
Cathepsin B p.Gly284Val Variant in Parkinson’s Disease Pathogenesis

 , , , , , ,
, , , , , ,  , , , , and add
Show full author list
, , , , and add
Show full author list
Abstract
:1. Introduction
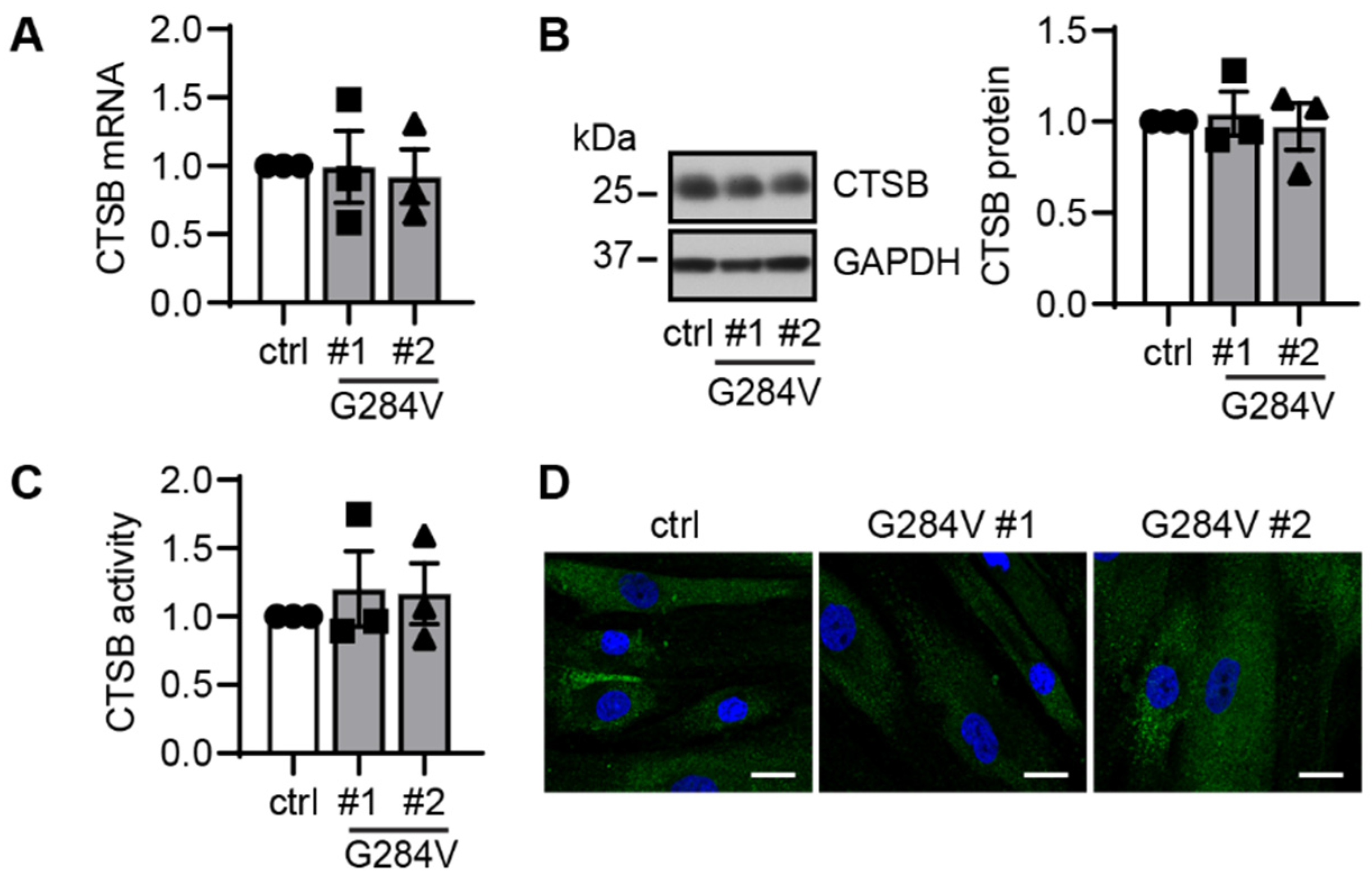
2. Results
3. Discussion
4. Materials and Methods
4.1. Clinical Examination
4.2. Exome Sequencing in Sib-Pairs
4.3. Replication Cohorts and Genotyping
4.4. Gene Burden Analysis
4.5. Protein Sequence and Structure Analysis
4.6. Generation of Fibroblasts and Cell Culture
4.7. RNA Extraction and qRT-PCR
4.8. Protein Extraction and Western Blot
4.9. CTSB Enzymatic Activity Assay
4.10. Immunocytochemistry
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Balestrino, R.; Schapira, A. Parkinson disease. Eur. J. Neurol. 2020, 27, 27–42. [Google Scholar] [CrossRef] [PubMed]
- Postuma, R.B.; Berg, D.; Stern, M.; Poewe, W.; Olanow, C.W.; Oertel, W.; Obeso, J.; Marek, K.; Litvan, I.; Lang, A.E. MDS clinical diagnostic criteria for Parkinson’s disease. Mov. Disord. 2015, 30, 1591–1601. [Google Scholar] [CrossRef] [PubMed]
- Dickson, D.W. Neuropathology of Parkinson disease. Parkinsonism Relat. Disord. 2018, 46, S30–S33. [Google Scholar] [CrossRef]
- Karimi-Moghadam, A.; Charsouei, S.; Bell, B.; Jabalameli, M.R. Parkinson disease from mendelian forms to genetic susceptibility: New molecular insights into the neurodegeneration process. Cell. Mol. Neurobiol. 2018, 38, 1153–1178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nalls, M.A.; Blauwendraat, C.; Vallerga, C.L.; Heilbron, K.; Bandres-Ciga, S.; Chang, D.; Tan, M.; Kia, D.A.; Noyce, A.J.; Xue, A. Identification of novel risk loci, causal insights, and heritable risk for Parkinson’s disease: A meta-analysis of genome-wide association studies. Lancet Neurol. 2019, 18, 1091–1102. [Google Scholar] [CrossRef]
- Milanowski, Ł.M.; Ross, O.A.; Friedman, A.; Hoffman-Zacharska, D.; Gorka-Skoczylas, P.; Jurek, M.; Koziorowski, D.; Wszolek, Z.K. Genetics of Parkinson’s disease in the Polish population. Neurol. Neurochir. Pol. 2021, 55, 241–252. [Google Scholar] [CrossRef]
- Siuda, J.; Boczarska-Jedynak, M.; Budrewicz, S.; Figura, M.; Fiszer, U.; Gajos, A.; Gorzkowska, A.; Koziorowska-Gawron, E.; Koziorowski, D.; Krygowska-Wajs, A.; et al. Validation of the Polish version of the Movement Disorder Society-Unified Parkinson’s Disease Rating Scale (MDS-UPDRS). Neurol. I Neurochir. Pol. 2020, 54, 416–425. [Google Scholar] [CrossRef]
- Wissemann, W.T.; Hill-Burns, E.M.; Zabetian, C.P.; Factor, S.A.; Patsopoulos, N.; Hoglund, B.; Holcomb, C.; Donahue, R.J.; Thomson, G.; Erlich, H.; et al. Association of Parkinson disease with structural and regulatory variants in the HLA region. Am. J. Hum. Genet. 2013, 93, 984–993. [Google Scholar] [CrossRef] [Green Version]
- Porrini, V.; Mota, M.; Parrella, E.; Bellucci, A.; Benarese, M.; Faggi, L.; Tonin, P.; Spano, P.F.; Pizzi, M. Mild Inflammatory Profile without Gliosis in the c-Rel Deficient Mouse Modeling a Late-Onset Parkinsonism. Front. Aging Neurosci. 2017, 9, 229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, S.J.; Armasu, S.M.; Biernacka, J.M.; Lesnick, T.G.; Rider, D.N.; Cunningham, J.M.; Maraganore, D.M. Variants in estrogen-related genes and risk of Parkinson’s disease. Mov. Disord. 2011, 26, 1234–1242. [Google Scholar] [CrossRef] [PubMed]
- Yuan, L.; Song, Z.; Deng, X.; Zheng, W.; Guo, Y.; Yang, Z.; Deng, H. Systematic analysis of genetic variants in Han Chinese patients with sporadic Parkinson’s disease. Sci. Rep. 2016, 6, 33850. [Google Scholar] [CrossRef] [PubMed]
- Germer, E.L.; Imhoff, S.; Vilariño-Güell, C.; Kasten, M.; Seibler, P.; Brüggemann, N.; Klein, C.; Trinh, J. The Role of Rare Coding Variants in Parkinson’s Disease GWAS Loci. Front. Neurol. 2019, 10, 1284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McGlinchey, R.P.; Lee, J.C. Cysteine cathepsins are essential in lysosomal degradation of α-synuclein. Proc. Natl. Acad. Sci. USA 2015, 112, 9322–9327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klein, C.L.; Rovelli, G.; Springer, W.; Schall, C.; Gasser, T.; Kahle, P.J. Homo- and heterodimerization of ROCO kinases: LRRK2 kinase inhibition by the LRRK2 ROCO fragment. J. Neurochem. 2009, 111, 703–715. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Lee, S.; Du, J.; Jain, K.; Ding, M.; Kadado, A.J.; Atteya, G.; Jaji, Z.; Tyagi, T.; Kim, W.H.; et al. Mitochondrial MsrB2 serves as a switch and transducer for mitophagy. EMBO Mol. Med. 2019, 11, e10409. [Google Scholar] [CrossRef]
- Pérez-Santamarina, E.; García-Ruiz, P.; Martínez-Rubio, D.; Ezquerra, M.; Pla-Navarro, I.; Puente, J.; Martí, M.J.; Palau, F.; Hoenicka, J. Regulatory rare variants of the dopaminergic gene ANKK1 as potential risk factors for Parkinson’s disease. Sci. Rep. 2021, 11, 9879. [Google Scholar] [CrossRef]
- Kedashiro, S.; Pastuhov, S.I.; Nishioka, T.; Watanabe, T.; Kaibuchi, K.; Matsumoto, K.; Hanafusa, H. LRRK1-phosphorylated CLIP-170 regulates EGFR trafficking by recruiting p150Glued to microtubule plus ends. J. Cell. Sci. 2015, 128, 385–396. [Google Scholar] [CrossRef] [Green Version]
- Greenbaum, L.; Rigbi, A.; Lipshtat, N.; Cilia, R.; Tesei, S.; Asselta, R.; Djaldetti, R.; Goldwurm, S.; Lerer, B. Association of nicotine dependence susceptibility gene, CHRNA5, with Parkinson’s disease age at onset: Gene and smoking status interaction. Parkinsonism Relat. Disord. 2013, 19, 72–76. [Google Scholar] [CrossRef]
- Dong, W.; Qiu, C.; Gong, D.; Jiang, X.; Liu, W.; Liu, W.; Zhang, L.; Zhang, W. Proteomics and bioinformatics approaches for the identification of plasma biomarkers to detect Parkinson’s disease. Exp. Med. 2019, 18, 2833–2842. [Google Scholar] [CrossRef]
- Jansen, I.E.; Ye, H.; Heetveld, S.; Lechler, M.C.; Michels, H.; Seinstra, R.I.; Lubbe, S.J.; Drouet, V.; Lesage, S.; Majounie, E.; et al. Discovery and functional prioritization of Parkinson’s disease candidate genes from large-scale whole exome sequencing. Genome Biol. 2017, 18, 22. [Google Scholar] [CrossRef] [Green Version]
- Farrow, S.L.; Schierding, W.; Gokuladhas, S.; Golovina, E.; Fadason, T.; Cooper, A.A.; O’Sullivan, J.M. Establishing gene regulatory networks from Parkinson’s disease risk loci. Brain 2022, awac022. [Google Scholar] [CrossRef] [PubMed]
- Blauwendraat, C.; Reed, X.; Krohn, L.; Heilbron, K.; Bandres-Ciga, S.; Tan, M.; Gibbs, J.R.; Hernandez, D.G.; Kumaran, R.; Langston, R. Genetic modifiers of risk and age at onset in GBA associated Parkinson’s disease and Lewy body dementia. Brain 2020, 143, 234–248. [Google Scholar] [CrossRef] [PubMed]
- McGlinchey, R.P.; Lacy, S.M.; Huffer, K.E.; Tayebi, N.; Sidransky, E.; Lee, J.C. C-terminal α-synuclein truncations are linked to cysteine cathepsin activity in Parkinson’s disease. J. Biol. Chem. 2019, 294, 9973–9984. [Google Scholar] [CrossRef] [Green Version]
- Ritonja, A.; Popovic, T.; Turk, V.; Wiedenmann, K.; Machleidt, W. Amino acid sequence of human liver cathepsin B. FEBS Lett. 1985, 181, 169–172. [Google Scholar] [CrossRef] [Green Version]
- Perera, R.M.; Zoncu, R. The lysosome as a regulatory hub. Annu. Rev. Cell Dev. Biol. 2016, 32, 223–253. [Google Scholar] [CrossRef] [Green Version]
- Vasiljeva, O.; Reinheckel, T.; Peters, C.; Turk, D.; Turk, V.; Turk, B. Emerging roles of cysteine cathepsins in disease and their potential as drug targets. Curr. Pharm. Des. 2007, 13, 387–403. [Google Scholar] [CrossRef] [PubMed]
- Cao, M.; Luo, X.; Wu, K.; He, X. Targeting lysosomes in human disease: From basic research to clinical applications. Signal Transduct. Target. Ther. 2021, 6, 1–28. [Google Scholar] [CrossRef]
- Cermak, S.; Kosicek, M.; Mladenovic-Djordjevic, A.; Smiljanic, K.; Kanazir, S.; Hecimovic, S. Loss of cathepsin B and L leads to lysosomal dysfunction, NPC-like cholesterol sequestration and accumulation of the key Alzheimer’s proteins. PLoS ONE 2016, 11, e0167428. [Google Scholar] [CrossRef] [Green Version]
- Jiang, M.; Meng, J.; Zeng, F.; Qing, H.; Hook, G.; Hook, V.; Wu, Z.; Ni, J. Cathepsin B inhibition blocks neurite outgrowth in cultured neurons by regulating lysosomal trafficking and remodeling. J. Neurochem. 2020, 155, 300–312. [Google Scholar] [CrossRef]
- Man, S.M.; Kanneganti, T.-D. Regulation of lysosomal dynamics and autophagy by CTSB/cathepsin B. Autophagy 2016, 12, 2504–2505. [Google Scholar] [CrossRef] [Green Version]
- Milanowski, Ł.; Hoffman-Zacharska, D.; Geremek, M.; Friedman, A.; Figura, M.; Koziorowski, D. The matter of significance–Has the p.(Glu121Lys) variant of TOR1A gene a pathogenic role in dystonia or Parkinson disease? J. Clin. Neurosci. 2020, 72, 501–503. [Google Scholar] [CrossRef] [PubMed]
- Konno, T.; Deutschländer, A.; Heckman, M.G.; Ossi, M.; Vargas, E.R.; Strongosky, A.J.; van Gerpen, J.A.; Uitti, R.J.; Ross, O.A.; Wszolek, Z.K. Comparison of clinical features among Parkinson’s disease subtypes: A large retrospective study in a single center. J. Neurol. Sci. 2018, 386, 39–45. [Google Scholar] [CrossRef]
- Notredame, C.; Higgins, D.G.; Heringa, J. T-Coffee: A novel method for fast and accurate multiple sequence alignment. J. Mol. Biol. 2000, 302, 205–217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Armougom, F.; Moretti, S.; Poirot, O.; Audic, S.; Dumas, P.; Schaeli, B.; Keduas, V.; Notredame, C. Expresso: Automatic incorporation of structural information in multiple sequence alignments using 3D-Coffee. Nucleic Acids Res. 2006, 34, W604–W608. [Google Scholar] [CrossRef]
- Robert, X.; Gouet, P. Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res. 2014, 42, W320–W324. [Google Scholar] [CrossRef] [Green Version]
- Wei, B.; Gunzner-Toste, J.; Yao, H.; Wang, T.; Wang, J.; Xu, Z.; Chen, J.; Wai, J.; Nonomiya, J.; Tsai, S.P.; et al. Discovery of Peptidomimetic Antibody-Drug Conjugate Linkers with Enhanced Protease Specificity. J. Med. Chem. 2018, 61, 989–1000. [Google Scholar] [CrossRef] [PubMed]
- McNicholas, S.; Potterton, E.; Wilson, K.S.; Noble, M.E. Presenting your structures: The CCP4mg molecular-graphics software. Acta Cryst. D Biol. Cryst. 2011, 67, 386–394. [Google Scholar] [CrossRef] [Green Version]
- Puschmann, A.; Fiesel, F.C.; Caulfield, T.R.; Hudec, R.; Ando, M.; Truban, D.; Hou, X.; Ogaki, K.; Heckman, M.G.; James, E.D.; et al. Heterozygous PINK1 p.G411S increases risk of Parkinson’s disease via a dominant-negative mechanism. Brain 2016, 140, 98–117. [Google Scholar] [CrossRef] [Green Version]
- Ando, M.; Fiesel, F.C.; Hudec, R.; Caulfield, T.R.; Ogaki, K.; Górka-Skoczylas, P.; Koziorowski, D.; Friedman, A.; Chen, L.; Dawson, V.L.; et al. The PINK1 p.I368N mutation affects protein stability and ubiquitin kinase activity. Mol. Neurodegener. 2017, 12, 32. [Google Scholar] [CrossRef] [Green Version]
- Watzlawik, J.O.; Hou, X.; Fricova, D.; Ramnarine, C.; Barodia, S.K.; Gendron, T.F.; Heckman, M.G.; DeTure, M.; Siuda, J.; Wszolek, Z.K.; et al. Sensitive ELISA-based detection method for the mitophagy marker p-S65-Ub in human cells, autopsy brain, and blood samples. Autophagy 2021, 17, 2613–2628. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Position (GRCh38) | rs | Ref | Alt | CADD Score | Classification | Gene | Evidence |
|---|---|---|---|---|---|---|---|
| Homozygous | |||||||
| chr6:g.32518589 | rs147439581 | C | T | 13.6 | Nonsyn SNV | HLA-DRB5 | GWAS [5] |
| chr6:g.32519397 | rs112872773 | C | G,T | 10.9 | Nonsyn SNV | HLA-DRB5 | GWAS [5] |
| chr6:g.32521967 | rs780328684 | G | TGG,- | Frameshift Del, Frameshift Sub | HLA-DRB5 | GWAS [5] | |
| chr6:g.32584262 | rs150747106 | C | T,G | 21.9 | Nonsyn SNV | HLA-DRB1 | GWAS [8] |
| chr10:g.17849736 | rs71497225 | G | C | 8.1 | Nonsyn SNV | MRC1 | Decrease the risk of PD [9] |
| chr10:g.17849710 | rs71497223 | A | G | 6.7 | Nonsyn SNV | MRC1 | Decrease the risk of PD [9] |
| Heterozygous | |||||||
| chr1:g.13782630 | rs140700877 | A | C | 22.7 | Nonsyn SNV | PRDM2 | SNP more frequent observed in PD [10] |
| chr2:g.182756345 | rs138065612 | C | T | 24.1 | Nonsyn SNV | DNAJC10 | Decrease the risk of PD [11] |
| chr3:g.52378778 | rs201064587 | G | A | 21.2 | Nonsyn SNV | DNAH1 | SNP more frequent observed in PD [12] |
| chr8:g.11845732 | Novel | C | A | 26.7 | Nonsyn SNV | CTSB | Confirmed PD/LBD GWAS loci [5,13] |
| chr9:g.87637944 | rs200255856 | G | A | 32 | Nonsyn SNV | DAPK1 | Positive impact on LRRK2 and synuclein expression in animal models [14] |
| chr10:g.23104162 | rs2296466 | A | G | 15.56 | Nonsyn SNV | MSRB2 | Important in regulation of mitophagy [15] |
| chr11:g.113387894 | rs35657708 | G | T | 10.01 | Nonsyn SNV | ANKK1 | SNP more frequent observed in PD [16] |
| chr12:g.122340779 | Novel | T | A | 17.03 | Nonsyn SNV | CLIP1 | Increase risk of PD in LRRK2 patients [17] |
| chr15:g.78593232 | rs76071148 | T | A | 15.39 | Nonsyn SNV | CHRNA5 | Decrease risk of PD [18] |
| chr16:g.1443261 | Novel | C | T | 36 | Nonsyn SNV | CCDC154 | Higher level of protein observed in serum of PD patients [19] |
| chr19:g.55185948 | rs140748270 | G | C | 23 | Nonsyn SNV | PTPRH | Positive association in single WES study (1000 patients) [20] |
| Country of Origin | PD (N Females) | Age of Onset (Mean ± SD) | Controls (N Females) |
|---|---|---|---|
| USA | 997 (358) | 65.2 (±11.9) | - |
| Poland | 610 (250) | 59.3 (±12.6) | 248 (125) |
| Ireland | 320 (142) | 57.0 (±11.9) | 343 (217) |
| Ukraine | 122 (57) | 59.9 (±11.6) | 24 (12) |
| Czech Republic | 28 (14) | 59.3 (±11.8) | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Milanowski, L.M.; Hou, X.; Bredenberg, J.M.; Fiesel, F.C.; Cocker, L.T.; Soto-Beasley, A.I.; Walton, R.L.; Strongosky, A.J.; Faroqi, A.H.; Barcikowska, M.; et al. Cathepsin B p.Gly284Val Variant in Parkinson’s Disease Pathogenesis. Int. J. Mol. Sci. 2022, 23, 7086. https://doi.org/10.3390/ijms23137086
Milanowski LM, Hou X, Bredenberg JM, Fiesel FC, Cocker LT, Soto-Beasley AI, Walton RL, Strongosky AJ, Faroqi AH, Barcikowska M, et al. Cathepsin B p.Gly284Val Variant in Parkinson’s Disease Pathogenesis. International Journal of Molecular Sciences. 2022; 23(13):7086. https://doi.org/10.3390/ijms23137086
Chicago/Turabian StyleMilanowski, Lukasz M., Xu Hou, Jenny M. Bredenberg, Fabienne C. Fiesel, Liam T. Cocker, Alexandra I. Soto-Beasley, Ronald L. Walton, Audrey J. Strongosky, Ayman H. Faroqi, Maria Barcikowska, and et al. 2022. "Cathepsin B p.Gly284Val Variant in Parkinson’s Disease Pathogenesis" International Journal of Molecular Sciences 23, no. 13: 7086. https://doi.org/10.3390/ijms23137086
APA StyleMilanowski, L. M., Hou, X., Bredenberg, J. M., Fiesel, F. C., Cocker, L. T., Soto-Beasley, A. I., Walton, R. L., Strongosky, A. J., Faroqi, A. H., Barcikowska, M., Boczarska-Jedynak, M., Dulski, J., Fedoryshyn, L., Janik, P., Potulska-Chromik, A., Karpinsky, K., Krygowska-Wajs, A., Lynch, T., Olszewska, D. A., ... Wszolek, Z. K. (2022). Cathepsin B p.Gly284Val Variant in Parkinson’s Disease Pathogenesis. International Journal of Molecular Sciences, 23(13), 7086. https://doi.org/10.3390/ijms23137086