
Systems Drug Discovery for Diffuse Large B Cell Lymphoma Based on Pathogenic Molecular Mechanism via Big Data Mining and Deep Learning Method


Abstract
:1. Introduction
2. Results
2.1. The Pathogenic Molecular Mechanisms in DLBCL ABC
2.2. The Carcinogenic Molecular Mechanism in DLBCL GCB
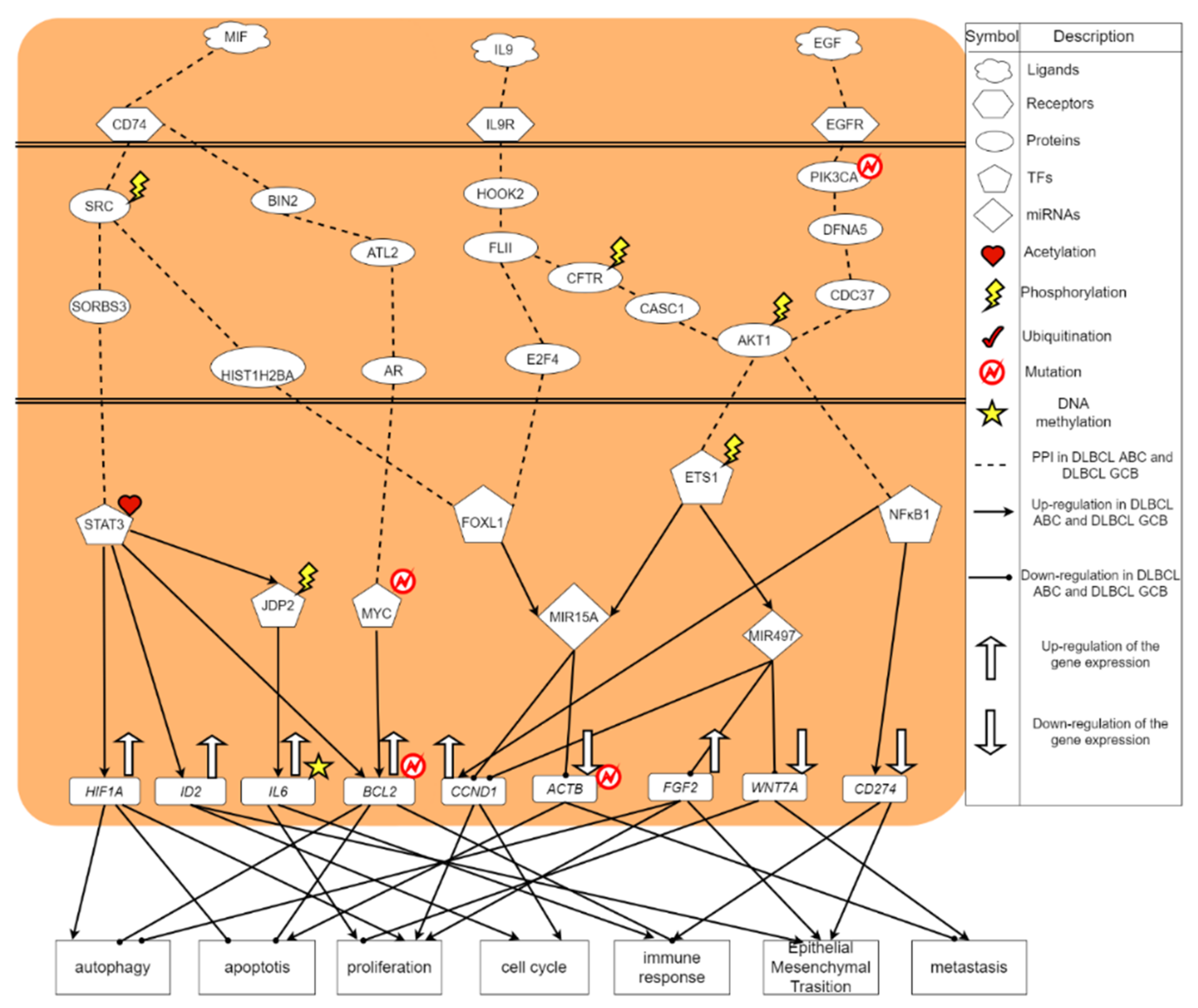
2.3. The Common and Specific Carcinogenic Molecular Mechanism between DLBCL ABC and DLBCL GCB
2.4. Systems Drug Design Procedure Considering Drug-Target Interaction, Drug Regulation Ability, and Drug Toxicity
3. Discussion
4. Materials and Methods
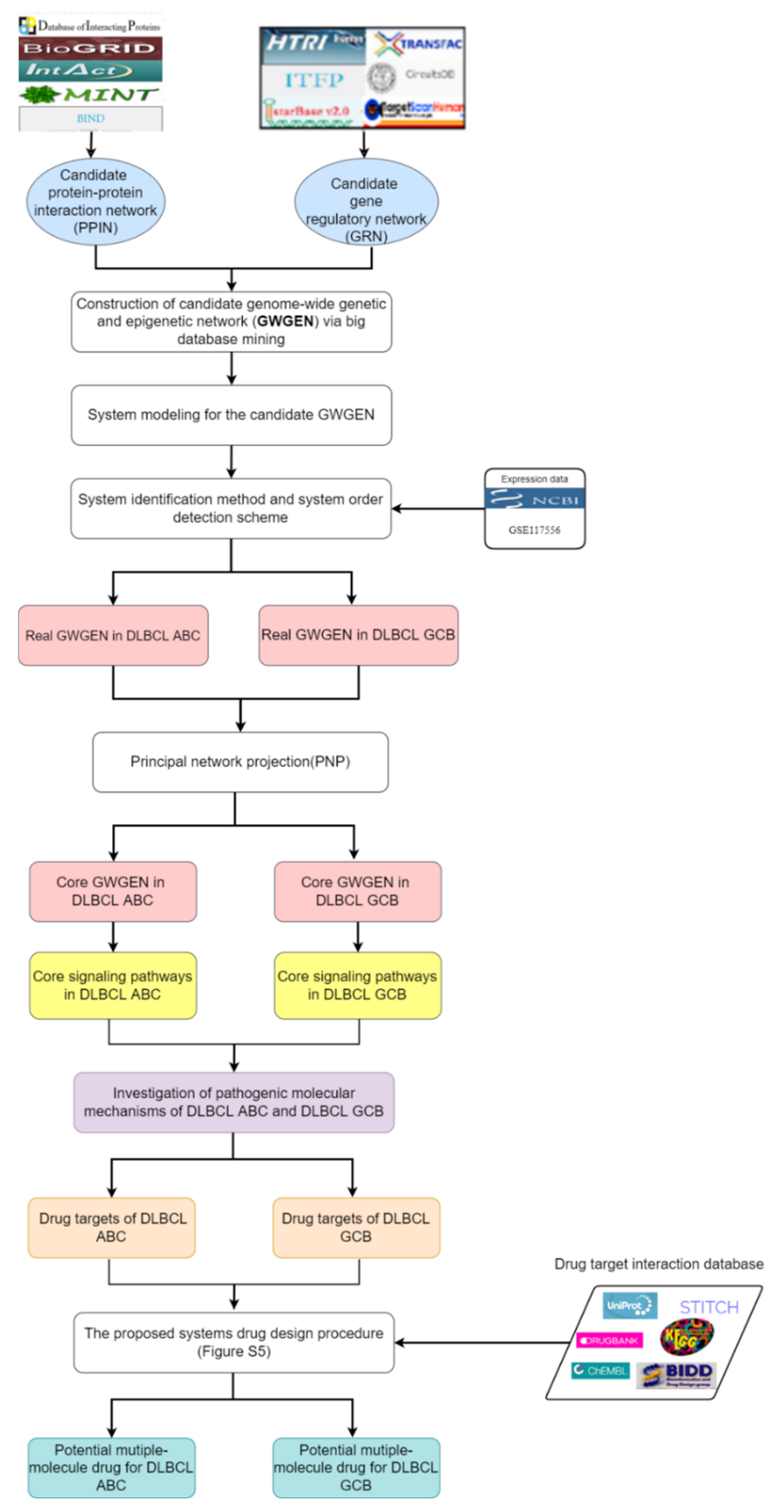
4.1. Overview of Systems Drug Discovery for DLBCL ABC and DLBCL GCB
4.2. Constructing the System Models in the GWGEN to Identify Real GWGEN of DLBCL GCB and DLBCL ABC
4.3. Using the System Identification Method and System Order Detection Approach to Build Real GWGENs of DLBCL GCB and DLBCL ABC
4.4. Extracting the Core GWGENs from the Real GWGENs by Principal Network Projection (PNP) Method
4.5. Deep Neural Netwok (DNN)-Based Drug-Target Interaction (DTI) Model for Multiple-Molecule Drug Design
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Clarke, C.A.; Glaser, S.L.; Dorfman, R.F.; Bracci, P.M.; Eberle, E.; Holly, E.A.J.C.E.; Biomarkers, P. Expert review of non-Hodgkin’s lymphomas in a population-based cancer registry: Reliability of diagnosis and subtype classifications. Cancer Epidemiol. Biomark. Prev. 2004, 13, 138–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prochazka, V.; Jarošová, M.; Prouzova, Z.; Nedomova, R.; Papajik, T.; Indrák, K. Immune escape mechanisms in diffuse large B-cell lymphoma. Int. Sch. Res. Not. 2012, 2012, 208903. [Google Scholar] [CrossRef] [Green Version]
- Sehn, L.H.; Gascoyne, R.D. Diffuse large B-cell lymphoma: Optimizing outcome in the context of clinical and biologic heterogeneity. Blood 2015, 125, 22–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salles, G.; Barrett, M.; Foà, R.; Maurer, J.; O’Brien, S.; Valente, N.; Wenger, M.; Maloney, D.G. Rituximab in B-cell hematologic malignancies: A review of 20 years of clinical experience. Adv. Ther. 2017, 34, 2232–2273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mok, C.C. Rituximab for the treatment of rheumatoid arthritis: An update. Drug Des. Dev. Ther. 2014, 8, 87. [Google Scholar] [CrossRef] [Green Version]
- Al-Homsi, A.S.; Roy, T.S.; Cole, K.; Feng, Y.; Duffner, U. Post-transplant high-dose cyclophosphamide for the prevention of graft-versus-host disease. Biol. Blood Marrow Transplant. 2015, 21, 604–611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Travis, L.B.; Curtis, R.E.; Glimelius, B.; Holowaty, E.J.; Van Leeuwen, F.E.; Lynch, C.F.; Hagenbeek, A.; Stovall, M.; Banks, P.M.; Adami, J.; et al. Bladder and Kidney Cancer Following Cyclophosphamide Therapy for Non-Hodgkin’s Lymphoma. JNCI J. Natl. Cancer Inst. 1995, 87, 524–531. [Google Scholar] [CrossRef] [PubMed]
- Thorn, C.F.; Oshiro, C.; Marsh, S.; Hernandez-Boussard, T.; McLeod, H.; Klein, T.E.; Altman, R.B. Doxorubicin pathways: Pharmacodynamics and adverse effects. Pharm. Genom. 2011, 21, 440. [Google Scholar] [CrossRef]
- Vayssade, M.; Faridoni-Laurens, L.; Bénard, J.; Ahomadegbe, J.-C. Expression of p53-family members and associated target molecules in breast cancer cell lines in response to vincristine treatment. Biochem. Pharmacol. 2002, 63, 1609–1617. [Google Scholar] [CrossRef]
- Samoon, Z.; Shabbir-Moosajee, M. Vincristine-induced vocal cord palsy and successful re-treatment in a patient with diffuse large B cell lymphoma: A case report. BMC Res. Notes 2014, 7, 318. [Google Scholar] [CrossRef] [Green Version]
- Kimberly, R.P. Mechanisms of action, dosage schedules, and side effects of steroid therapy. Curr. Opin. Rheumatol. 1991, 3, 373–379. [Google Scholar] [CrossRef]
- Modlinski, R.; Fields, K.B. The effect of anabolic steroids on the gastrointestinal system, kidneys, and adrenal glands. Curr. Sports Med. Rep. 2006, 5, 104–109. [Google Scholar] [CrossRef]
- Papageorgiou, S.G.; Thomopoulos, T.P.; Liaskas, A.; Vassilakopoulos, T.P. Monoclonal Antibodies in the Treatment of Diffuse Large B-Cell Lymphoma: Moving beyond Rituximab. Cancers 2022, 14, 1917. [Google Scholar] [CrossRef]
- Sehn, L.H.; Herrera, A.F.; Flowers, C.R.; Kamdar, M.K.; McMillan, A.; Hertzberg, M.; Assouline, S.; Kim, T.M.; Kim, W.S.; Ozcan, M. Polatuzumab vedotin in relapsed or refractory diffuse large B-cell lymphoma. J. Clin. Oncol. 2020, 38, 155. [Google Scholar] [CrossRef]
- Kalakonda, N.; Maerevoet, M.; Cavallo, F.; Follows, G.; Goy, A.; Vermaat, J.S.; Casasnovas, O.; Hamad, N.; Zijlstra, J.M.; Bakhshi, S. Selinexor in patients with relapsed or refractory diffuse large B-cell lymphoma (SADAL): A single-arm, multinational, multicentre, open-label, phase 2 trial. Lancet Haematol. 2020, 7, e511–e522. [Google Scholar] [CrossRef]
- Salles, G.; Duell, J.; Barca, E.G.; Tournilhac, O.; Jurczak, W.; Liberati, A.M.; Nagy, Z.; Obr, A.; Gaidano, G.; André, M. Tafasitamab plus lenalidomide in relapsed or refractory diffuse large B-cell lymphoma (L-MIND): A multicentre, prospective, single-arm, phase 2 study. Lancet Oncol. 2020, 21, 978–988. [Google Scholar] [CrossRef]
- Cheson, B.D.; Nowakowski, G.; Salles, G. Diffuse large B-cell lymphoma: New targets and novel therapies. Blood Cancer J. 2021, 11, 68. [Google Scholar] [CrossRef]
- Mohs, R.C.; Greig, N.H. Drug discovery and development: Role of basic biological research. Alzheimer’s Dement. Transl. Res. Clin. Interv. 2017, 3, 651–657. [Google Scholar] [CrossRef]
- Takebe, T.; Imai, R.; Ono, S. The current status of drug discovery and development as originated in United States academia: The influence of industrial and academic collaboration on drug discovery and development. Clin. Transl. Sci. 2018, 11, 597–606. [Google Scholar] [CrossRef]
- Bailón-Moscoso, N.; Romero-Benavides, J.C.; Ostrosky-Wegman, P. Development of anticancer drugs based on the hallmarks of tumor cells. Tumor Biol. 2014, 35, 3981–3995. [Google Scholar] [CrossRef]
- Sachdev, K.; Gupta, M.K. A comprehensive review of feature based methods for drug target interaction prediction. J. Biomed. Inform. 2019, 93, 103159. [Google Scholar] [CrossRef]
- Butina, D.; Segall, M.D.; Frankcombe, K. Predicting ADME properties in silico: Methods and models. Drug Discov. Today 2002, 7, S83–S88. [Google Scholar] [CrossRef]
- Li, H.; Gao, Z.; Kang, L.; Zhang, H.; Yang, K.; Yu, K.; Luo, X.; Zhu, W.; Chen, K.; Shen, J. TarFisDock: A web server for identifying drug targets with docking approach. Nucleic Acids Res. 2006, 34, W219–W224. [Google Scholar] [CrossRef] [Green Version]
- Cheng, A.C.; Coleman, R.G.; Smyth, K.T.; Cao, Q.; Soulard, P.; Caffrey, D.R.; Salzberg, A.C.; Huang, E.S. Structure-based maximal affinity model predicts small-molecule druggability. Nat. Biotechnol. 2007, 25, 71–75. [Google Scholar] [CrossRef]
- Pujadas, G.; Vaque, M.; Ardevol, A.; Blade, C.; Salvado, M.; Blay, M.; Fernandez-Larrea, J.; Arola, L. Protein-ligand docking: A review of recent advances and future perspectives. Curr. Pharm. Anal. 2008, 4, 1–19. [Google Scholar] [CrossRef]
- Bagherian, M.; Sabeti, E.; Wang, K.; Sartor, M.A.; Nikolovska-Coleska, Z.; Najarian, K. Machine learning approaches and databases for prediction of drug–target interaction: A survey paper. Brief. Bioinform. 2021, 22, 247–269. [Google Scholar] [CrossRef] [Green Version]
- Nath, A.; Kumari, P.; Chaube, R. Prediction of human drug targets and their interactions using machine learning methods: Current and future perspectives. Comput. Drug Discov. Des. 2018, 1762, 21–30. [Google Scholar]
- Wen, M.; Zhang, Z.; Niu, S.; Sha, H.; Yang, R.; Yun, Y.; Lu, H.J. Deep-learning-based drug–target interaction prediction. J. Proteome Res. 2017, 16, 1401–1409. [Google Scholar] [CrossRef]
- Gao, K.Y.; Fokoue, A.; Luo, H.; Iyengar, A.; Dey, S.; Zhang, P. Interpretable drug target prediction using deep neural representation. In Proceedings of the IJCAI, Stockholm, Sweden, 13–19 July 2018; pp. 3371–3377. [Google Scholar]
- Öztürk, H.; Özgür, A.; Ozkirimli, E.J.B. DeepDTA: Deep drug–target binding affinity prediction. Bioinformatics 2018, 34, i821–i829. [Google Scholar] [CrossRef] [Green Version]
- Lee, I.; Keum, J.; Nam, H. DeepConv-DTI: Prediction of drug-target interactions via deep learning with convolution on protein sequences. PLoS Comput. Biol. 2019, 15, e1007129. [Google Scholar] [CrossRef] [Green Version]
- You, J.; McLeod, R.D.; Hu, P. Predicting drug-target interaction network using deep learning model. Comput. Biol. Chem. 2019, 80, 90–101. [Google Scholar] [CrossRef] [PubMed]
- Ezzat, A.; Wu, M.; Li, X.-L.; Kwoh, C.-K. Drug-target interaction prediction via class imbalance-aware ensemble learning. BMC Bioinform. 2016, 17, 509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Qiao, G.; Wang, K.; Wang, G. Drug–target interaction predication via multi-channel graph neural networks. Brief. Bioinform. 2021, 23, bbab346. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Z.; Yan, C.; Wu, F.; Wang, J. Drug-target interaction prediction using multi-head self-attention and graph attention network. IEEE/ACM Trans. Comput. Biol. Bioinform. 2021, 1, 33956632. [Google Scholar] [CrossRef]
- Zhao, T.; Hu, Y.; Valsdottir, L.R.; Zang, T.; Peng, J. Identifying drug–target interactions based on graph convolutional network and deep neural network. Brief. Bioinform. 2020, 22, 2141–2150. [Google Scholar] [CrossRef]
- Tillmann, S.; Bernhagen, J.; Noels, H. Arrest Functions of the MIF Ligand/Receptor Axes in Atherogenesis. Front. Immunol. 2013, 4, 115. [Google Scholar] [CrossRef] [Green Version]
- Figueiredo, C.R.; Azevedo, R.A.; Mousdell, S.; Resende-Lara, P.T.; Ireland, L.; Santos, A.; Girola, N.; Cunha, R.L.O.R.; Schmid, M.C.; Polonelli, L.; et al. Blockade of MIF-CD74 Signalling on Macrophages and Dendritic Cells Restores the Antitumour Immune Response against Metastatic Melanoma. Front. Immunol. 2018, 9, 1132. [Google Scholar] [CrossRef]
- Gil-Yarom, N.; Radomir, L.; Sever, L.; Kramer, M.P.; Lewinsky, H.; Bornstein, C.; Blecher-Gonen, R.; Barnett-Itzhaki, Z.; Mirkin, V.; Friedlander, G.; et al. CD74 is a novel transcription regulator. Proc. Natl. Acad. Sci. USA 2017, 114, 562–567. [Google Scholar] [CrossRef] [Green Version]
- Roskoski, R. Src protein–tyrosine kinase structure and regulation. Biochem. Biophys. Res. Commun. 2004, 324, 1155–1164. [Google Scholar] [CrossRef]
- Huang, X.; Meng, B.; Iqbal, J.; Ding, B.B.; Perry, A.M.; Cao, W.; Smith, L.M.; Bi, C.; Jiang, C.; Greiner, T.C.; et al. Activation of the STAT3 signaling pathway is associated with poor survival in diffuse large B-cell lymphoma treated with R-CHOP. J. Clin. Oncol. 2013, 31, 4520–4528. [Google Scholar] [CrossRef]
- Pawlus, M.R.; Wang, L.; Hu, C.J. STAT3 and HIF1α cooperatively activate HIF1 target genes in MDA-MB-231 and RCC4 cells. Oncogene 2014, 33, 1670–1679. [Google Scholar] [CrossRef] [Green Version]
- Carmeliet, P.; Dor, Y.; Herbert, J.-M.; Fukumura, D.; Brusselmans, K.; Dewerchin, M.; Neeman, M.; Bono, F.; Abramovitch, R.; Maxwell, P.; et al. Role of HIF-1α in hypoxia-mediated apoptosis, cell proliferation and tumour angiogenesis. Nature 1998, 394, 485–490. [Google Scholar] [CrossRef]
- Wein, F.; Otto, T.; Lambertz, P.; Fandrey, J.; Hansmann, M.-L.; Küppers, R. Potential role of hypoxia in early stages of Hodgkin lymphoma pathogenesis. Haematologica 2015, 100, 1320–1326. [Google Scholar] [CrossRef] [Green Version]
- Burger, R. Impact of interleukin-6 in hematological malignancies. Transfus. Med. Hemotherapy 2013, 40, 336–343. [Google Scholar] [CrossRef] [Green Version]
- Sánchez-Barrena, M.J.; Vallis, Y.; Clatworthy, M.R.; Doherty, G.J.; Veprintsev, D.B.; Evans, P.R.; McMahon, H.T. Bin2 is a membrane sculpting N-BAR protein that influences leucocyte podosomes, motility and phagocytosis. PLoS ONE 2012, 7, e52401. [Google Scholar] [CrossRef] [Green Version]
- Davey, R.A.; Grossmann, M. Androgen Receptor Structure, Function and Biology: From Bench to Bedside. Clin. Biochem. Rev. 2016, 37, 3–15. [Google Scholar]
- Nguyen, L.; Papenhausen, P.; Shao, H. The Role of c-MYC in B-Cell Lymphomas: Diagnostic and Molecular Aspects. Genes 2017, 8, 116. [Google Scholar] [CrossRef] [Green Version]
- Tjin, E.P.M.; Groen, R.W.J.; Vogelzang, I.; Derksen, P.W.B.; Klok, M.D.; Meijer, H.P.; van Eeden, S.; Pals, S.T.; Spaargaren, M. Functional analysis of HGF/MET signaling and aberrant HGF-activator expression in diffuse large B-cell lymphoma. Blood 2006, 107, 760–768. [Google Scholar] [CrossRef] [Green Version]
- Lam, B.Q.; Dai, L.; Qin, Z. The role of HGF/c-MET signaling pathway in lymphoma. J. Hematol. Oncol. 2016, 9, 135. [Google Scholar] [CrossRef] [Green Version]
- Haycraft, C.J.; Banizs, B.; Aydin-Son, Y.; Zhang, Q.; Michaud, E.J.; Yoder, B.K. Gli2 and Gli3 localize to cilia and require the intraflagellar transport protein polaris for processing and function. PLoS Genet. 2005, 1, e53. [Google Scholar] [CrossRef]
- Lentjes, M.H.F.M.; Niessen, H.E.C.; Akiyama, Y.; de Bruïne, A.P.; Melotte, V.; van Engeland, M. The emerging role of GATA transcription factors in development and disease. Expert Rev. Mol. Med. 2016, 18, e3. [Google Scholar] [CrossRef]
- Crispino, J.D.; Horwitz, M.S. GATA factor mutations in hematologic disease. Blood 2017, 129, 2103–2110. [Google Scholar] [CrossRef]
- Chakrama, F.Z.; Seguin-Py, S.; Le Grand, J.N.; Fraichard, A.; Delage-Mourroux, R.; Despouy, G.; Perez, V.; Jouvenot, M.; Boyer-Guittaut, M. GABARAPL1 (GEC1) associates with autophagic vesicles. Autophagy 2010, 6, 495–505. [Google Scholar] [CrossRef] [Green Version]
- Zhong, X.; Xiao, Y.; Chen, C.; Wei, X.; Hu, C.; Ling, X.; Liu, X. MicroRNA-203-mediated posttranscriptional deregulation of CPEB4 contributes to colorectal cancer progression. Biochem. Biophys. Res. Commun. 2015, 466, 206–213. [Google Scholar] [CrossRef]
- Béguelin, W.; Popovic, R.; Teater, M.; Jiang, Y.; Bunting, K.L.; Rosen, M.; Shen, H.; Yang, S.N.; Wang, L.; Ezponda, T.; et al. EZH2 is required for germinal center formation and somatic EZH2 mutations promote lymphoid transformation. Cancer Cell 2013, 23, 677–692. [Google Scholar] [CrossRef] [Green Version]
- Bisserier, M.; Wajapeyee, N. Mechanisms of resistance to EZH2 inhibitors in diffuse large B-cell lymphomas. Blood 2018, 131, 2125–2137. [Google Scholar] [CrossRef]
- Kim, K.H.; Roberts, C.W. Targeting EZH2 in cancer. Nat. Med. 2016, 22, 128–134. [Google Scholar] [CrossRef]
- Elian, F.A.; Yan, E.; Walter, M.A. FOXC1, the new player in the cancer sandbox. Oncotarget 2017, 9, 8165–8178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, S.; Han, S.-H.; Kim, H.-G.; Jeong, J.; Choi, M.; Kim, H.-Y.; Kim, M.-G.; Park, J.-K.; Han, J.E.; Cho, G.-J.; et al. Suppression of PRPF4 regulates pluripotency, proliferation, and differentiation in mouse embryonic stem cells. Cell Biochem. Funct. 2019, 37, 608–617. [Google Scholar] [CrossRef] [PubMed]
- Song, H.; Dang, X.; He, Y.-Q.; Zhang, T.; Wang, H.-Y. Selection of housekeeping genes as internal controls for quantitative RT-PCR analysis of the veined rapa whelk (Rapana venosa). PeerJ 2017, 5, e3398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, T.-P.; Oh, S.P.; Fuchs, M.; Zhou, N.-D.; Ch’ng, L.-E.; Newsome, D.; Bronson, R.T.; Li, E.; Livingston, D.M.; Eckner, R. Gene Dosage–Dependent Embryonic Development and Proliferation Defects in Mice Lacking the Transcriptional Integrator p300. Cell 1998, 93, 361–372. [Google Scholar] [CrossRef] [Green Version]
- Pasqualucci, L.; Dominguez-Sola, D.; Chiarenza, A.; Fabbri, G.; Grunn, A.; Trifonov, V.; Kasper, L.H.; Lerach, S.; Tang, H.; Ma, J.; et al. Inactivating mutations of acetyltransferase genes in B-cell lymphoma. Nature 2011, 471, 189–195. [Google Scholar] [CrossRef] [Green Version]
- Dang, C.V. MYC on the Path to Cancer. Cell 2012, 149, 22–35. [Google Scholar] [CrossRef] [Green Version]
- Ortega, M.; Bhatnagar, H.; Lin, A.P.; Wang, L.; Aster, J.C.; Sill, H.; Aguiar, R.C.T. A microRNA-mediated regulatory loop modulates NOTCH and MYC oncogenic signals in B- and T-cell malignancies. Leukemia 2015, 29, 968–976. [Google Scholar] [CrossRef] [Green Version]
- Lv, X.; Feng, L.; Ge, X.; Lu, K.; Wang, X. Interleukin-9 promotes cell survival and drug resistance in diffuse large B-cell lymphoma. J. Exp. Clin. Cancer Res. 2016, 35, 106. [Google Scholar] [CrossRef] [Green Version]
- Chen, A.; Zhong, L.; Lv, J. FOXL1 overexpression is associated with poor outcome in patients with glioma. Oncol. Lett. 2019, 18, 751–757. [Google Scholar] [CrossRef] [Green Version]
- Ni, H.; Tong, R.; Zou, L.; Song, G.; Cho, W.C. MicroRNAs in diffuse large B-cell lymphoma. Oncol. Lett. 2016, 11, 1271–1280. [Google Scholar] [CrossRef] [Green Version]
- Lohr, J.G.; Stojanov, P.; Lawrence, M.S.; Auclair, D.; Chapuy, B.; Sougnez, C.; Cruz-Gordillo, P.; Knoechel, B.; Asmann, Y.W.; Slager, S.L.; et al. Discovery and prioritization of somatic mutations in diffuse large B-cell lymphoma (DLBCL) by whole-exome sequencing. Proc. Natl. Acad. Sci. USA 2012, 109, 3879–3884. [Google Scholar] [CrossRef] [Green Version]
- Vela-Chávez, T.; Adam, P.; Kremer, M.; Bink, K.; Bacon, C.M.; Menon, G.; Ferry, J.A.; Fend, F.; Jaffe, E.S.; Quintanilla-Martínez, L. Cyclin D1 positive diffuse large B-cell lymphoma is a post-germinal center-type lymphoma without alterations in the CCND1 gene locus. Leuk Lymphoma 2011, 52, 458–466. [Google Scholar] [CrossRef]
- Vermeulen, K.; Van Bockstaele, D.R.; Berneman, Z.N. The cell cycle: A review of regulation, deregulation and therapeutic targets in cancer. Cell Prolif. 2003, 36, 131–149. [Google Scholar] [CrossRef]
- Gennaro, V.J.; Stanek, T.J.; Peck, A.R.; Sun, Y.; Wang, F.; Qie, S.; Knudsen, K.E.; Rui, H.; Butt, T.; Diehl, J.A.; et al. Control of CCND1 ubiquitylation by the catalytic SAGA subunit USP22 is essential for cell cycle progression through G1 in cancer cells. Proc. Natl. Acad. Sci. USA 2018, 115, E9298. [Google Scholar] [CrossRef] [Green Version]
- Hwang, T.-C.; Kirk, K.L. The CFTR ion channel: Gating, regulation, and anion permeation. Cold Spring Harb. Perspect Med. 2013, 3, a009498. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Xu-Monette, Z.Y.; Jabbar, K.J.; Shen, Q.; Manyam, G.C.; Tzankov, A.; Visco, C.; Wang, J.; Montes-Moreno, S.; Dybkær, K.; et al. AKT Hyperactivation and the Potential of AKT-Targeted Therapy in Diffuse Large B-Cell Lymphoma. Am. J. Pathol. 2017, 187, 1700–1716. [Google Scholar] [CrossRef] [Green Version]
- Dittmer, J. The Biology of the Ets1 Proto-Oncogene. Mol. Cancer 2003, 2, 29. [Google Scholar] [CrossRef] [Green Version]
- Bonetti, P.; Testoni, M.; Scandurra, M.; Ponzoni, M.; Piva, R.; Mensah, A.A.; Rinaldi, A.; Kwee, I.; Tibiletti, M.G.; Iqbal, J.; et al. Deregulation of ETS1 and FLI1 contributes to the pathogenesis of diffuse large B-cell lymphoma. Blood 2013, 122, 2233–2241. [Google Scholar] [CrossRef] [Green Version]
- Ochoa-Hernández, A.B.; Ramos-Solano, M.; Meza-Canales, I.D.; García-Castro, B.; Rosales-Reynoso, M.A.; Rosales-Aviña, J.A.; Barrera-Chairez, E.; Ortíz-Lazareno, P.C.; Hernández-Flores, G.; Bravo-Cuellar, A.; et al. Peripheral T-lymphocytes express WNT7A and its restoration in leukemia-derived lymphoblasts inhibits cell proliferation. BMC Cancer 2012, 12, 60. [Google Scholar] [CrossRef] [Green Version]
- Yuan, H.; Li, Z.-M.; Shao, J.; Ji, W.-X.; Xia, W.; Lu, S. FGF2/FGFR1 regulates autophagy in FGFR1-amplified non-small cell lung cancer cells. J. Exp. Clin. Cancer Res. 2017, 36, 72. [Google Scholar] [CrossRef] [Green Version]
- Parzych, K.R.; Klionsky, D.J. An overview of autophagy: Morphology, mechanism, and regulation. Antioxid. Redox Signal. 2014, 20, 460–473. [Google Scholar] [CrossRef] [Green Version]
- Cui, W.; Zheng, S.; Liu, Z.; Wang, W.; Cai, Y.; Bi, R.; Cao, B.; Zhou, X. PIK3CA expression in diffuse large B cell lymphoma tissue and the effect of its knockdown in vitro. OncoTargets Ther. 2017, 10, 2239–2247. [Google Scholar] [CrossRef] [Green Version]
- Compagno, M.; Lim, W.K.; Grunn, A.; Nandula, S.V.; Brahmachary, M.; Shen, Q.; Bertoni, F.; Ponzoni, M.; Scandurra, M.; Califano, A.; et al. Mutations of multiple genes cause deregulation of NF-kappaB in diffuse large B-cell lymphoma. Nature 2009, 459, 717–721. [Google Scholar] [CrossRef] [Green Version]
- Dong, P.; Xiong, Y.; Yue, J.; Hanley, S.J.B.; Watari, H. Tumor-Intrinsic PD-L1 Signaling in Cancer Initiation, Development and Treatment: Beyond Immune Evasion. Front. Oncol. 2018, 8, 386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamb, J. The Connectivity Map: A new tool for biomedical research. Nat. Rev. Cancer 2007, 7, 54–60. [Google Scholar] [CrossRef] [PubMed]
- Wishart, D.S.; Feunang, Y.D.; Guo, A.C.; Lo, E.J.; Marcu, A.; Grant, J.R.; Sajed, T.; Johnson, D.; Li, C.; Sayeeda, Z.; et al. DrugBank 5.0: A major update to the DrugBank database for 2018. Nucleic Acids Res. 2018, 46, D1074–D1082. [Google Scholar] [CrossRef] [PubMed]
- Roy, P.; Ghosh, A. Mechanochemical cocrystallization to improve the physicochemical properties of chlorzoxazone. CrystEngComm 2020, 22, 4611–4620. [Google Scholar] [CrossRef]
- Sogawa, C.; Eguchi, T.; Tran, M.T.; Ishige, M.; Trin, K.; Okusha, Y.; Taha, E.A.; Lu, Y.; Kawai, H.; Sogawa, N.; et al. Antiparkinson Drug Benztropine Suppresses Tumor Growth, Circulating Tumor Cells, and Metastasis by Acting on SLC6A3/DAT and Reducing STAT3. Cancers 2020, 12, 523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prusila, R.E.I.; Peroja, P.; Jantunen, E.; Turpeenniemi-Hujanen, T.; Kuittinen, O.J.H.O. Treatment of diffuse large B-cell lymphoma in elderly patients: Replacing doxorubicin with either epirubicin or etoposide (VP-16). Hematol. Oncol. 2019, 37, 136–142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moccia, A.A.; Schaff, K.; Hoskins, P.; Klasa, R.; Savage, K.J.; Shenkier, T.; Gascoyne, R.D.; Connors, J.M.; Sehn, L.H. R-CHOP with Etoposide Substituted for Doxorubicin (R-CEOP): Excellent Outcome in Diffuse Large B Cell Lymphoma for Patients with a Contraindication to Anthracyclines. Blood 2009, 114, 408. [Google Scholar] [CrossRef]
- Huang, W.-Y.; Yang, P.-M.; Chang, Y.-F.; Marquez, V.E.; Chen, C.-C. Methotrexate induces apoptosis through p53/p21-dependent pathway and increases E-cadherin expression through downregulation of HDAC/EZH2. Biochem. Pharmacol. 2011, 81, 510–517. [Google Scholar] [CrossRef]
- Han Li, C.; Chen, Y. Targeting EZH2 for cancer therapy: Progress and perspective. Curr. Protein Pept. Sci. 2015, 16, 559–570. [Google Scholar] [CrossRef]
- Salwinski, L.; Miller, C.S.; Smith, A.J.; Pettit, F.K.; Bowie, J.U.; Eisenberg, D. The database of interacting proteins: 2004 update. Nucleic Acids Res. 2004, 32, D449–D451. [Google Scholar] [CrossRef] [Green Version]
- Orchard, S.; Ammari, M.; Aranda, B.; Breuza, L.; Briganti, L.; Broackes-Carter, F.; Campbell, N.H.; Chavali, G.; Chen, C.; Del-Toro, N. The MIntAct project—IntAct as a common curation platform for 11 molecular interaction databases. Nucleic Acids Res. 2014, 42, D358–D363. [Google Scholar] [CrossRef] [Green Version]
- Chatr-Aryamontri, A.; Breitkreutz, B.-J.; Oughtred, R.; Boucher, L.; Heinicke, S.; Chen, D.; Stark, C.; Breitkreutz, A.; Kolas, N.; O’Donnell, L. The BioGRID interaction database: 2015 update. Nucleic Acids Res. 2015, 43, D470–D478. [Google Scholar] [CrossRef]
- Bader, G.D.; Betel, D.; Hogue, C.W. BIND: The biomolecular interaction network database. Nucleic Acids Res. 2003, 31, 248–250. [Google Scholar] [CrossRef]
- Licata, L.; Briganti, L.; Peluso, D.; Perfetto, L.; Iannuccelli, M.; Galeota, E.; Sacco, F.; Palma, A.; Nardozza, A.P.; Santonico, E. MINT, the molecular interaction database: 2012 update. Nucleic Acids Res. 2012, 40, D857–D861. [Google Scholar] [CrossRef]
- Zheng, G.; Tu, K.; Yang, Q.; Xiong, Y.; Wei, C.; Xie, L.; Zhu, Y.; Li, Y. ITFP: An integrated platform of mammalian transcription factors. Bioinformatics 2008, 24, 2416–2417. [Google Scholar] [CrossRef]
- Bovolenta, L.A.; Acencio, M.L.; Lemke, N.J. HTRIdb: An open-access database for experimentally verified human transcriptional regulation interactions. BMC Genom. 2012, 13, 405. [Google Scholar] [CrossRef] [Green Version]
- Agarwal, V.; Bell, G.W.; Nam, J.-W.; Bartel, D.P. Predicting effective microRNA target sites in mammalian mRNAs. elife 2015, 4, e05005. [Google Scholar] [CrossRef]
- Friard, O.; Re, A.; Taverna, D.; De Bortoli, M.; Corá, D. CircuitsDB: A database of mixed microRNA/transcription factor feed-forward regulatory circuits in human and mouse. BMC Bioinform. 2010, 11, 435. [Google Scholar] [CrossRef] [Green Version]
- Li, J.-H.; Liu, S.; Zhou, H.; Qu, L.-H.; Yang, J.-H. starBase v2. 0: Decoding miRNA-ceRNA, miRNA-ncRNA and protein–RNA interaction networks from large-scale CLIP-Seq data. Nucleic Acids Res. 2014, 42, D92–D97. [Google Scholar] [CrossRef] [Green Version]
- Sakamoto, Y.; Ishiguro, M.; Kitagawa, G.J.D. Akaike Information Criterion Statistics; Reidel, D., Ed.; Springer: Dordrecht, The Netherlands, 1986; Volume 81, p. 26853. [Google Scholar]
- Gilson, M.K.; Liu, T.; Baitaluk, M.; Nicola, G.; Hwang, L.; Chong, J. BindingDB in 2015: A public database for medicinal chemistry, computational chemistry and systems pharmacology. Nucleic Acids Res. 2016, 44, D1045–D1053. [Google Scholar] [CrossRef] [PubMed]
- Dong, J.; Yao, Z.J.; Zhang, L.; Luo, F.; Lin, Q.; Lu, A.P.; Chen, A.F.; Cao, D.S. PyBioMed: A python library for various molecular representations of chemicals, proteins and DNAs and their interactions. J. Cheminform. 2018, 10, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ringnér, M. What is principal component analysis? Nat. Biotechnol. 2008, 26, 303–304. [Google Scholar] [CrossRef] [PubMed]
- Nwankpa, C.; Ijomah, W.; Gachagan, A.; Marshall, S. Activation functions: Comparison of trends in practice and research for deep learning. arXiv 2018, arXiv:1811.03378. [Google Scholar]
- Hecht-Nielsen, R. Theory of the backpropagation neural network. In Neural Networks for Perception; Elsevier: Amsterdam, The Netherlands, 1992; pp. 65–93. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cancer | Biomarkers (Drug Targets) |
|---|---|
| DLBCL ABC | FOXL1 NFκB1 AKT1 MYC STAT3 |
| DLBCL GCB | FOXL1 NFκB1 AKT1 MYC EZH2 |
| Targets | FOXL1 | NFκB1 | AKT1 | MYC | STAT3 | |
|---|---|---|---|---|---|---|
| Drugs | ||||||
| Famotidine | O | O | O | |||
| Chlorzoxazone | O | O | O | |||
| Etoposide | O | O | O | |||
| Targets | FOXL1 | NFκB1 | AKT1 | MYC | EZH2 | |
|---|---|---|---|---|---|---|
| Drugs | ||||||
| Famotidine | O | O | O | |||
| Chlorzoxazone | O | O | O | |||
| Methotrexate | O | O | O | |||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yeh, S.-J.; Yeh, T.-Y.; Chen, B.-S. Systems Drug Discovery for Diffuse Large B Cell Lymphoma Based on Pathogenic Molecular Mechanism via Big Data Mining and Deep Learning Method. Int. J. Mol. Sci. 2022, 23, 6732. https://doi.org/10.3390/ijms23126732
Yeh S-J, Yeh T-Y, Chen B-S. Systems Drug Discovery for Diffuse Large B Cell Lymphoma Based on Pathogenic Molecular Mechanism via Big Data Mining and Deep Learning Method. International Journal of Molecular Sciences. 2022; 23(12):6732. https://doi.org/10.3390/ijms23126732
Chicago/Turabian StyleYeh, Shan-Ju, Tsun-Yung Yeh, and Bor-Sen Chen. 2022. "Systems Drug Discovery for Diffuse Large B Cell Lymphoma Based on Pathogenic Molecular Mechanism via Big Data Mining and Deep Learning Method" International Journal of Molecular Sciences 23, no. 12: 6732. https://doi.org/10.3390/ijms23126732
APA StyleYeh, S. -J., Yeh, T. -Y., & Chen, B. -S. (2022). Systems Drug Discovery for Diffuse Large B Cell Lymphoma Based on Pathogenic Molecular Mechanism via Big Data Mining and Deep Learning Method. International Journal of Molecular Sciences, 23(12), 6732. https://doi.org/10.3390/ijms23126732