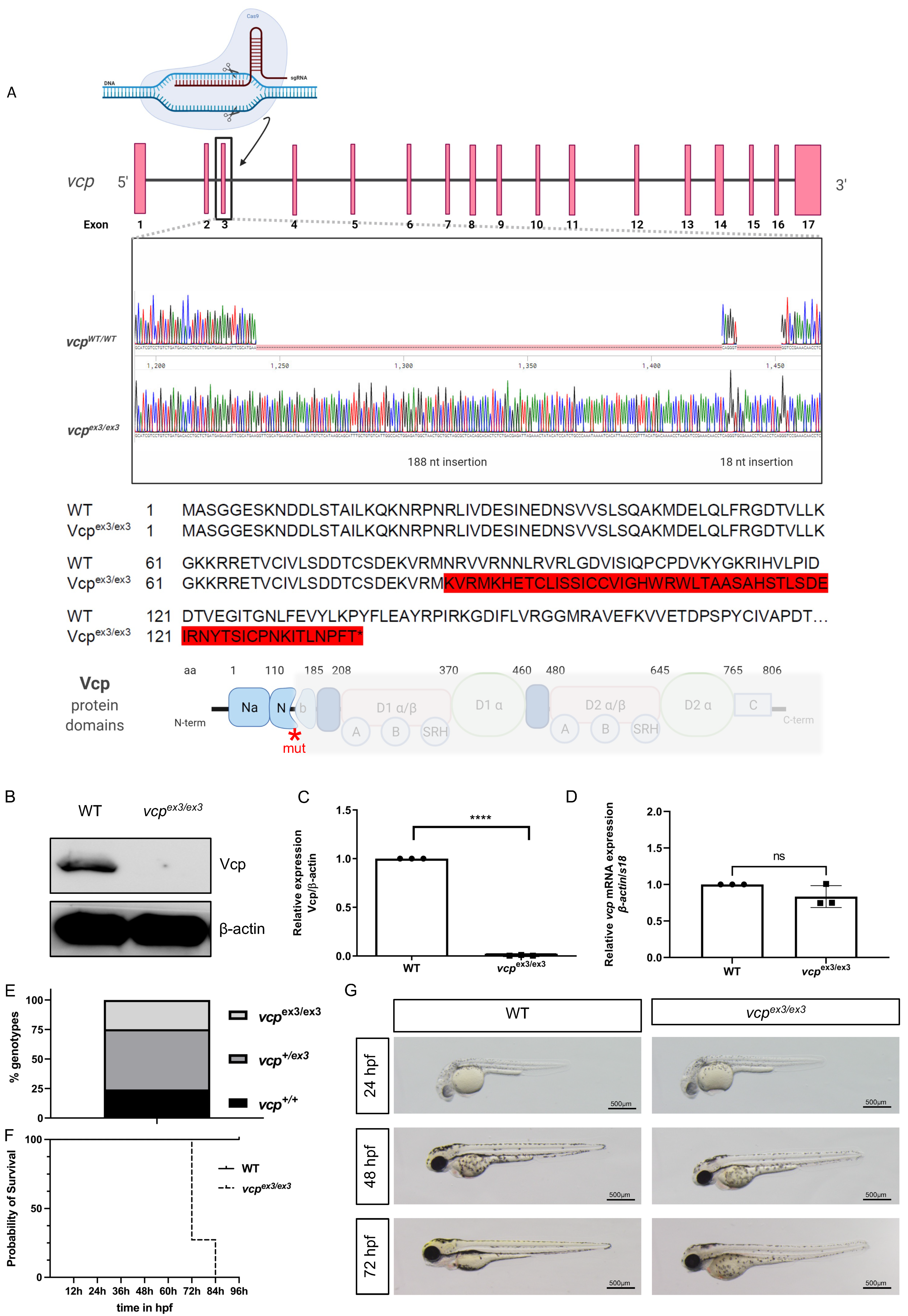
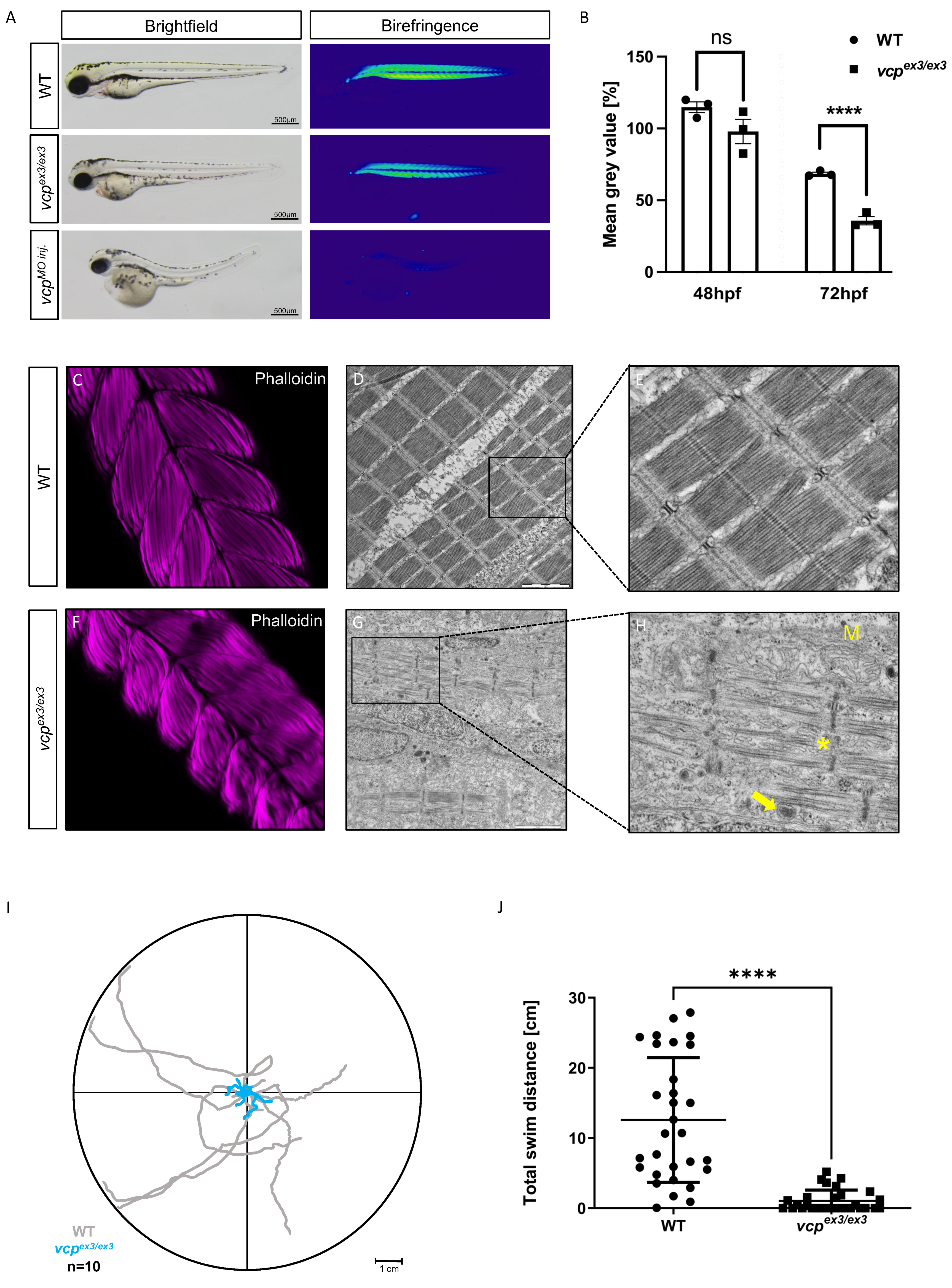
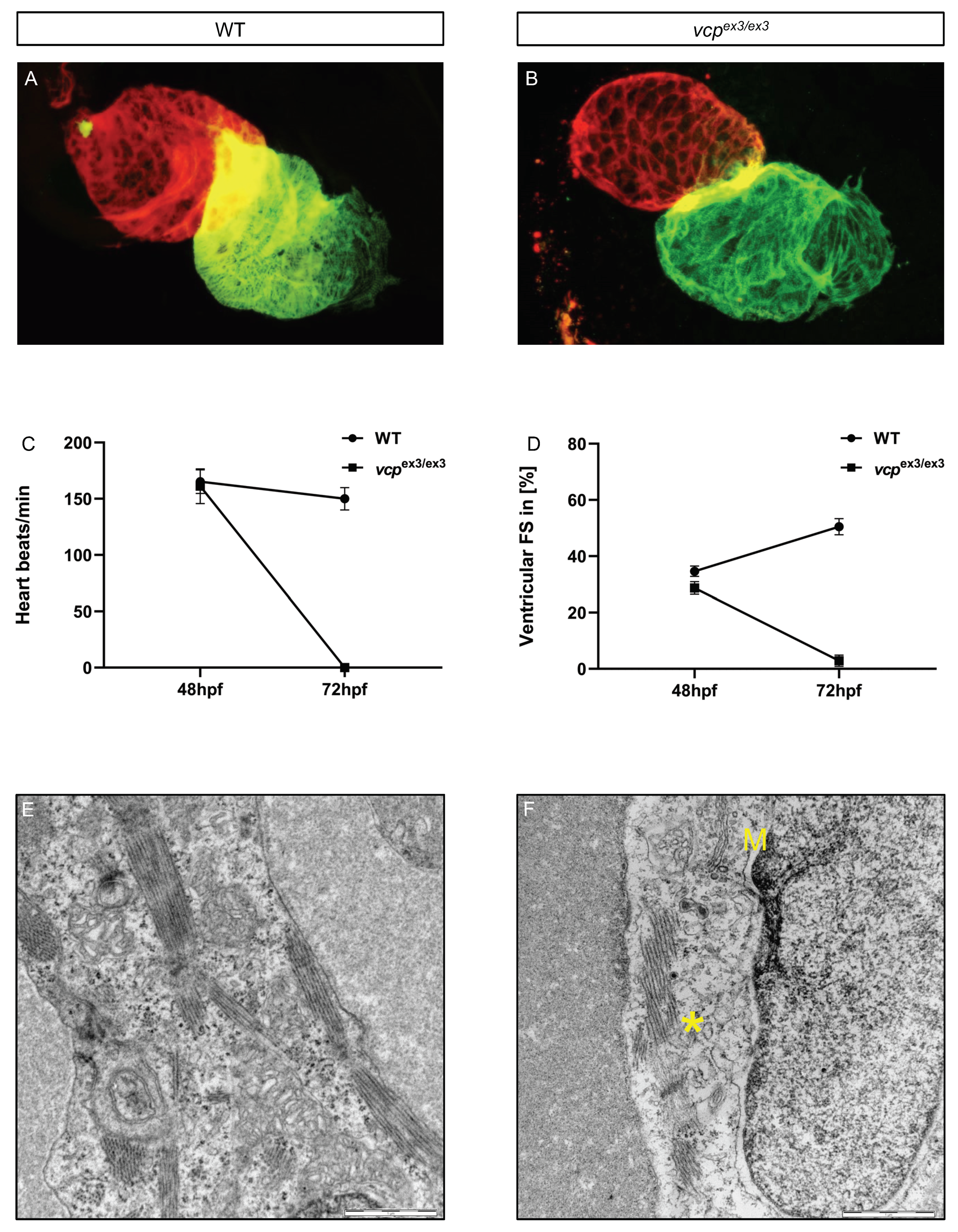
2.1. Targeted Constitutive Knockout of Zebrafish Vcp Leads to Defective Heart and Skeletal Muscle Structure and Function
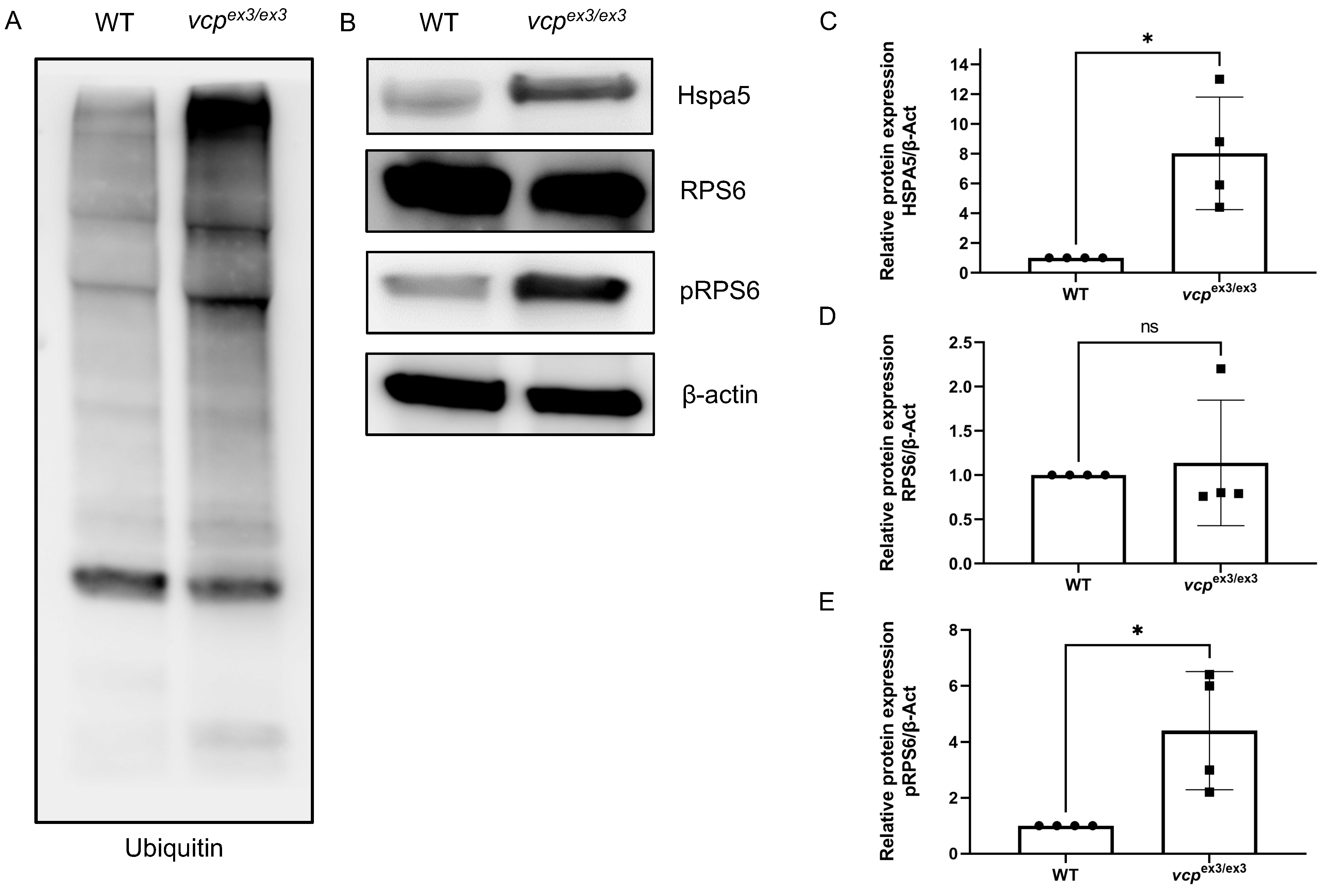
2.2. Loss of Vcp Interferes with Protein Homeostasis in Developing Zebrafish
3. Discussion
4. Materials and Methods
4.1. Zebrafish Strains and Injection Procedures
4.2. Genotyping
4.3. RNA Extraction and Quantitative Real-Time PCR
4.4. Immunoblot Analysis
4.5. Light Microscopy Analysis
4.6. Transmission Electron Microscopy
4.7. Immunostaining
4.8. Touch-Evoked Escape Response Assay (TEER)
4.9. Cardiac Functional Assessment and Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Van Den Boom, J.; Meyer, H. VCP/p97-Mediated Unfolding as a Principle in Protein Homeostasis and Signaling. Mol. Cell 2018, 69, 182–194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, S.H.; Liu, J.; Kobayashi, R.; Tonks, N.K. Identification of the cell cycle regulator VCP (p97/CDC48) as a substrate of the band 4.1-related protein-tyrosine phosphatase PTPH1. J. Biol. Chem. 1999, 274, 17806–17812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, K.; Cheng, H.H.; Zhou, R.J. Molecular mechanisms and functions of autophagy and the ubiquitin-proteasome pathway. Yi Chuan 2012, 34, 5–18. [Google Scholar] [CrossRef] [PubMed]
- Watts, G.D.J.; Wymer, J.; Kovach, M.J.; Mehta, S.G.; Mumm, S.; Darvish, D.; Pestronk, A.; Whyte, M.P.; Kimonis, V.E. Inclusion body myopathy associated with Paget disease of bone and frontotemporal dementia is caused by mutant valosin-containing protein. Nat. Genet. 2004, 36, 377–381. [Google Scholar] [CrossRef] [PubMed]
- Segawa, M.; Hoshi, A.; Naruse, H.; Kuroda, M.; Bujo, H.; Ugawa, Y. A patient with familial amyotrophic lateral sclerosis associated with a new valosin-containing protein (VCP) gene mutation. Rinsho Shinkeigaku 2015, 55, 914–920. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrer, I.; Olivé, M. Molecular pathology of myofibrillar myopathies. Expert Rev. Mol. Med. 2008, 10, e25. [Google Scholar] [CrossRef]
- Hübbers, C.U.; Clemen, C.S.; Kesper, K.; Böddrich, A.; Hofmann, A.; Kämäräinen, O.; Tolksdorf, K.; Stumpf, M.; Reichelt, J.; Roth, U.; et al. Pathological consequences of VCP mutations on human striated muscle. Brain 2007, 130, 381–393. [Google Scholar] [CrossRef] [Green Version]
- Wani, A.; Zhu, J.; Ulrich, J.D.; Eteleeb, A.; Sauerbeck, A.D.; Reitz, S.J.; Arhzaouy, K.; Ikenaga, C.; Yuede, C.M.; Pittman, S.K.; et al. Neuronal VCP loss of function recapitulates FTLD-TDP pathology. Cell Rep. 2021, 36, 109399. [Google Scholar] [CrossRef]
- Arhzaouy, K.; Papadopoulos, C.; Schulze, N.; Pittman, S.K.; Meyer, H.; Weihl, C.C. VCP maintains lysosomal homeostasis and TFEB activity in differentiated skeletal muscle. Autophagy 2019, 15, 1082–1099. [Google Scholar] [CrossRef]
- Wani, A.; Weihl, C.C. Loss-of-function mutation in VCP mimics the characteristic pathology as in FTLD-TARDBP. Autophagy 2021, 17, 4502–4503. [Google Scholar] [CrossRef]
- Brody, M.J.; Vanhoutte, D.; Bakshi, C.V.; Liu, R.; Correll, R.N.; Sargent, M.A.; Molkentin, J.D. Disruption of valosin-containing protein activity causes cardiomyopathy and reveals pleiotropic functions in cardiac homeostasis. J. Biol. Chem. 2019, 294, 8918–8929. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Zhou, N.; Ma, B.; Wu, W.; Stoll, S.; Lai, L.; Qin, G.; Qiu, H. Functional Inhibition of Valosin-Containing Protein Induces Cardiac Dilation and Dysfunction in a New Dominant-Negative Transgenic Mouse Model. Cells 2021, 10, 2891. [Google Scholar] [CrossRef] [PubMed]
- Viswanathan, M.C.; Blice-Baum, A.C.; Sang, T.K.; Cammarato, A. Cardiac-Restricted Expression of VCP/TER94 RNAi or Disease Alleles Perturbs Drosophila Heart Structure and Impairs Function. J. Cardiovasc. Dev. Dis. 2016, 3, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Obeidi, E.; Al-Tahan, S.; Surampalli, A.; Goyal, N.; Wang, A.K.; Hermann, A.; Omizo, M.; Smith, C.; Mozaffar, T.; Kimonis, V. Genotype-phenotype study in patients with valosin-containing protein mutations associated with multisystem proteinopathy. Clin. Genet. 2018, 93, 119–125. [Google Scholar] [CrossRef] [PubMed]
- Müller, J.M.; Deinhardt, K.; Rosewell, I.; Warren, G.; Shima, D.T. Targeted deletion of p97 (VCP/CDC48) in mouse results in early embryonic lethality. Biochem. Biophys. Res. Commun. 2007, 354, 459–465. [Google Scholar] [CrossRef] [PubMed]
- Higashiyama, H.; Hirose, F.; Yamaguchi, M.; Inoue, Y.H.; Fujikake, N.; Matsukage, A.; Kakizuka, A. Identification of ter94, Drosophila VCP, as a modulator of polyglutamine-induced neurodegeneration. Cell Death Differ. 2002, 9, 264–273. [Google Scholar] [CrossRef] [Green Version]
- Kustermann, M.; Manta, L.; Paone, C.; Kustermann, J.; Lausser, L.; Wiesner, C.; Eichinger, L.; Clemen, C.S.; Schröder, R.; Kestler, H.A.; et al. Loss of the novel Vcp (valosin containing protein) interactor Washc4 interferes with autophagy-mediated proteostasis in striated muscle and leads to myopathy in vivo. Autophagy 2018, 14, 1911–1927. [Google Scholar] [CrossRef] [Green Version]
- Sztal, T.E.; Stainier, D.Y.R. Transcriptional adaptation: A mechanism underlying genetic robustness. Development 2020, 147, dev186452. [Google Scholar] [CrossRef]
- Diofano, F.; Weinmann, K.; Schneider, I.; Thiessen, K.D.; Rottbauer, W.; Just, S. Genetic compensation prevents myopathy and heart failure in an in vivo model of Bag3 deficiency. PLoS Genet. 2020, 16, e1009088. [Google Scholar] [CrossRef]
- Smith, L.L.; Beggs, A.H.; Gupta, V.A. Analysis of skeletal muscle defects in larval zebrafish by birefringence and touch-evoke escape response assays. J. Vis. Exp. 2013, 82, e50925. [Google Scholar] [CrossRef] [Green Version]
- Glickman, N.S.; Yelon, D. Cardiac development in zebrafish: Coordination of form and function. Semin Cell Dev. Biol. 2002, 13, 507–513. [Google Scholar] [CrossRef] [PubMed]
- Imamura, S.; Yabu, T.; Yamashita, M. Protective role of cell division cycle 48 (CDC48) protein against neurodegeneration via ubiquitin-proteasome system dysfunction during zebrafish development. J. Biol. Chem. 2012, 287, 23047–23056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hetz, C. The unfolded protein response: Controlling cell fate decisions under ER stress and beyond. Nat. Rev. Mol. Cell Biol. 2012, 13, 89–102. [Google Scholar] [CrossRef] [PubMed]
- Hay, N.; Sonenberg, N. Upstream and downstream of mTOR. Genes Dev. 2004, 18, 1926–1945. [Google Scholar] [CrossRef] [Green Version]
- Ju, J.-S.; Fuentealba, R.A.; Miller, S.E.; Jackson, E.; Piwnica-Worms, D.; Baloh, R.H.; Weihl, C.C. Valosin-containing protein (VCP) is required for autophagy and is disrupted in VCP disease. J. Cell Biol. 2009, 187, 875–888. [Google Scholar] [CrossRef]
- Gingras, A.C.; Raught, B.; Sonenberg, N. Regulation of translation initiation by FRAP/mTOR. Genes Dev. 2001, 15, 807–826. [Google Scholar] [CrossRef] [Green Version]
- Sfakianos, A.P.; Mellor, L.E.; Pang, Y.F.; Kritsiligkou, P.; Needs, H.; Abou-Hamdan, H.; Désaubry, L.; Poulin, G.B.; Ashe, M.P.; Whitmarsh, A.J. The mTOR-S6 kinase pathway promotes stress granule assembly. Cell Death Differ. 2018, 25, 1766–1780. [Google Scholar] [CrossRef] [Green Version]
- Zhou, N.; Ma, B.; Stoll, S.; Hays, T.T.; Qiu, H. The valosin-containing protein is a novel repressor of cardiomyocyte hypertrophy induced by pressure overload. Aging Cell 2017, 16, 1168–1179. [Google Scholar] [CrossRef] [Green Version]
- Zhou, N.; Stoll, S.; Qiu, H. VCP represses pathological cardiac hypertrophy. Aging (Albany NY) 2017, 9, 2469–2470. [Google Scholar] [CrossRef] [Green Version]
- Shu, H.; Peng, Y.; Hang, W.; Zhou, N.; Wang, D.W. Emerging role of VCP/p97 in cardiovascular diseases: Novel insights and therapeutic opportunities. Biochem. Soc. Trans. 2021, 49, 485–494. [Google Scholar] [CrossRef]
- Rabanal-Ruiz, Y.; Otten, E.G.; Korolchuk, V.I. mTORC1 as the main gateway to autophagy. Essays Biochem. 2017, 61, 565–584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korb, M.; Peck, A.; Alfano, L.N.; Berger, K.I.; James, M.K.; Ghoshal, N.; Healzer, E.; Henchcliffe, C.; Khan, S.; Mammen, P.P.A.; et al. Development of a standard of care for patients with valosin-containing protein associated multisystem proteinopathy. Orphanet J. Rare Dis. 2022, 17, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Kimonis, V.E.; Fulchiero, E.; Vesa, J.; Watts, G. VCP disease associated with myopathy, Paget disease of bone and frontotemporal dementia: Review of a unique disorder. Biochim. Biophys. Acta 2008, 1782, 744–748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tresse, E.; Salomons, F.A.; Vesa, J.; Bott, L.C.; Kimonis, V.; Yao, T.P.; Dantuma, N.P.; Taylor, J.P. VCP/p97 is essential for maturation of ubiquitin-containing autophagosomes and this function is impaired by mutations that cause IBMPFD. Autophagy 2010, 6, 217–227. [Google Scholar] [CrossRef] [Green Version]
- Nalbandian, A.; Llewellyn, K.J.; Kitazawa, M.; Yin, H.Z.; Badadani, M.; Khanlou, N.; Edwards, R.; Nguyen, C.; Mukherjee, J.; Mozaffar, T.; et al. The homozygote VCP(R¹⁵⁵H/R¹⁵⁵H) mouse model exhibits accelerated human VCP-associated disease pathology. PLoS ONE 2012, 7, e46308. [Google Scholar] [CrossRef]
- Bühler, A.; Kustermann, M.; Bummer, T.; Rottbauer, W.; Sandri, M.; Just, S. Atrogin-1 Deficiency Leads to Myopathy and Heart Failure in Zebrafish. Int. J. Mol. Sci. 2016, 17, 187. [Google Scholar] [CrossRef] [Green Version]
- Kimmel, C.B.; Ballard, W.W.; Kimmel, S.R.; Ullmann, B.; Schilling, T.F. Stages of embryonic development of the zebrafish. Dev. Dyn. 1995, 203, 253–310. [Google Scholar] [CrossRef]
- Alexandre-Moreno, S.; Bonet-Fernández, J.-M.; Atienzar-Aroca, R.; Aroca-Aguilar, J.-D.; Escribano, J. Null cyp1b1 Activity in Zebrafish Leads to Variable Craniofacial Defects Associated with Altered Expression of Extracellular Matrix and Lipid Metabolism Genes. Int. J. Mol. Sci. 2021, 22, 6430. [Google Scholar] [CrossRef]
- Hirth, S.; Bühler, A.; Bührdel, J.B.; Rudeck, S.; Dahme, T.; Rottbauer, W.; Just, S. Paxillin and Focal Adhesion Kinase (FAK) Regulate Cardiac Contractility in the Zebrafish Heart. PLoS ONE 2016, 11, e0150323. [Google Scholar] [CrossRef] [Green Version]
- Schnabel, D.; Castillo-Robles, J.; Lomeli, H. Protein Purification and Western Blot Detection from Single Zebrafish Embryo. Zebrafish 2019, 16, 505–507. [Google Scholar] [CrossRef] [Green Version]
- Kessler, M.; Berger, I.M.; Just, S.; Rottbauer, W. Loss of dihydrolipoyl succinyltransferase (DLST) leads to reduced resting heart rate in the zebrafish. Basic Res. Cardiol. 2015, 110, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buhrdel, J.B.; Hirth, S.; Kessler, M.; Westphal, S.; Forster, M.; Manta, L.; Wiche, G.; Schoser, B.; Schessl, J.; Schroder, R.; et al. In vivo characterization of human myofibrillar myopathy genes in zebrafish. Biochem. Biophys. Res. Commun. 2015, 461, 217–223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bose, P.; Armstrong, G.A.B.; Drapeau, P. Neuromuscular junction abnormalities in a zebrafish loss-of-function model of TDP-43. J. Neurophysiol. 2019, 121, 285–297. [Google Scholar] [CrossRef] [PubMed]
- Benslimane, F.M.; Zakaria, Z.Z.; Shurbaji, S.; Abdelrasool, M.K.A.; Al-Badr, M.A.H.I.; Al Absi, E.S.K.; Yalcin, H.C. Cardiac function and blood flow hemodynamics assessment of zebrafish (Danio rerio) using high-speed video microscopy. Micron 2020, 136, 102876. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Voisard, P.; Diofano, F.; Glazier, A.A.; Rottbauer, W.; Just, S. CRISPR/Cas9-Mediated Constitutive Loss of VCP (Valosin-Containing Protein) Impairs Proteostasis and Leads to Defective Striated Muscle Structure and Function In Vivo. Int. J. Mol. Sci. 2022, 23, 6722. https://doi.org/10.3390/ijms23126722
Voisard P, Diofano F, Glazier AA, Rottbauer W, Just S. CRISPR/Cas9-Mediated Constitutive Loss of VCP (Valosin-Containing Protein) Impairs Proteostasis and Leads to Defective Striated Muscle Structure and Function In Vivo. International Journal of Molecular Sciences. 2022; 23(12):6722. https://doi.org/10.3390/ijms23126722
Chicago/Turabian StyleVoisard, Philipp, Federica Diofano, Amelia A. Glazier, Wolfgang Rottbauer, and Steffen Just. 2022. "CRISPR/Cas9-Mediated Constitutive Loss of VCP (Valosin-Containing Protein) Impairs Proteostasis and Leads to Defective Striated Muscle Structure and Function In Vivo" International Journal of Molecular Sciences 23, no. 12: 6722. https://doi.org/10.3390/ijms23126722
APA StyleVoisard, P., Diofano, F., Glazier, A. A., Rottbauer, W., & Just, S. (2022). CRISPR/Cas9-Mediated Constitutive Loss of VCP (Valosin-Containing Protein) Impairs Proteostasis and Leads to Defective Striated Muscle Structure and Function In Vivo. International Journal of Molecular Sciences, 23(12), 6722. https://doi.org/10.3390/ijms23126722