
Glycation of Tie-2 Inhibits Angiopoietin-1 Signaling Activation and Angiopoietin-1-Induced Angiogenesis

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
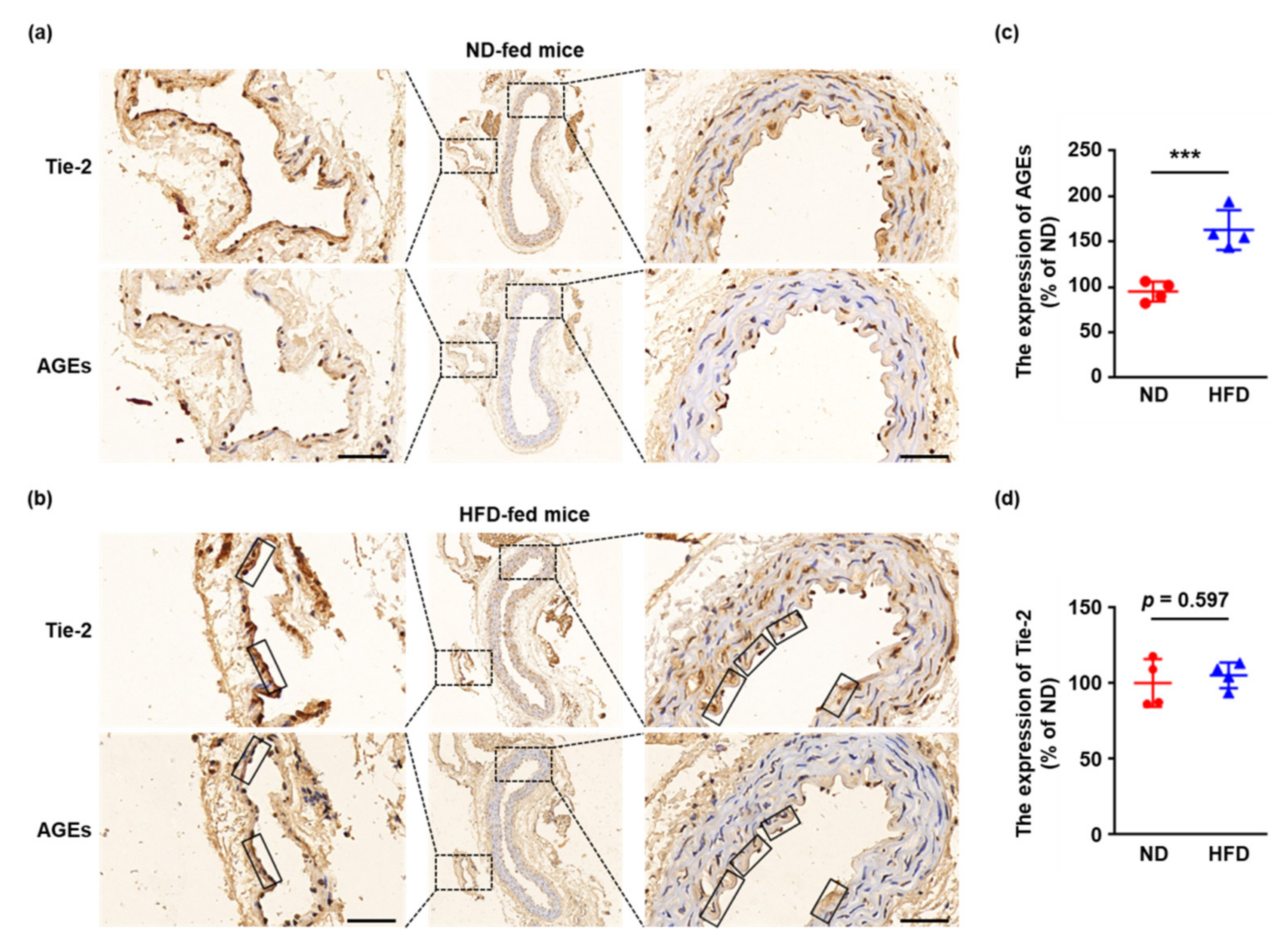
2.1. Tie-2 Is Glycated in HFD-Fed Mice
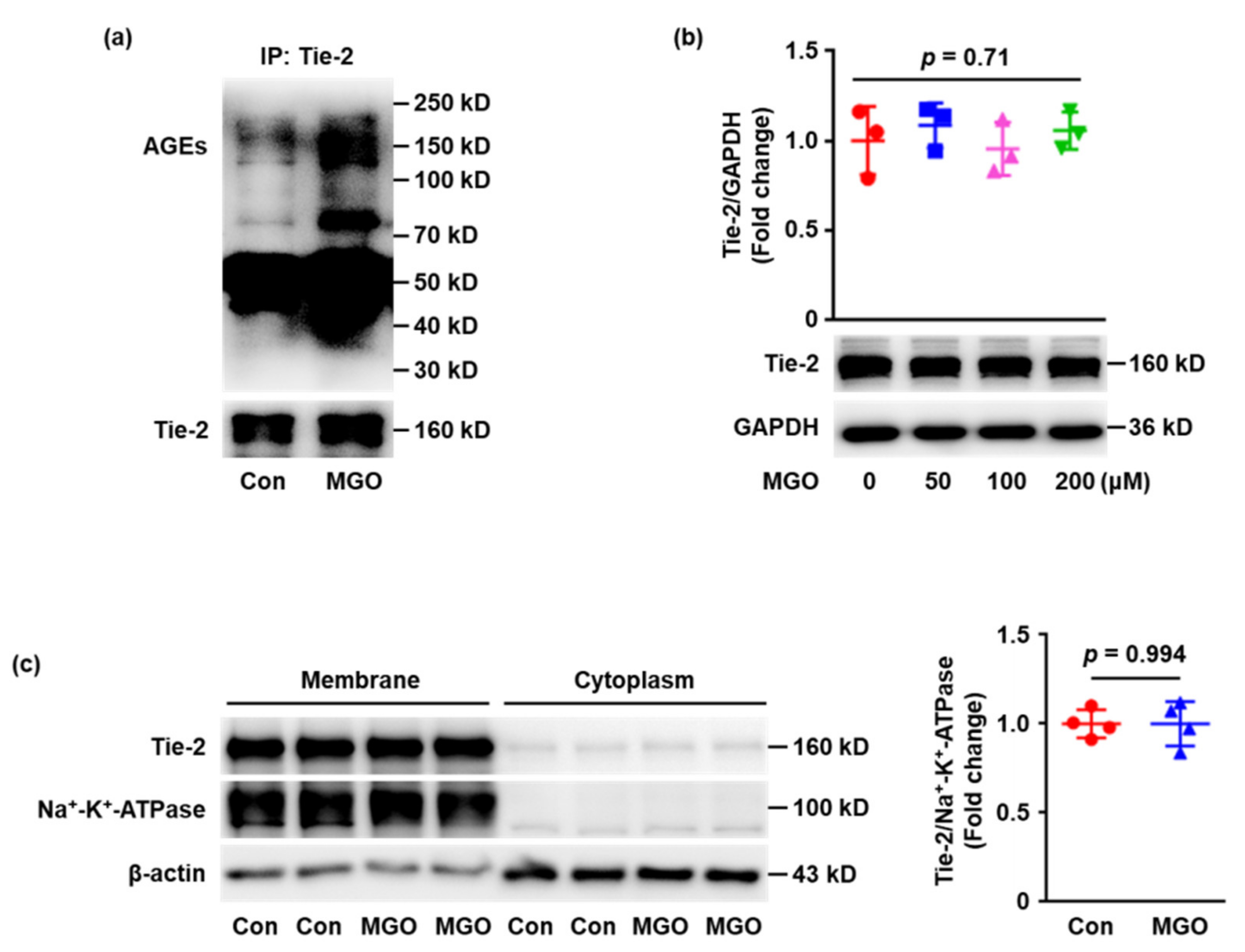
2.2. Tie-2 Is Glycated in MGO-Treated Human Umbilical Vein Endothelial Cells (HUVECs) In Vitro
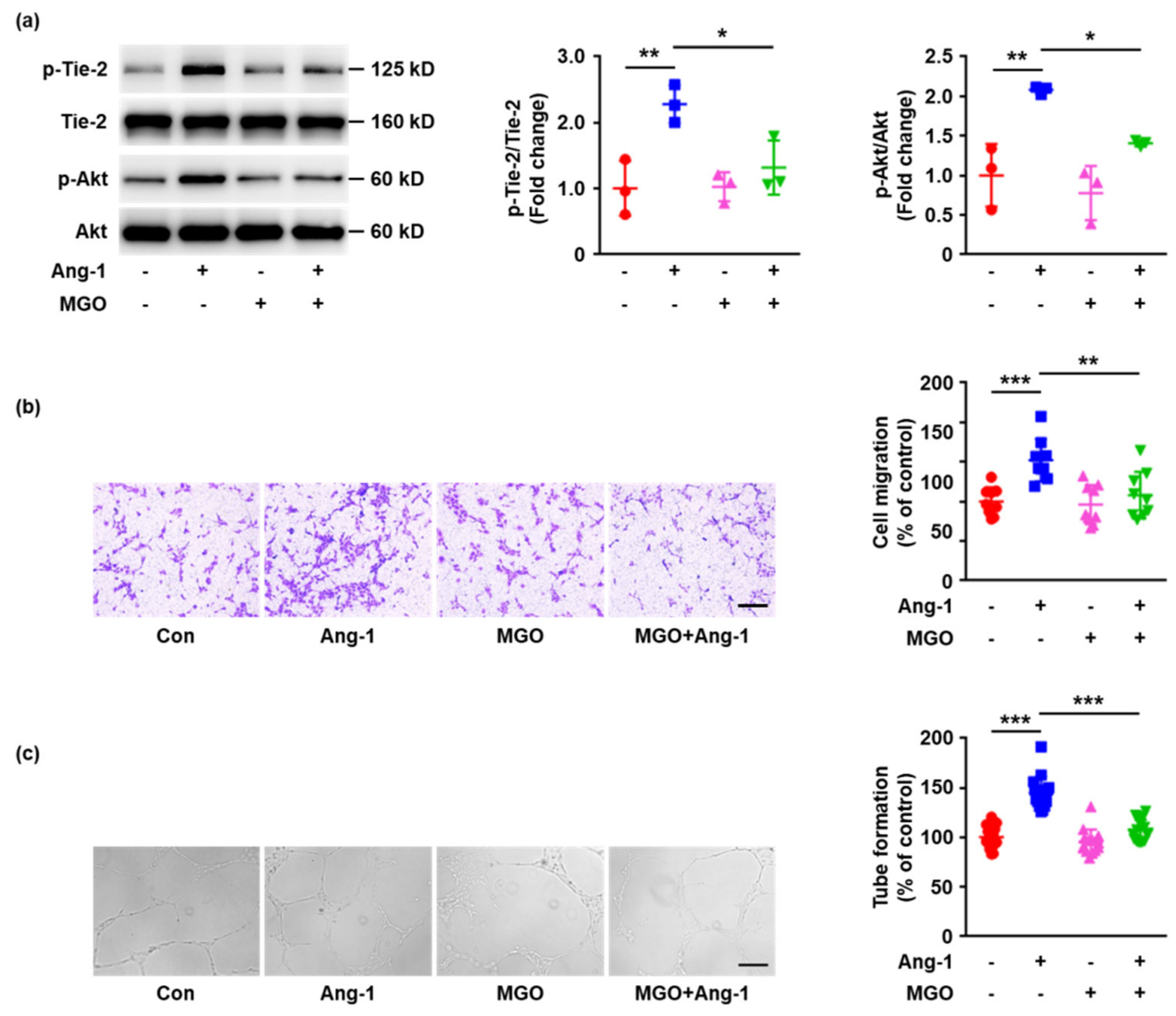
2.3. MGO-Induced Tie-2 Glycation Impairs Ang-1 Signaling Activation and Ang-1 Induced Angiogenesis In Vitro
2.4. The Inhibition of Tie-2 Glycation Rescues Ang-1 Signaling Activation and Ang-1-Induced Angiogenesis In Vitro
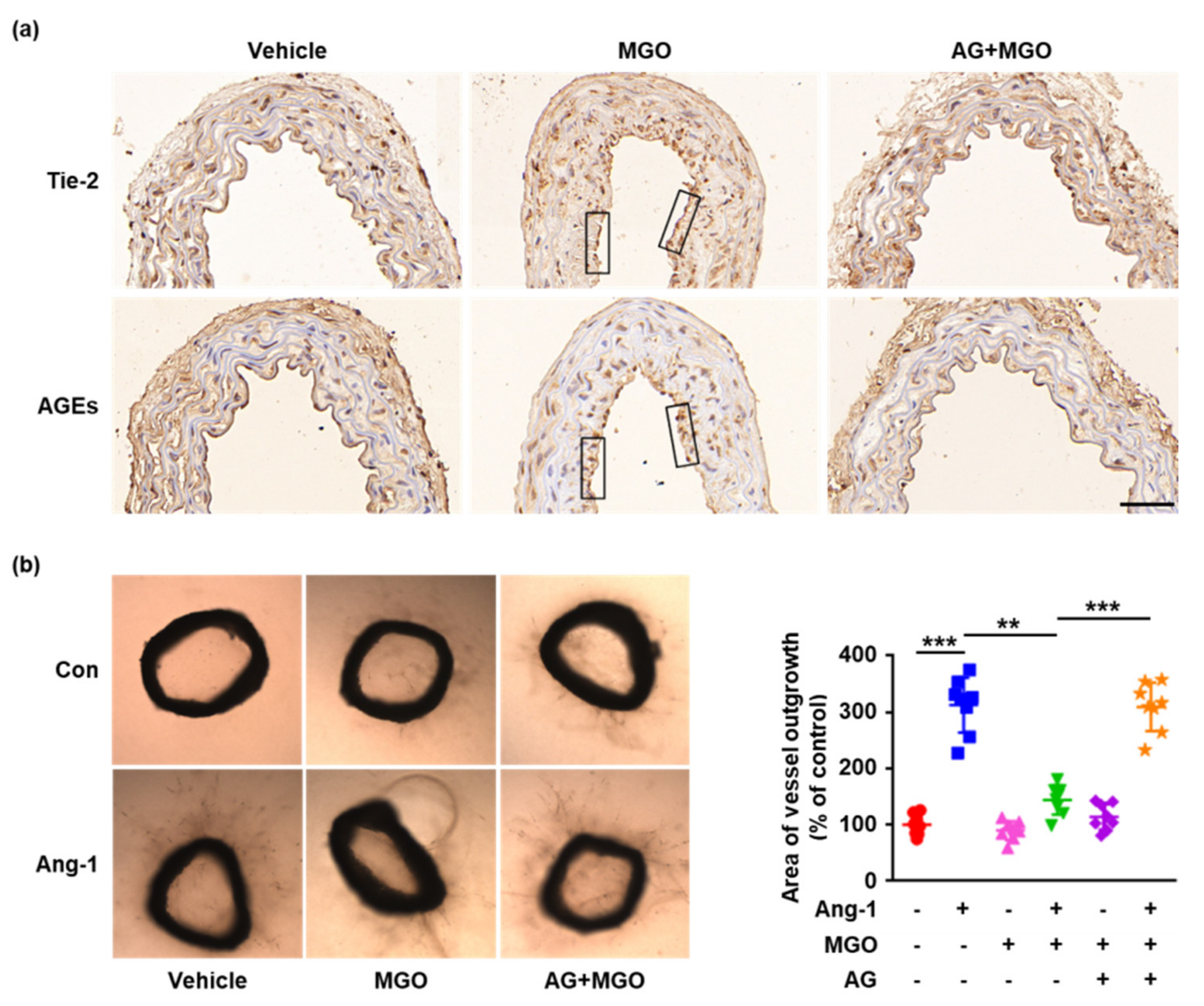
2.5. Glycation of Tie-2 Inhibits Ang-1-Induced Vessel Outgrowth Ex Vivo
3. Discussion
4. Materials and Methods
4.1. Chemicals and Reagents
4.2. Animals
4.3. HFD-Fed Mouse Model
4.4. Histological Analysis
4.5. Cell Culture
4.6. Cell Scratch Wound Healing Assay
4.7. Cell Migration Assay
4.8. Tube Formation Assay
4.9. Western Blotting
4.10. Immunoprecipitation
4.11. Ex Vivo Angiogenesis Assay
4.12. Data Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fadini, G.P.; Albiero, M.; Bonora, B.M.; Avogaro, A. Angiogenic Abnormalities in Diabetes Mellitus: Mechanistic and Clinical Aspects. J. Clin. Endocrinol. Metab. 2019, 104, 5431–5444. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Z.; Kishore, R. Potential role of hydrogen sulfide in diabetes-impaired angiogenesis and ischemic tissue repair. Redox Biol. 2020, 37, 101704. [Google Scholar] [CrossRef]
- Tanabe, K.; Wada, J.; Sato, Y. Targeting angiogenesis and lymphangiogenesis in kidney disease. Nat. Rev. Nephrol. 2020, 16, 289–303. [Google Scholar] [CrossRef] [PubMed]
- Duran, C.L.; Borriello, L.; Karagiannis, G.S.; Entenberg, D.; Oktay, M.H.; Condeelis, J.S. Targeting Tie2 in the Tumor Microenvironment: From Angiogenesis to Dissemination. Cancers 2021, 13, 5730. [Google Scholar] [CrossRef] [PubMed]
- Waltenberger, J. VEGF resistance as a molecular basis to explain the angiogenesis paradox in diabetes mellitus. Biochem. Soc. Trans. 2009, 37, 1167–1170. [Google Scholar] [CrossRef]
- Ishikura, K.; Misu, H.; Kumazaki, M.; Takayama, H.; Nagata, N.; Tajima, N.; Chikamoto, K.; Lan, F.; Ando, H.; Ota, T.; et al. Selenoprotein P as a diabetes-associated hepatokine that impairs angiogenesis by inducing VEGF resistance in vascular endothelial cells. Diabetologia 2014, 57, 1968–1976. [Google Scholar] [CrossRef]
- Wang, L.; Zhang, X.; Pang, N.; Xiao, L.; Li, Y.; Chen, N.; Ren, M.; Deng, X.; Wu, J. Glycation of vitronectin inhibits VEGF-induced angiogenesis by uncoupling VEGF receptor-2-αvβ3 integrin cross-talk. Cell Death Dis. 2015, 6, e1796. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.X.; Tuo, Q.; Liao, D.F.; Zeng, H. Inhibition of protein tyrosine phosphatase improves angiogenesis via enhancing Ang-1/Tie-2 signaling in diabetes. Exp. Diabetes Res. 2012, 2012, 836759. [Google Scholar] [CrossRef] [Green Version]
- Tuo, Q.-H.; Xiong, G.-Z.; Zeng, H.; Yu, H.-D.; Sun, S.-W.; Ling, H.-Y.; Zhu, B.-Y.; Liao, D.-F.; Chen, J.-X. Angiopoietin-1 protects myocardial endothelial cell function blunted by angiopoietin-2 and high glucose condition. Acta Pharmacol. Sin. 2010, 32, 45–51. [Google Scholar] [CrossRef] [Green Version]
- Chen, T.; Dong, J.; Zhou, H.; Deng, X.; Li, R.; Chen, N.; Wang, L. Glycation of fibronectin inhibits VEGF-induced angiogenesis by uncoupling VEGF receptor-2-c-Src crosstalk. J. Cell. Mol. Med. 2020, 24, 9154–9164. [Google Scholar] [CrossRef]
- Sato, T.N.; Tozawa, Y.; Deutsch, U.; Wolburg-Buchholz, K.; Fujiwara, Y.; Gendron-Maguire, M.; Gridley, T.; Wolburg, H.; Risau, W.; Qin, Y. Distinct roles of the receptor tyrosine kinases Tie-1 and Tie-2 in blood vessel formation. Nature 1995, 376, 70–74. [Google Scholar] [CrossRef] [PubMed]
- Isidori, A.M.; Venneri, M.A.; Fiore, D. Angiopoietin-1 and Angiopoietin-2 in metabolic disorders: Therapeutic strategies to restore the highs and lows of angiogenesis in diabetes. J. Endocrinol. Investig. 2016, 39, 1235–1246. [Google Scholar] [CrossRef] [PubMed]
- Fukuhara, S.; Sako, K.; Noda, K.; Zhang, J.; Minami, M.; Mochizuki, N. Angiopoietin-1/Tie-2 receptor signaling in vascular quiescence and angiogenesis. Histol. Histopathol. 2010, 25, 387–396. [Google Scholar] [PubMed]
- Hernandez-Castillo, C.; Shuck, S.C. Diet and obesity-induced methylglyoxal production and links to metabolic disease. Chem. Res. Toxicol. 2021, 34, 2424–2440. [Google Scholar] [CrossRef]
- Schalkwijk, C.; Stehouwer, C.D.A. Methylglyoxal, a Highly Reactive Dicarbonyl Compound, in Diabetes, Its Vascular Complications, and Other Age-Related Diseases. Physiol. Rev. 2020, 100, 407–461. [Google Scholar] [CrossRef]
- Bellier, J.; Nokin, M.-J.; Lardé, E.; Karoyan, P.; Peulen, O.; Castronovo, V.; Bellahcène, A. Methylglyoxal, a potent inducer of AGEs, connects between diabetes and cancer. Diabetes Res. Clin. Pract. 2019, 148, 200–211. [Google Scholar] [CrossRef]
- Wetzels, S.; Wouters, K.; Schalkwijk, C.G.; Vanmierlo, T.; Hendriks, J.J.A. Methylglyoxal-Derived Advanced Glycation Endproducts in Multiple Sclerosis. Int. J. Mol. Sci. 2017, 18, 421. [Google Scholar] [CrossRef] [Green Version]
- Khalid, M.; Petroianu, G.; Adem, A. Advanced Glycation End Products and Diabetes Mellitus: Mechanisms and Perspectives. Biomolecules 2022, 12, 542. [Google Scholar] [CrossRef]
- Sakamoto, H.; Mashima, T.; Yamamoto, K.; Tsuruo, T. Modulation of Heat-shock Protein 27 (Hsp27) Anti-apoptotic Activity by Methylglyoxal Modification. J. Biol. Chem. 2002, 277, 45770–45775. [Google Scholar] [CrossRef] [Green Version]
- Fukunaga, M.; Miyata, S.; Higo, S.; Hamada, Y.; Ueyama, S.; Kasuga, M. Methylglyoxal Induces Apoptosis through Oxidative Stress-Mediated Activation of p38 Mitogen-Activated Protein Kinase in Rat Schwann Cells. Ann. N. Y. Acad. Sci. 2005, 1043, 151–157. [Google Scholar] [CrossRef]
- Portero-Otín, M.; Pamplona, R.; Bellmunt, M.J.; Ruiz, M.C.; Prat, J.; Salvayre, R.; Nègre-Salvayre, A. Advanced Glycation End Product Precursors Impair Epidermal Growth Factor Receptor Signaling. Diabetes 2002, 51, 1535–1542. [Google Scholar] [CrossRef] [Green Version]
- Jia, X.; Olson, D.J.H.; Ross, A.R.S.; Wu, L. Structural and functional changes in human insulin induced by methylglyoxal. FASEB J. 2006, 20, 1555–1557. [Google Scholar] [CrossRef] [PubMed]
- Riboulet-Chavey, A.; Pierron, A.; Durand, I.; Murdaca, J.; Giudicelli, J.; Van Obberghen, E. Methylglyoxal Impairs the Insulin Signaling Pathways Independently of the Formation of Intracellular Reactive Oxygen Species. Diabetes 2006, 55, 1289–1299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cantero, A.V.; Portero-Otín, M.; Ayala, V.; Auge, N.; Sanson, M.; Elbaz, M. Methylglyoxal induces advanced glycation end product (AGEs) formation and dysfunction of PDGF receptor-beta: Implications for diabetic atherosclerosis. FASEB J. 2007, 21, 3096–3106. [Google Scholar] [CrossRef] [PubMed]
- Monu Agnihotri, P.; Biswas, S. AGE/non-AGE glycation: An important event in rheumatoid arthritis athophysiology. Inflammation 2022, 45, 477–496. [Google Scholar] [CrossRef]
- Sirangelo, I.; Iannuzzi, C. Understanding the Role of Protein Glycation in the Amyloid Aggregation Process. Int. J. Mol. Sci. 2021, 22, 6609. [Google Scholar] [CrossRef]
- Rabbani, N.; Xue, M.; Thornalley, P.J. Dicarbonyl stress, protein glycation and the unfolded protein response. Glycoconj. J. 2021, 38, 331–340. [Google Scholar] [CrossRef]
- Padival, A.K.; Crabb, J.W.; Nagaraj, R.H. Methylglyoxal modifies heat shock protein 27 in glomerular mesangial cells. FEBS Lett. 2003, 551, 113–118. [Google Scholar] [CrossRef]
- Krämer, A.C.; Davies, M.J. Effect of Methylglyoxal-Induced Glycation on the Composition and Structure of β-Lactoglobulin and α-Lactalbumin. J. Agric. Food Chem. 2018, 67, 699–710. [Google Scholar] [CrossRef]
- Fiory, F.; Lombardi, A.; Miele, C.; Giudicelli, J.; Beguinot, F.; Van Obberghen, E. Methylglyoxal impairs insulin signalling and insulin action on glucose-induced insulin secretion in the pancreatic beta cell line INS-1E. Diabetologia 2011, 54, 2941–2952. [Google Scholar] [CrossRef] [Green Version]
- Prasad, K.; Tiwari, S. Therapeutic Interventions for Advanced Glycation-End Products and its Receptor- Mediated Cardiovascular Disease. Curr. Pharm. Des. 2017, 23, 937–943. [Google Scholar] [CrossRef] [PubMed]
- Wolosowicz, M.; Prokopiuk, S.; Kaminski, T.W. Recent advances in the treatment ofinsulin resistance targeting molecular and metabolic pathways: Fighting a losing battle? Medicina 2022, 58, 472. [Google Scholar] [CrossRef] [PubMed]
- Feng, Z.; Du, Z.; Shu, X.; Zhu, L.; Wu, J.; Gao, Q.; Wang, L.; Chen, N.; Li, Y.; Luo, M.; et al. Role of RAGE in obesity-induced adipose tissue inflammation and insulin resistance. Cell Death Discov. 2021, 7, 305. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Feng, Z.; Shu, X.; Gao, Q.; Wu, J.; Du, Z.; Wu, J. In situ transplantation of adipose-derived stem cells via photoactivation improves glucose metabolism in obese mice. Stem Cell Res. Ther. 2021, 12, 408. [Google Scholar] [CrossRef]
- Lee, J.H.; Parveen, A.; Do, M.H.; Kang, M.C.; Yumnam, S.; Kim, S.Y. Molecular mechanisms of methylglyoxal-induced aortic endothelial dysfunction in human vascular endothelial cells. Cell Death Dis. 2020, 11, 403. [Google Scholar] [CrossRef]
- Kim, J.S.; Lee, J.H.; Hong, S.M.; Cho, K.H.; Kim, S.Y. Salvia miltiorrhiza Prevents Methylglyoxal-Induced Glucotoxicity via the Regulation of Apoptosis-Related Pathways and the Glyoxalase System in Human Umbilical Vein Endothelial Cells. Biol. Pharm. Bull. 2022, 45, 51–62. [Google Scholar] [CrossRef]
- Dong, J.; Zhou, H.; Li, Y.; Li, R.; Chen, N.; Zheng, Y.; Deng, X.; Luo, M.; Wu, J.; Wang, L. MG53 inhibits angiogenesis through regulating focal adhesion kinase signalling. J. Cell. Mol. Med. 2021, 25, 7462–7471. [Google Scholar] [CrossRef]
- Baker, M.; Robinson, S.; Lechertier, T.; Barber, P.R.; Tavora, B.; D’Amico, G.; Jones, D.T.; Vojnovic, B.; Hodivala-Dilke, K. Use of the mouse aortic ring assay to study angiogenesis. Nat. Protoc. 2011, 7, 89–104. [Google Scholar] [CrossRef]
- Chen, L.; Cui, Y.; Li, B.; Weng, J.; Wang, W.; Zhang, S.; Huang, X.; Guo, X.; Huang, Q. Advanced glycation end products induce immature angiogenesis in in vivo and ex vivo mouse models. Am. J. Physiol. Circ. Physiol. 2020, 318, H519–H533. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, H.; Chen, T.; Li, Y.; You, J.; Deng, X.; Chen, N.; Li, T.; Zheng, Y.; Li, R.; Luo, M.; et al. Glycation of Tie-2 Inhibits Angiopoietin-1 Signaling Activation and Angiopoietin-1-Induced Angiogenesis. Int. J. Mol. Sci. 2022, 23, 7137. https://doi.org/10.3390/ijms23137137
Zhou H, Chen T, Li Y, You J, Deng X, Chen N, Li T, Zheng Y, Li R, Luo M, et al. Glycation of Tie-2 Inhibits Angiopoietin-1 Signaling Activation and Angiopoietin-1-Induced Angiogenesis. International Journal of Molecular Sciences. 2022; 23(13):7137. https://doi.org/10.3390/ijms23137137
Chicago/Turabian StyleZhou, Haiyan, Tangting Chen, Yongjie Li, Jingcan You, Xin Deng, Ni Chen, Tian Li, Youkun Zheng, Rong Li, Mao Luo, and et al. 2022. "Glycation of Tie-2 Inhibits Angiopoietin-1 Signaling Activation and Angiopoietin-1-Induced Angiogenesis" International Journal of Molecular Sciences 23, no. 13: 7137. https://doi.org/10.3390/ijms23137137
APA StyleZhou, H., Chen, T., Li, Y., You, J., Deng, X., Chen, N., Li, T., Zheng, Y., Li, R., Luo, M., Wu, J., & Wang, L. (2022). Glycation of Tie-2 Inhibits Angiopoietin-1 Signaling Activation and Angiopoietin-1-Induced Angiogenesis. International Journal of Molecular Sciences, 23(13), 7137. https://doi.org/10.3390/ijms23137137