
Adaptation of Oxidative Phosphorylation Machinery Compensates for Hepatic Lipotoxicity in Early Stages of MAFLD

 , , , and
, , , and
Abstract
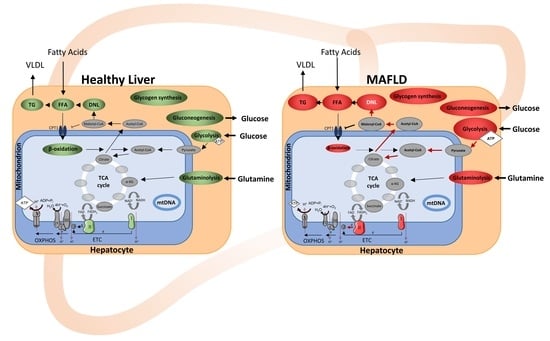
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
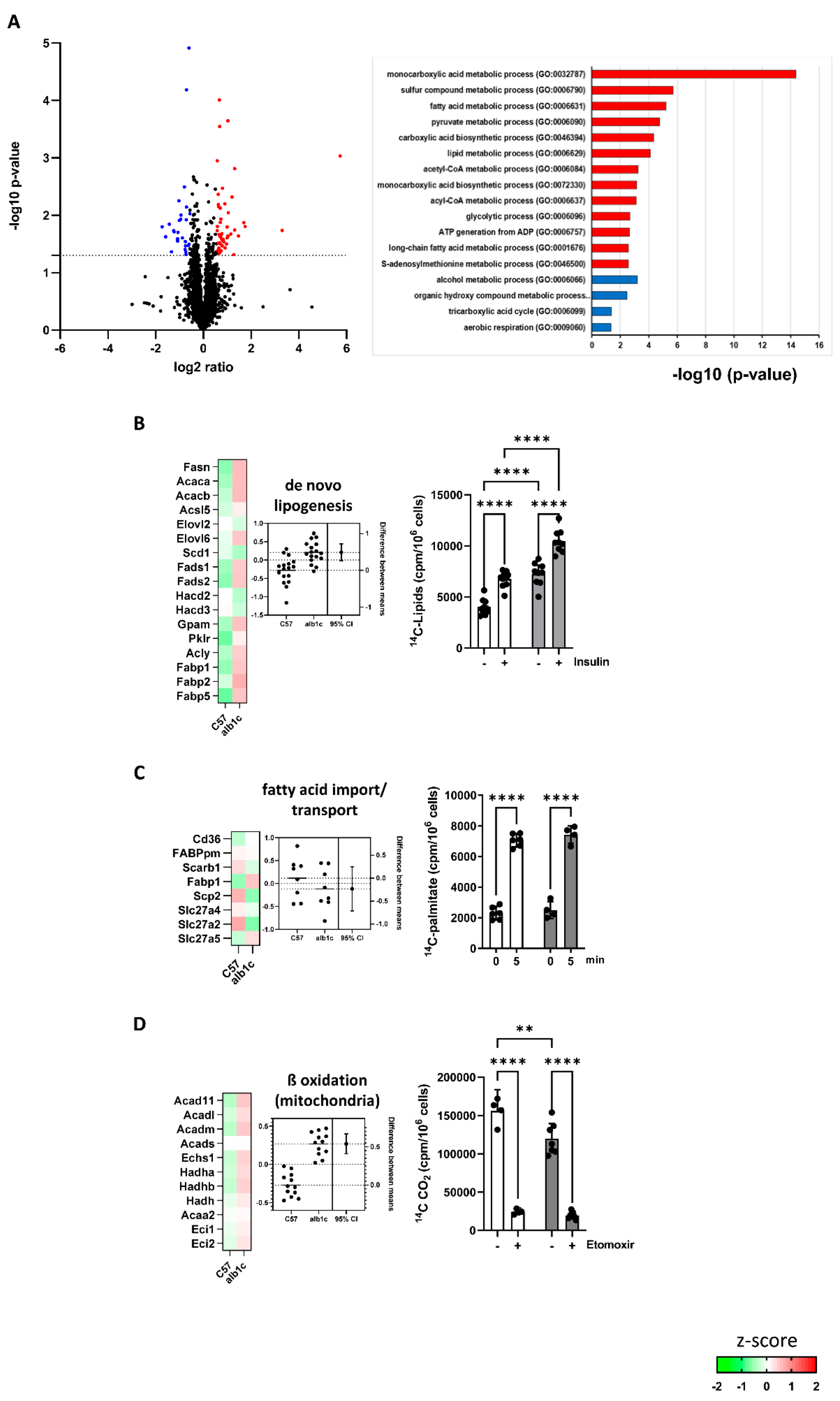{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. MAFLD and Mitochondrial Capacity
2.1.1. Alterations of Protein Abundance in Mitochondrial ETC Complexes
2.1.2. Altered ETC Subunit Abundance Interfered with Function
2.1.3. Increased Mitochondrial Function Related to Physiological Changes in Cellular Metabolism
2.2. Mechanisms of Mitochondrial and Energy Metabolism in Early-Stage MAFLD
2.2.1. Impact of SREBP-1c-Forced DNL in Early-Stage MAFLD on Transcriptome of Primary Hepatocytes
2.2.2. High Mitochondrial Activity in Early-Stage MAFLD Was Mainly Driven by SREBP-1c-Forced DNL and Glucose Oxidation
2.2.3. Altered Protein Abundance of Main Lipid and Glucose Metabolism Pathways in Early-Stage MAFLD
2.2.4. A Shift in Substrate Flux Pathways and Preferences Accompanied Early Stage MAFLD
3. Discussion
3.1. Mechanisms in Early-Stage MAFLD to Alter Mitochondria and Energy Metabolism
3.2. MAFLD Is Accompanied by a Shift in Substrate Flux Pathways and Preferences
4. Materials and Methods
4.1. Animals
4.2. Hepatocyte Analyses
4.2.1. Isolation of Primary Hepatocytes
4.2.2. Analyses of Lipid and Carbohydrate Metabolism
4.3. Analyses in Isolated Mitochondria
Analyses of Mitochondrial Respiratory Dynamics
4.4. Molecular Analyses
4.4.1. Proteome Analyses
4.4.2. Gene Expression and Trancriptome Analysis
4.4.3. Mitochondrial Copy Number
4.5. Enzyme Assays
4.6. Data Analyses
4.7. Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Jelenik, T.; Kaul, K.; Sequaris, G.; Flogel, U.; Phielix, E.; Kotzka, J.; Knebel, B.; Fahlbusch, P.; Horbelt, T.; Lehr, S.; et al. Mechanisms of Insulin Resistance in Primary and Secondary Nonalcoholic Fatty Liver. Diabetes 2017, 66, 2241–2253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knebel, B.; Haas, J.; Hartwig, S.; Jacob, S.; Kollmer, C.; Nitzgen, U.; Muller-Wieland, D.; Kotzka, J. Liver-specific expression of transcriptionally active SREBP-1c is associated with fatty liver and increased visceral fat mass. PLoS ONE 2012, 7, e31812. [Google Scholar] [CrossRef] [Green Version]
- Knebel, B.; Fahlbusch, P.; Dille, M.; Wahlers, N.; Hartwig, S.; Jacob, S.; Kettel, U.; Schiller, M.; Herebian, D.; Koellmer, C.; et al. Fatty Liver Due to Increased de novo Lipogenesis: Alterations in the Hepatic Peroxisomal Proteome. Front. Cell Dev. Biol. 2019, 7, 248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horton, J.D.; Bashmakov, Y.; Shimomura, I.; Shimano, H. Regulation of sterol regulatory element binding proteins in livers of fasted and refed mice. Proc. Natl. Acad. Sci. USA 1998, 95, 5987–5992. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horton, J.D.; Goldstein, J.L.; Brown, M.S. SREBPs: Activators of the complete program of cholesterol and fatty acid synthesis in the liver. J. Clin. Investig. 2002, 109, 1125–1131. [Google Scholar] [CrossRef] [PubMed]
- Shimomura, I.; Bashmakov, Y.; Horton, J.D. Increased levels of nuclear SREBP-1c associated with fatty livers in two mouse models of diabetes mellitus. J Biol. Chem. 1999, 274, 30028–30032. [Google Scholar] [CrossRef] [Green Version]
- Heeren, J.; Scheja, L. Metabolic-associated fatty liver disease and lipoprotein metabolism. Mol. Metab. 2021, 50, 101238. [Google Scholar] [CrossRef]
- Eslam, M.; Sanyal, A.J.; George, J. MAFLD: A Consensus-Driven Proposed Nomenclature for Metabolic Associated Fatty Liver Disease. Gastroenterology 2020, 158, 1999–2014.e1. [Google Scholar] [CrossRef]
- Brady, L.J.; Silverstein, L.J.; Hoppel, C.L.; Brady, P.S. Hepatic mitochondrial inner membrane properties and carnitine palmitoyltransferase A and B. Effect of diabetes and starvation. Biochem. J. 1985, 232, 445–450. [Google Scholar] [CrossRef] [Green Version]
- Iozzo, P.; Bucci, M.; Roivainen, A.; Någren, K.; Järvisalo, M.J.; Kiss, J.; Guiducci, L.; Fielding, B.; Naum, A.G.; Borra, R.; et al. Fatty acid metabolism in the liver, measured by positron emission tomography, is increased in obese individuals. Gastroenterology 2010, 139, 846–856.e6. [Google Scholar] [CrossRef]
- Koliaki, C.; Szendroedi, J.; Kaul, K.; Jelenik, T.; Nowotny, P.; Jankowiak, F.; Herder, C.; Carstensen, M.; Krausch, M.; Knoefel, W.T.; et al. Adaptation of hepatic mitochondrial function in humans with non-alcoholic fatty liver is lost in steatohepatitis. Cell Metab. 2015, 21, 739–746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sunny, N.E.; Parks, E.J.; Browning, J.D.; Burgess, S.C. Excessive hepatic mitochondrial TCA cycle and gluconeogenesis in humans with nonalcoholic fatty liver disease. Cell Metab. 2011, 14, 804–810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rath, S.; Sharma, R.; Gupta, R.; Ast, T.; Chan, C.; Durham, T.J.; Goodman, R.P.; Grabarek, Z.; Haas, M.E.; Hung, W.H.W.; et al. MitoCarta3.0: An updated mitochondrial proteome now with sub-organelle localization and pathway annotations. Nucleic Acids Res. 2021, 49, D1541–D1547. [Google Scholar] [CrossRef] [PubMed]
- Moon, Y.A.; Liang, G.; Xie, X.; Frank-Kamenetsky, M.; Fitzgerald, K.; Koteliansky, V.; Brown, M.S.; Goldstein, J.L.; Horton, J.D. The Scap/SREBP pathway is essential for developing diabetic fatty liver and carbohydrate-induced hypertriglyceridemia in animals. Cell Metab. 2012, 15, 240–246. [Google Scholar] [CrossRef] [Green Version]
- Smith, G.I.; Shankaran, M.; Yoshino, M.; Schweitzer, G.G.; Chondronikola, M.; Beals, J.W.; Okunade, A.L.; Patterson, B.W.; Nyangau, E.; Field, T.; et al. Insulin resistance drives hepatic de novo lipogenesis in nonalcoholic fatty liver disease. J. Clin. Investig. 2020, 130, 1453–1460. [Google Scholar] [CrossRef]
- Brown, M.S.; Goldstein, J.L. Selective versus total insulin resistance: A pathogenic paradox. Cell Metab. 2008, 7, 95–96. [Google Scholar] [CrossRef] [Green Version]
- Softic, S.; Cohen, D.E.; Kahn, C.R. Role of Dietary Fructose and Hepatic De Novo Lipogenesis in Fatty Liver Disease. Dig. Dis. Sci. 2016, 61, 1282–1293. [Google Scholar] [CrossRef] [Green Version]
- Campbell, C.T.; Kolesar, J.E.; Kaufman, B.A. Mitochondrial transcription factor A regulates mitochondrial transcription initiation, DNA packaging, and genome copy number. Biochim. Biophys. Acta 2012, 1819, 921–929. [Google Scholar] [CrossRef]
- Chew, K.; Zhao, L. Interactions of Mitochondrial Transcription Factor A with DNA Damage: Mechanistic Insights and Functional Implications. Genes 2021, 12, 1246. [Google Scholar] [CrossRef]
- Finck, B.N.; Kelly, D.P. PGC-1 coactivators: Inducible regulators of energy metabolism in health and disease. J. Clin. Investig. 2006, 116, 615–622. [Google Scholar] [CrossRef]
- Puigserver, P.; Spiegelman, B.M. Peroxisome proliferator-activated receptor-gamma coactivator 1 alpha (PGC-1 alpha): Transcriptional coactivator and metabolic regulator. Endocr. Rev. 2003, 24, 78–90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scarpulla, R.C. Metabolic control of mitochondrial biogenesis through the PGC-1 family regulatory network. Biochim. Biophys. Acta 2011, 1813, 1269–1278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujii, N.; Narita, T.; Okita, N.; Kobayashi, M.; Furuta, Y.; Chujo, Y.; Sakai, M.; Yamada, A.; Takeda, K.; Konishi, T.; et al. Sterol regulatory element-binding protein-1c orchestrates metabolic remodeling of white adipose tissue by caloric restriction. Aging Cell 2017, 16, 508–517. [Google Scholar] [CrossRef]
- Scarpulla, R.C. Transcriptional paradigms in mammalian mitochondrial biogenesis and function. Physiol. Rev. 2008, 88, 611–638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piantadosi, C.A.; Suliman, H.B. Transcriptional Regulation of SDHa flavoprotein by nuclear respiratory factor-1 prevents pseudo-hypoxia in aerobic cardiac cells. J. Biol. Chem. 2008, 283, 10967–10977. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knebel, B.; Goddeke, S.; Hartwig, S.; Horbelt, T.; Fahlbusch, P.; Al-Hasani, H.; Jacob, S.; Koellmer, C.; Nitzgen, U.; Schiller, M.; et al. Alteration of Liver Peroxisomal and Mitochondrial Functionality in the NZO Mouse Model of Metabolic Syndrome. Proteom. Clin. Appl. 2018, 12, 1700028. [Google Scholar] [CrossRef]
- Knebel, B.; Hartwig, S.; Haas, J.; Lehr, S.; Goeddeke, S.; Susanto, F.; Bohne, L.; Jacob, S.; Koellmer, C.; Nitzgen, U.; et al. Peroxisomes compensate hepatic lipid overflow in mice with fatty liver. Biochim. Biophys. Acta 2015, 1851, 965–976. [Google Scholar] [CrossRef]
- Fransen, M.; Nordgren, M.; Wang, B.; Apanasets, O. Role of peroxisomes in ROS/RNS-metabolism: Implications for human disease. Biochim. Biophys. Acta 2012, 1822, 1363–1373. [Google Scholar] [CrossRef] [Green Version]
- Kondrup, J.; Lazarow, P.B. Flux of palmitate through the peroxisomal and mitochondrial beta-oxidation systems in isolated rat hepatocytes. Biochim. Biophys. Acta 1985, 835, 147–153. [Google Scholar] [CrossRef]
- Lodhi, I.J.; Semenkovich, C.F. Peroxisomes: A nexus for lipid metabolism and cellular signaling. Cell Metab. 2014, 19, 380–392. [Google Scholar] [CrossRef] [Green Version]
- Wanders, R.J. Metabolic functions of peroxisomes in health and disease. Biochimie 2014, 98, 36–44. [Google Scholar] [CrossRef] [PubMed]
- Wanders, R.J.; Waterham, H.R.; Ferdinandusse, S. Metabolic Interplay between Peroxisomes and Other Subcellular Organelles Including Mitochondria and the Endoplasmic Reticulum. Front. Cell Dev. Biol. 2015, 3, 83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, L.; Yu, M.; Arshad, M.; Wang, W.; Lu, Y.; Gong, J.; Gu, Y.; Li, P.; Xu, L. Coordination Among Lipid Droplets, Peroxisomes, and Mitochondria Regulates Energy Expenditure Through the CIDE-ATGL-PPARα Pathway in Adipocytes. Diabetes 2018, 67, 1935–1948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lake, A.D.; Novak, P.; Shipkova, P.; Aranibar, N.; Robertson, D.; Reily, M.D.; Lu, Z.; Lehman-McKeeman, L.D.; Cherrington, N.J. Decreased hepatotoxic bile acid composition and altered synthesis in progressive human nonalcoholic fatty liver disease. Toxicol. Appl. Pharmacol. 2013, 268, 132–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nordgren, M.; Fransen, M. Peroxisomal metabolism and oxidative stress. Biochimie 2014, 98, 56–62. [Google Scholar] [CrossRef] [Green Version]
- Guo, W.; Lian, S.; Zhen, L.; Zang, S.; Chen, Y.; Lang, L.; Xu, B.; Guo, J.; Ji, H.; Wang, J.; et al. The Favored Mechanism for Coping with Acute Cold Stress: Upregulation of miR-210 in Rats. Cell Physiol. Biochem. 2018, 46, 2090–2102. [Google Scholar] [CrossRef] [Green Version]
- Lu, Q.; Tian, X.; Wu, H.; Huang, J.; Li, M.; Mei, Z.; Zhou, L.; Xie, H.; Zheng, S. Metabolic Changes of Hepatocytes in NAFLD. Front. Physiol. 2021, 12, 710420. [Google Scholar] [CrossRef]
- Chao, H.W.; Chao, S.W.; Lin, H.; Ku, H.C.; Cheng, C.F. Homeostasis of Glucose and Lipid in Non-Alcoholic Fatty Liver Disease. Int. J. Mol. Sci. 2019, 20, 298. [Google Scholar] [CrossRef] [Green Version]
- Mota, M.; Banini, B.A.; Cazanave, S.C.; Sanyal, A.J. Molecular mechanisms of lipotoxicity and glucotoxicity in nonalcoholic fatty liver disease. Metabolism 2016, 65, 1049–1061. [Google Scholar] [CrossRef] [Green Version]
- Hoang, S.A.; Oseini, A.; Feaver, R.E.; Cole, B.K.; Asgharpour, A.; Vincent, R.; Siddiqui, M.; Lawson, M.J.; Day, N.C.; Taylor, J.M.; et al. Gene Expression Predicts Histological Severity and Reveals Distinct Molecular Profiles of Nonalcoholic Fatty Liver Disease. Sci. Rep. 2019, 9, 12541. [Google Scholar] [CrossRef] [Green Version]
- Suppli, M.P.; Rigbolt, K.T.G.; Veidal, S.S.; Heebøll, S.; Eriksen, P.L.; Demant, M.; Bagger, J.I.; Nielsen, J.C.; Oró, D.; Thrane, S.W.; et al. Hepatic transcriptome signatures in patients with varying degrees of nonalcoholic fatty liver disease compared with healthy normal-weight individuals. Am. J. Physiol. Gastrointest. Liver Physiol. 2019, 316, G462–G472. [Google Scholar] [CrossRef] [PubMed]
- Arendt, B.M.; Comelli, E.M.; Ma, D.W.L.; Lou, W.; Teterina, A.; Kim, T.; Fung, S.K.; Wong, D.K.H.; McGilvray, I.; Fischer, S.E.; et al. Altered hepatic gene expression in nonalcoholic fatty liver disease is associated with lower hepatic n-3 and n-6 polyunsaturated fatty acids. Hepatology 2015, 61, 1565–1578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lefebvre, P.; Lalloyer, F.; Baugé, E.; Pawlak, M.; Gheeraert, C.; Dehondt, H.; Vanhoutte, J.; Woitrain, E.; Hennuyer, N.; Mazuy, C.; et al. Interspecies NASH disease activity whole-genome profiling identifies a fibrogenic role of PPARα-regulated dermatopontin. JCI Insight 2017, 2, e92264. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.M.; Gorreta, F.; Ong, J.P.; Schlauch, K.; Del Giacco, L.; Elariny, H.; Van Meter, A.; Younoszai, A.; Goodman, Z.; Baranova, A.; et al. Hepatic gene expression in patients with obesity-related non-alcoholic steatohepatitis. Liver Int. 2005, 25, 760–771. [Google Scholar] [CrossRef]
- Zhu, R.; Baker, S.S.; Moylan, C.A.; Abdelmalek, M.F.; Guy, C.D.; Zamboni, F.; Wu, D.; Lin, W.; Liu, W.; Baker, R.D.; et al. Systematic transcriptome analysis reveals elevated expression of alcohol-metabolizing genes in NAFLD livers. J. Pathol. 2016, 238, 531–542. [Google Scholar] [CrossRef]
- Gerhard, G.S.; Legendre, C.; Still, C.D.; Chu, X.; Petrick, A.; DiStefano, J.K. Transcriptomic Profiling of. Obesity-Related Nonalcoholic Steatohepatitis Reveals a Core Set of Fibrosis-Specific Genes. J. Endocr. Soc. 2018, 13, 710–726. [Google Scholar] [CrossRef] [Green Version]
- Besse-Patin, A.; Léveillé, M.; Oropeza, D.; Nguyen, B.N.; Prat, A.; Estall, J.L. Estrogen Signals Through Peroxisome Proliferator-Activated Receptor-γ Coactivator 1α to Reduce Oxidative Damage Associated With Diet-Induced Fatty Liver Disease. Gastroenterology 2017, 152, 243–256. [Google Scholar] [CrossRef]
- Clouet, P.; Henninger, C.; Bézard, J. Study of some factors controlling fatty acid oxidation in liver mitochondria of obese Zucker rats. Biochem. J. 1986, 239, 103–108. [Google Scholar] [CrossRef] [Green Version]
- McGarry, J.D.; Foster, D.W. Regulation of hepatic fatty acid oxidation and ketone body production. Annu. Rev. Biochem. 1980, 49, 395–420. [Google Scholar] [CrossRef]
- Owen, O.E.; Kalhan, S.C.; Hanson, R.W. The key role of anaplerosis and cataplerosis for citric acid cycle function. J. Biol. Chem. 2002, 277, 30409–30412. [Google Scholar] [CrossRef] [Green Version]
- Fletcher, J.A.; Deja, S.; Satapati, S.; Fu, X.; Burgess, S.C.; Browning, J.D. Impaired ketogenesis and increased acetyl-CoA oxidation promote hyperglycemia in human fatty liver. JCI Insight 2019, 5, e127737. [Google Scholar] [CrossRef] [PubMed]
- Yoo, H.C.; Yu, Y.C.; Sung, Y.; Han, J.M. Glutamine reliance in cell metabolism. Exp. Mol. Med. 2020, 52, 1496–1516. [Google Scholar] [CrossRef] [PubMed]
- Akie, T.E.; Cooper, M.P. Determination of Fatty Acid Oxidation and Lipogenesis in Mouse Primary Hepatocytes. J. Vis. Exp. 2015, 102, e52982. [Google Scholar] [CrossRef] [Green Version]
- Benninghoff, T.; Espelage, L.; Eickelschulte, S.; Zeinert, I.; Sinowenka, I.; Müller, F.; Schöndeling, C.; Batchelor, H.; Cames, S.; Zhou, Z.; et al. The RabGAPs TBC1D1 and TBC1D4 Control Uptake of Long-Chain Fatty Acids Into Skeletal Muscle via Fatty Acid Transporter SLC27A4/FATP4. Diabetes 2020, 69, 2281–2293. [Google Scholar] [CrossRef] [PubMed]
- Horbelt, T.; Tacke, C.; Markova, M.; Herzfeld de Wiza, D.; Van de Velde, F.; Bekaert, M.; Van Nieuwenhove, Y.; Hornemann, S.; Rodiger, M.; Seebeck, N.; et al. The novel adipokine WISP1 associates with insulin resistance and impairs insulin action in human myotubes and mouse hepatocytes. Diabetologia 2018, 61, 2054–2065. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartwig, S.; Kotzka, J.; Lehr, S. Isolation and Quality Control of Functional Mitochondria. Methods Mol. Biol. 2021, 2276, 41–55. [Google Scholar] [CrossRef]
- Perez-Riverol, Y.; Csordas, A.; Bai, J.; Bernal-Llinares, M.; Hewapathirana, S.; Kundu, D.J.; Inuganti, A.; Griss, J.; Mayer, G.; Eisenacher, M.; et al. The PRIDE database and related tools and resources in 2019: Improving support for quantification data. Nucleic Acids Res. 2019, 47, D442–D450. [Google Scholar] [CrossRef]
- Barbosa, D.M.; Fahlbusch, P.; Herzfeld de Wiza, D.; Jacob, S.; Kettel, U.; Al-Hasani, H.; Krüger, M.; Ouwens, D.M.; Hartwig, S.; Lehr, S.; et al. Rhein, a novel Histone Deacetylase (HDAC) inhibitor with antifibrotic potency in human myocardial fibrosis. Sci. Rep. 2020, 10, 4888. [Google Scholar] [CrossRef]
- Dille, M.; Nikolic, A.; Wahlers, N.; Fahlbusch, P.; Jacob, S.; Hartwig, S.; Lehr, S.; Kabra, D.; Klymenko, O.; Al-Hasani, H.; et al. Long-term adjustment of hepatic lipid metabolism after chronic stress and the role of FGF21. Biochim. Biophys. Acta Mol. Basis Dis. 2022, 1868, 166286. [Google Scholar] [CrossRef]
- Fahlbusch, P.; Knebel, B.; Hörbelt, T.; Barbosa, D.M.; Nikolic, A.; Jacob, S.; Al-Hasani, H.; Van de Velde, F.; Van Nieuwenhove, Y.; Müller-Wieland, D.; et al. Physiological Disturbance in Fatty Liver Energy Metabolism Converges on IGFBP2 Abundance and Regulation in Mice and Men. Int. J. Mol. Sci. 2020, 21, 4144. [Google Scholar] [CrossRef]
- Horbelt, T.; Knebel, B.; Fahlbusch, P.; Barbosa, D.; de Wiza, D.H.; Van De Velde, F.; Van Nieuwenhove, Y.; Lapauw, B.; Thoresen, G.H.; Al-Hasani, H.; et al. The adipokine sFRP4 induces insulin resistance and lipogenesis in the liver. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 2671–2684. [Google Scholar] [CrossRef] [PubMed]
- Krämer, A.; Green, J.; Pollard, J.J.; Tugendreich, S. Causal analysis approaches in ingenuity pathway analysis. Bioinformatics 2014, 30, 523–530. [Google Scholar] [CrossRef]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fahlbusch, P.; Nikolic, A.; Hartwig, S.; Jacob, S.; Kettel, U.; Köllmer, C.; Al-Hasani, H.; Lehr, S.; Müller-Wieland, D.; Knebel, B.; et al. Adaptation of Oxidative Phosphorylation Machinery Compensates for Hepatic Lipotoxicity in Early Stages of MAFLD. Int. J. Mol. Sci. 2022, 23, 6873. https://doi.org/10.3390/ijms23126873
Fahlbusch P, Nikolic A, Hartwig S, Jacob S, Kettel U, Köllmer C, Al-Hasani H, Lehr S, Müller-Wieland D, Knebel B, et al. Adaptation of Oxidative Phosphorylation Machinery Compensates for Hepatic Lipotoxicity in Early Stages of MAFLD. International Journal of Molecular Sciences. 2022; 23(12):6873. https://doi.org/10.3390/ijms23126873
Chicago/Turabian StyleFahlbusch, Pia, Aleksandra Nikolic, Sonja Hartwig, Sylvia Jacob, Ulrike Kettel, Cornelia Köllmer, Hadi Al-Hasani, Stefan Lehr, Dirk Müller-Wieland, Birgit Knebel, and et al. 2022. "Adaptation of Oxidative Phosphorylation Machinery Compensates for Hepatic Lipotoxicity in Early Stages of MAFLD" International Journal of Molecular Sciences 23, no. 12: 6873. https://doi.org/10.3390/ijms23126873
APA StyleFahlbusch, P., Nikolic, A., Hartwig, S., Jacob, S., Kettel, U., Köllmer, C., Al-Hasani, H., Lehr, S., Müller-Wieland, D., Knebel, B., & Kotzka, J. (2022). Adaptation of Oxidative Phosphorylation Machinery Compensates for Hepatic Lipotoxicity in Early Stages of MAFLD. International Journal of Molecular Sciences, 23(12), 6873. https://doi.org/10.3390/ijms23126873