
Iron, Neuroinflammation and Neurodegeneration


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Iron Metabolism and Homeostasis
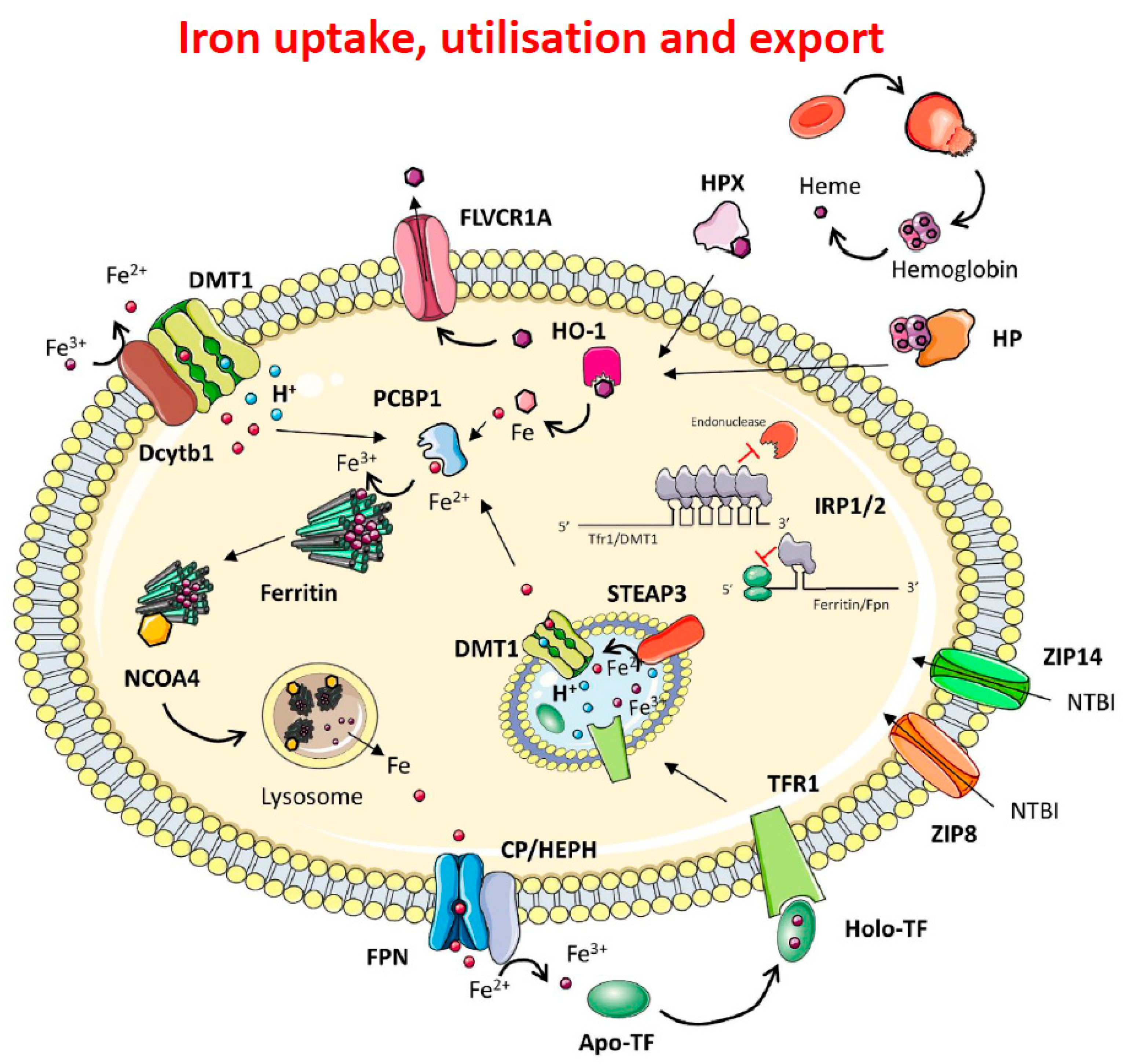
1.1. Iron Metabolism
1.2. Intracellular Iron Homeostasis
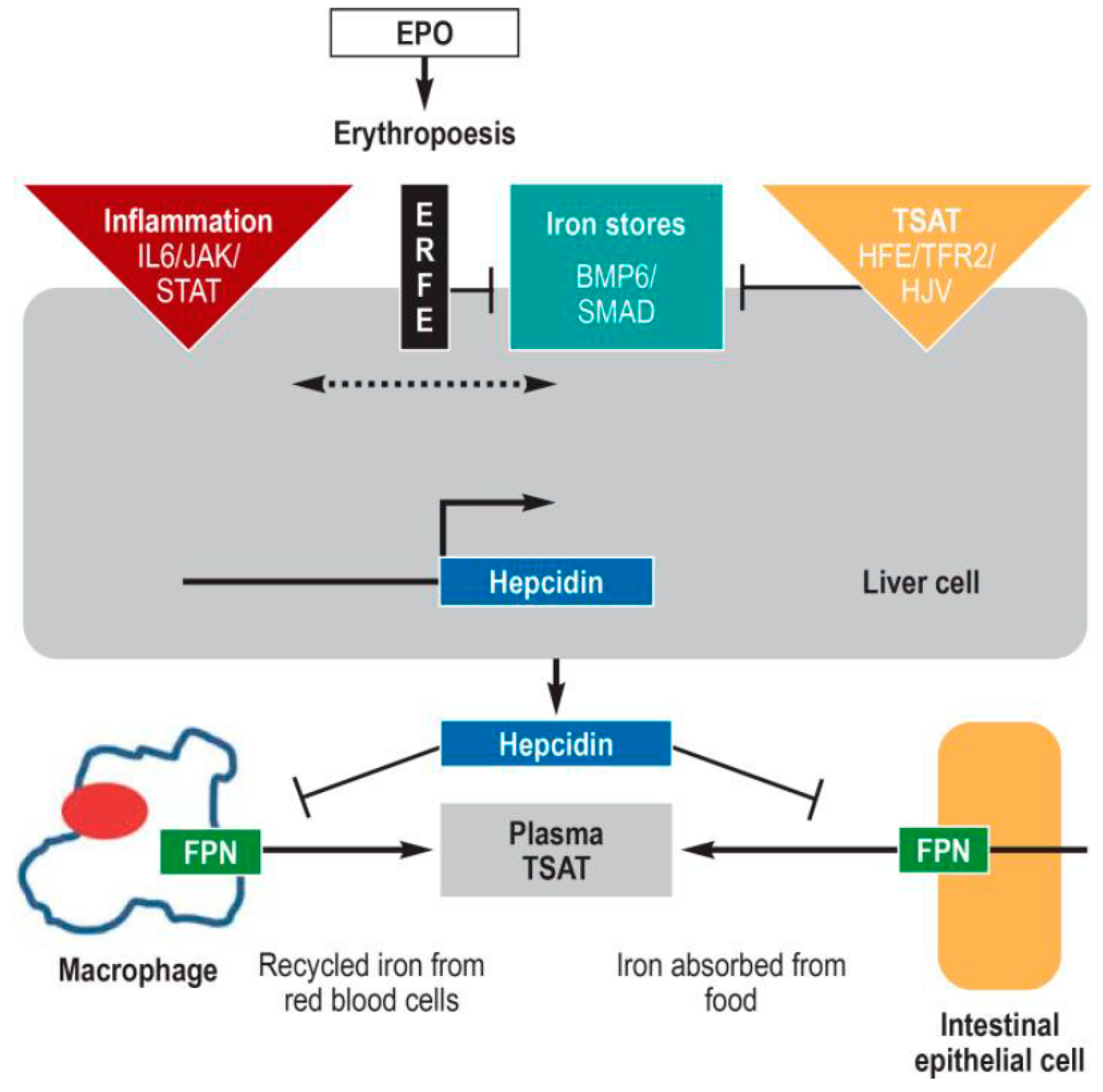
1.3. The Hepcidin-Ferroportin Regulatory System
1.4. Brain Iron Metabolism
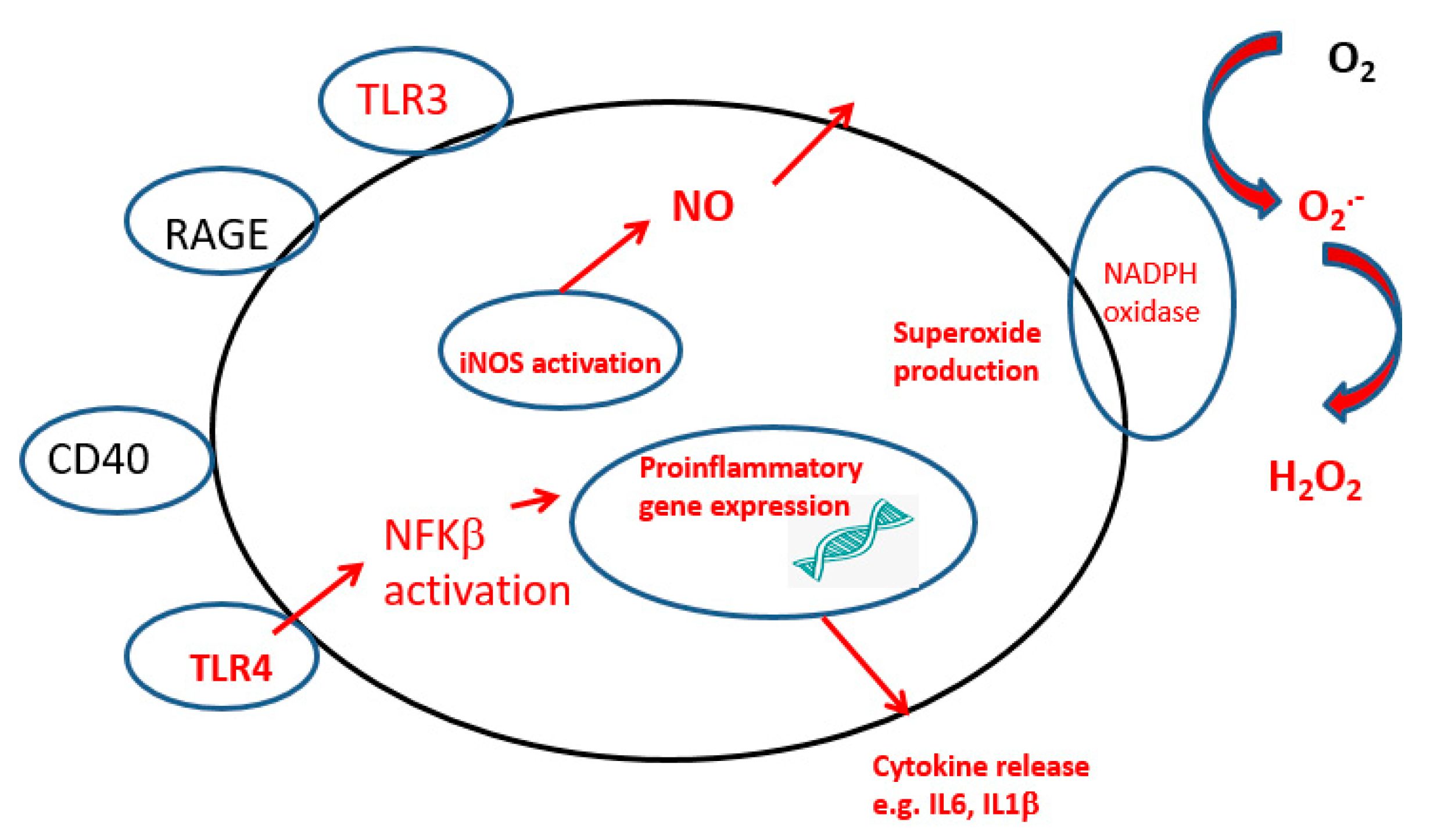
2. Neuroinflammation
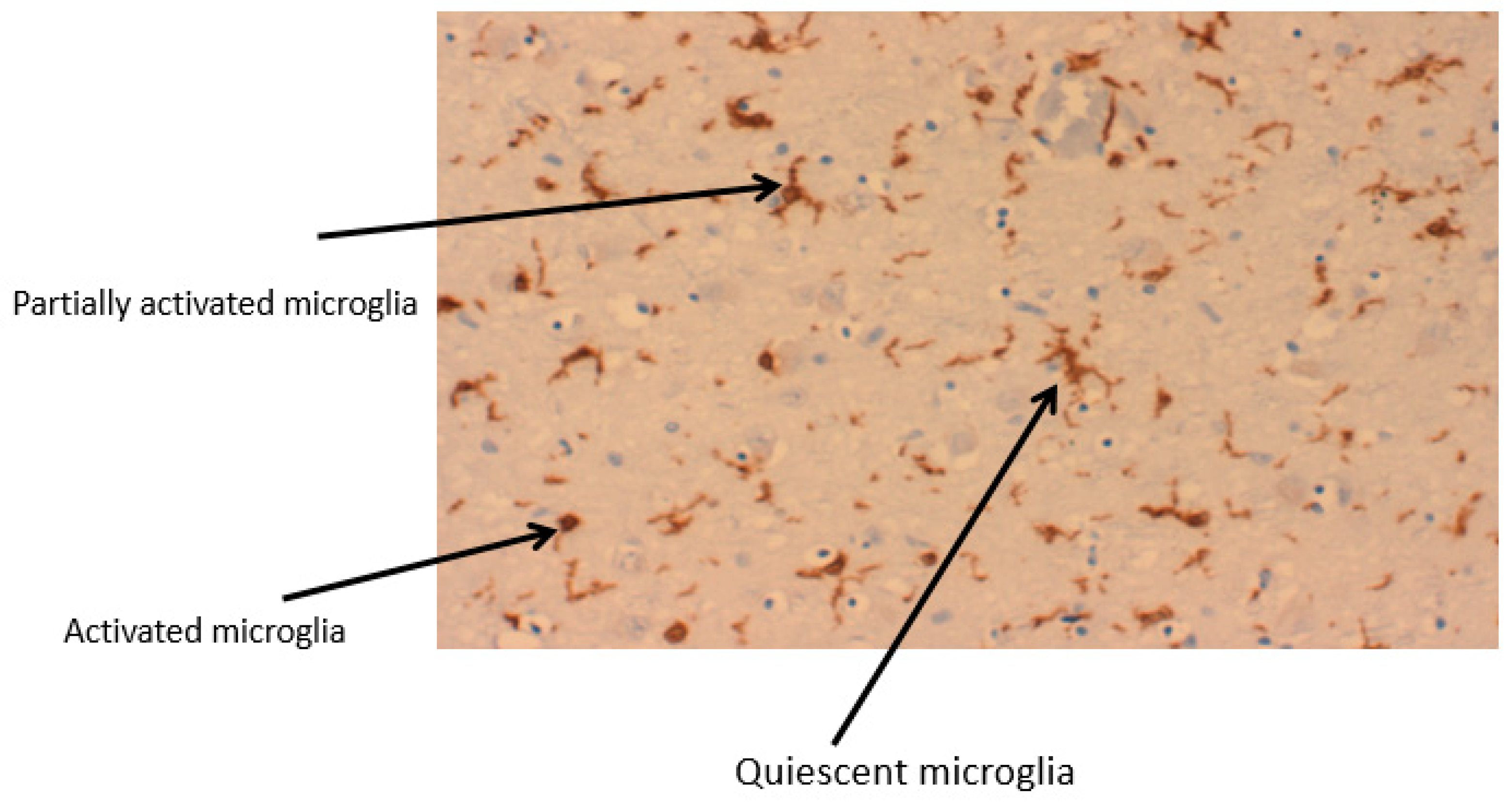
2.1. Microglia
2.2. Astrocytes
2.3. Oligodendrocytes
2.4. Neurons
3. Neuroinflammation Mediated Neurodegeneration in the Brain
3.1. Aging
3.2. Protein Misfolding
3.3. Mitochondria
3.4. Gut
3.5. Peripheral Inflammatory Markers
3.6. Blood–Brain Barrier
4. Neurodegenerative Diseases with Special Emphasis on PD and AD
4.1. Parkinson Disease
4.1.1. Peripheral Circulation
4.1.2. Mitochondria Function
4.1.3. Blood–Brain Barrier, BBB
4.1.4. Neuroinflammation
4.1.5. Iron in PD and Changes in Iron Proteins
4.2. Alzheimers Disease
4.2.1. Peripheral Circulation
4.2.2. Neuroinflammation in AD
4.2.3. Mitochondria
4.2.4. Blood–Brain Barrier
4.2.5. Iron Loading and Iron Proteins in AD
5. Therapeutic Approaches
5.1. Non-Steroidal Inflammatory Drugs, NSAI
5.2. N-Acetyl Cysteine
5.3. Iron Chelation
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Crichton, R.R. Iron Metabolism. From Molecular Mechanisms to Cellular Consequences, 4th ed.; John Wiley and Sons: Chichester, UK, 2016; p. 556. [Google Scholar]
- Santana-Codina, N.; Gikandi, A.; Mancias, J.D. The Role of NCOA4-Mediated Ferritinophagy in Ferroptosis. Adv. Exp. Med. Biol. 2021, 1301, 41–57. [Google Scholar] [PubMed]
- Yanatori, I.; Richardson, D.R.; Toyokuni, S.; Kishi, F. The new role of poly (rC)-binding proteins as iron transport chaperones: Proteins that could couple with inter-organelle interactions to safely traffic iron. Biochim. Biophys. Acta Gen. Subj. 2020, 1864, 129685. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.; Bencze, K.Z.; Stemmler, T.L.; Philpott, C.C. A cytosolic iron chaperone that delivers iron to ferritin. Science 2008, 320, 1207–1210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Philpott, C.C.; Jadhav, S. The ins and outs of iron: Escorting iron through the mammalian cytosol. Free Radic. Biol. Med. 2019, 133, 112–117. [Google Scholar] [CrossRef]
- Altamura, S.; Marques, O.; Colucci, S.; Mertens, C.; Alikhanyan, K.; Muckenthaler, M.U. Regulation of iron homeostasis: Lessons from mouse models. Mol. Aspects Med. 2020, 75, 100872. [Google Scholar] [CrossRef]
- Kühn, L.C. Iron regulatory proteins and their role in controlling iron metabolism. Metallomics 2015, 7, 232–243. [Google Scholar] [CrossRef]
- Muckenthaler, M.; Gray, N.K.; Hentze, M.W. IRP-1 binding to ferritin mRNA prevents the recruitment of the small ribosomal subunit by the cap-binding complex eIF4F. Mol. Cell. 1998, 2, 383–388. [Google Scholar] [CrossRef]
- Yoshinaga, M.; Nakatsuka, Y.; Vandenbon, A.; Ori, D.; Uehata, T.; Tsujimura, T.; Suzuki, Y.; Mino, T.; Takeuchi, O. Regnase-1 maintains iron homeostasis via the degradation of transferrin receptor 1 and prolyl-hydroxylase-domain-containing protein 3 mRNAs. Cell Rep. 2017, 19, 1614–1630. [Google Scholar] [CrossRef] [Green Version]
- Muckenthaler, M.U.; Rivella, S.; Hentze, M.W.; Galy, B. A red carpet for iron metabolism. Cell 2017, 168, 344–361. [Google Scholar] [CrossRef] [Green Version]
- Srole, D.N.; Ganz, T. Erythroferrone structure, function, and physiology: Iron homeostasis and beyond. J. Cell Physiol. 2021, 236, 4888–4901. [Google Scholar] [CrossRef]
- Arezes, J.; Foy, N.; McHugh, K.; Sawant, A.; Quinkert, D.; Terraube, V.; Brinth, A.; Tam, M.; LaVallie, E.R.; Taylor, S.; et al. Erythroferrone inhibits the induction of hepcidin by BMP6. Blood 2018, 132, 1473–1477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gattermann, N.; Muckenthaler, M.U.; Kulozik, A.E.; Metzgeroth, G.; Hastka, J. The evaluation of iron deficiency and iron overload. J. Dtsch. Arztebl. Int. 2021, 118, 847–856. [Google Scholar] [CrossRef] [PubMed]
- Ward, R.J.; Crichton, R.R. Ironing out the brain. Met. Ions Life Sci. 2019, 14, 19. [Google Scholar]
- Ward, R.J.; Zucca, F.A.; Duyn, J.H.; Crichton, R.R.; Zecca, L. The role of iron in brain ageing and neurodegenerative disorders. Lancet Neurol. 2014, 13, 1045–1060. [Google Scholar] [CrossRef] [Green Version]
- Thomsen, M.S.; Routhe, L.; Moos, T. The vascular basement membrane in the healthy and pathological brain. J. Cereb. Blood Flow Metab. 2017, 37, 3300–3317. [Google Scholar] [CrossRef]
- Burkhart, A.; Skjørringe, T.; Johnsen, K.B.; Siupka, P.; Thomsen, L.B.; Nielsen, M.S.; Thomsen, L.L.; Moos, T. Expression of iron-related proteins at the neurovascular unit supports reduction and reoxidation of iron through the blood-brain barrier. Mol. Neurobiol. 2016, 53, 7237–7253. [Google Scholar] [CrossRef]
- Mleczko-Sanecka, K.; Silvestri, L. Cell-type-specific insights into iron regulatory processes. Am. J. Haematol. 2021, 196, 110–127. [Google Scholar] [CrossRef]
- Abe, N.; Nishihara, T.; Yorozuya, T.; Tanaka, J. Microglia and Macrophages in the Pathological Central and Peripheral Nervous Systems. Cells 2020, 9, 2132. [Google Scholar] [CrossRef]
- Crichton, R.R.; Ward, R.J. Metal Based Neurodegeneration; from Molecular Mechansisms to Therapeutic Strategies, 2nd ed.; John Wiley and Sons: Chichester, UK, 2014. [Google Scholar]
- Kotwica-Mojzych, K.; Jodlowska-Jedrych, B.; Mojzych, M. CD200:CD200R Interactions and Their Importance in Immunoregulation. Int. J. Mol. Sci. 2021, 22, 1602. [Google Scholar] [CrossRef]
- Fan, Y.; Xie, L.; Chung, C.Y. Signaling Pathways Controlling Microglia Chemotaxis. Mol. Cells 2017, 40, 163–168. [Google Scholar]
- Moeller, H.E.; Bossoni, L.; Connor, J.R.; Crichton, R.R.; Does, M.D.; Ward, R.J.; Zecca, L.; Zucca, F.A.; Romen, I. Iron, Myelin, and the Brain: Neuroimaging Meets Neurobiology. Trends Neurosci. 2019, 42, 384–401. [Google Scholar] [CrossRef] [PubMed]
- Jessen, N.A.; Finmann-Munk, A.S.; Lundgaard, I.; Nedergaard, M. The glymphatic system: A beginner’s guide. Neurochem. Res. 2015, 40, 258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khakh, B.S.; Sofroniew, M.V. Diversity of astrocyte functions and phenotypes in neural circuits. Nat. Neurosci. 2015, 18, 942–952. [Google Scholar] [CrossRef] [PubMed]
- Moos, T.; Nielsen, T.R. Ferroportin in the postnatal rat brain: Implications for axonal transport and neuronal export of iron. Semin. Pediatr. Neurol. 2006, 13, 149–157. [Google Scholar] [CrossRef]
- Bishop, G.M.; Dang, T.N.; Dringen, R.; Robinson, S.R. Accumulation of transferrin of non–bound iron by neurons, astrocytes, and microglia. Neurotox. Res. 2011, 19, 443–451. [Google Scholar] [CrossRef]
- Sarkar, D.; Fischer, P.B. Molecular mechanisms of aging-associated inflammation. Cancer Letters 2006, 236, 13–23. [Google Scholar] [CrossRef]
- Fernández-Mendívil, C.; Luengo, E.; Trigo-Alonso, P.; Nuria García-Magro, N.; Pilar Negredo, P.; Lopez, M.G. Protective role of microglial HO-1 blockade in aging: Implication of iron metabolism. Redox. Biol. 2021, 38, 101789. [Google Scholar] [CrossRef]
- Hunter, R.L.; Liu, M.; Young, D.Y.; Cass, W.A.; Bing, G. Inflammation and age related iron accumulation in F344 rats. Curr. Aging Sci. 2008, 1, 2. [Google Scholar] [CrossRef]
- Stefanatos, R.; Sanz, A. The role of mitochondrial ROS in the aging brain. FEBS Lett. 2018, 743–754. [Google Scholar] [CrossRef] [Green Version]
- Mulak, A.; Bonaz, B. Brain-gut-microbiota axis in Parkinson’s disease. World J. Gastroenterol. 2015, 21, 10609–10620. [Google Scholar] [CrossRef]
- Basak, J.M.; Ferreiro, A.; Cohen, L.S.; Sheehan, P.W.; Nafarajah, C.J.; Kanan, M.F.; Sukhum, K.V.; Dantas, G.; Musiek, E.S. Bacterial sepsis increases hippocampal fibrillar amyloid plaque load and neuroinflammation in a mouse model of Alzheimer’s disease. Neurobiol. Dis. 2021, 152, 105292. [Google Scholar] [CrossRef] [PubMed]
- Hoogland, C.M.; Houbolt, C.; Westerloo, D.J.; van Gool, D.; Diederik van de Beek, D. Systemic inflammation and microglial activation: Systematic review of animal experiments. J. Inflamm. 2015, 12, 114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pavlov, V.A.; Tracey, K.J. The vagus nerve and the inflammatory reflex—linking immunity and metabolism. Nat. Rev. Endocrinol. 2012, 8, 743–754. [Google Scholar] [CrossRef] [PubMed]
- Liuzzi, J.P.; Lichten, L.A.; Rivera, S.; Blanchard, R.K.; Aydemir, B.T.; Knutson, M.D.; Ganz, T.; Cousins, R.J. Interleukin-6 regulates the zinc transporter Zip14 in liver and contributes to the hypozincemia of the acute-phase response. Proc. Natl. Acad. Sci. USA 2005, 102, 6843–6848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuhlmann, C.R.; Librizzi, L.; Closhen, D.; Pflanzner, T.; Lessmann, V.; Pietrzik, C.U.; de Curtis, M.; Luhmann, H.J. Mechanisms of C-reactive protein-induced blood-brain barrier disruption. Stroke 2009, 40, 1458–1466. [Google Scholar] [CrossRef] [Green Version]
- Moos, T.; Rosengren Nielsen, T.; Skjørringe, T.; Morgan, E.H. Iron trafficking inside the brain. J. Neurochem. 2007, 103, 1730–1740. [Google Scholar] [CrossRef]
- Wessling-Resnick, M. Iron homeostasis and the inflammatory response. Annu. Rev. Nutr. 2010, 30, 105–122. [Google Scholar] [CrossRef] [Green Version]
- Scheiblich, H.; Dansokho, C.; Mercan, D.; Schmidt, S.V.; Bousset, L.; Lena Wischhof, L.; Eikens, F.; Odainic, A.; Spitzer, J.; Griep, A.; et al. Microglia jointly degrade fibrillar alpha-synuclein cargo by distribution through tunneling nanotubes. Cell 2021, 184, 5089–5106. [Google Scholar] [CrossRef]
- Jin, H.; Gu, H.Y.; Mao, C.J.; Chen, J.; Liu, C.F. Association of inflammatory factors and aging in Parkinson’s disease. Neurosci. Lett. 2020, 736, 135259. [Google Scholar] [CrossRef]
- Martin-Bastida, A.; Ward, R.J.; Newbould, R.; Piccini, P.; Sharp, D.; Kabba, C.; Patel, M.; Spino, M.; Connelly, J.; Tricta, F.; et al. Brain iron chelation by deferiprone in a phase 2 randomised double-blinded placebo controlled clinical trial in Parkinson’s disease. Sci. Rep. 2017, 7, 139. [Google Scholar] [CrossRef]
- Brochard, C.; Combadière, B.; Prigent, A.; Laouar, Y.; Perrin, A.; Beray-Berthat, V.; Bonduelle, O.; Alvarez-Fischer, D.; Callebert, J.; Launay, J.-M.; et al. Infiltration of CD4+lymphocytes into the brain contributes to neurodegeneration in a mouse model of Parkinson disease. J. Clin. Investig. 2009, 119, 182–192. [Google Scholar] [CrossRef] [PubMed]
- Jang, H.; Boltz, D.A.; Webster, G.W.; Smeyne, J. Viral parkinsonism. Biochim. Biophys. Acta Mol. Basis Dis. 2009, 1792, 714–772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, M.; Yamada, T.; Setsuko, N.; Nakajima, K.; Takayuki Yamamoto, T.; Okada, H. The substantia nigra is a major target for neurovirulent Influenza A virus. J. Exp. Med. 1995, 181, 2161–2169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, K.-J.; Lin, K.-L.; Chen, S.-D.; Liou, C.-W.; Chuang, Y.-C.; Lin, H.-Y.; Lin, T.-K. The Overcrowded Crossroads: Mitochondria, Alpha-Synuclein, and the Endo-Lysosomal System Interaction in Parkinson’s Disease. Int. J. Mol. Sci. 2019, 20, 5312. [Google Scholar] [CrossRef] [Green Version]
- Bachari, S.; Naish, J.H.; Parker, G.J.M.; Emsley, H.C.A.; Parkes, L.M. Blood–Brain Barrier Leakage Is Increased in Parkinson’s Disease. Front. Physiol. 2020, 11, 593026. [Google Scholar] [CrossRef]
- McGeer, P.L.; McGeer, E.G. Inflammation and neurodegeneration in Parkinson’s disease. Parkinsonism Rel. Dis. 2004, 10 (Suppl. S1), S3–S7. [Google Scholar] [CrossRef]
- Zecca, L.; Wilms, H.; Geick, S.; Claasen, J.-H.; Brandenberg, L.-O.; Holzknecht, C.; Panizza, M.L.; Zucca, F.; Deuschl, G.; Sievers, J.; et al. Human Neuromelanin induces neuroinflammation and neurodegeneration in the rat substantia nigra: Implications for Parkinson’s disease. Acta Neuropathol. 2008, 116, 47–55. [Google Scholar] [CrossRef]
- Booth, H.D.E.; Hurst, W.D.; Wade-Martins, R. The Role of Astrocyte Dysfunction in Parkinson’s Disease Pathogenesis. Trends Neurosci. 2017, 40, 358–370. [Google Scholar] [CrossRef] [Green Version]
- Martin-Bastida, A.; Tilley, B.S.; Bansal, S.; Gentleman, S.M.; Dexter, D.T.; Ward, R.J. Iron and inflammation: In vivo and post-mortem studies in Parkinson’s disease. J. Neural. Transm. 2020, 128, 15–25. [Google Scholar] [CrossRef]
- Friedrich, I.; Reimann, K.; Jankuhn, S.; Kirilina, E.; Steiler, J.; Sonntag, M.; Meijer, J.; Weiskopf, N.; Reinert, T.; Arendt, T.; et al. Cell specific quantitative iron mapping on brain slices by immuno-µPIXE in healthy elderly and Parkinson’s disease. Acta Neuropathol. Comm. 2021, 9, 47. [Google Scholar] [CrossRef]
- Salazar, J.; Mena, N.; Hunot, S.; Prigent, A.; Alvarez-Fischer, D.; Arredondo, M.; Duyckaerts, C.; Sazdovitch, V.; Zhao, L.; Garrick, L.M.; et al. Divalent metal transporter 1 (DMT1) contributes to neurodegeneration in animal models of Parkinson’s disease. Proc. Natl. Acad. Sci. USA 2008, 105, 18578–18583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faucheux, B.A.; Martin, M.; Beaumont, C.; Hunot, S.; Hauw, J.-J.; Agid, Y.; Hirsch, E.C. Lack of up-regulation of ferritin is associated with sustained iron regulatory protein-1 binding activity in the substantia nigra of patients with Parkinson’s disease. J. Neurochem. 2002, 83, 320–330. [Google Scholar] [CrossRef] [PubMed]
- Pascoal, T.A.; Benedet, A.L.; Ashton, N.J.; Kang, M.S.; Therriault, J. Microglial activation and tau propagate jointly across Braak stages. Nat. Med. 2021, 27, 1592–1599. [Google Scholar] [CrossRef]
- Leng, F.; Edison, P. Neuroinflammation and microglial activation in Alzheimer disease: Where do we go from here? Nat. Rev. Neurol. 2021, 17, 157–172. [Google Scholar] [CrossRef]
- Tao, Y.; Wang, Y.; Rogers, J.T. Perturbed Iron Distribution in Alzheimer’s Disease Serum, Cerebrospinal Fluid, and Selected Brain Regions: A Systematic Review and Meta-Analysis. J. Alzheim. Dis. 2014, 42, 679–690. [Google Scholar] [CrossRef] [PubMed]
- Yokokura, M.; Terada, T.; Bunai, T.; Nakaizumi, K.; Takebayashi, K.; Iwate, Y.; Yoshikawa, E.; Futatsubashi, M.; Suzuki, K.; Mori, N.; et al. Depiction of microglial activation in aging and dementia: Positron emission tomography with [11C]DPA713 versus [11C](R)PK11195. J. Cereb. Blood Flow. Metab. 2017, 37, 877–889. [Google Scholar] [CrossRef] [Green Version]
- Zimmer, E.R.; Leuzy, A.; Benedet, A.L.; Breitner, J.; Gauthier, S.; Rosa-Neto, P. Tracking neuroinflammation in Alzheimer’s disease: The role of positron emission tomography imaging. J. Neuroinflammation 2014, 11, 120. [Google Scholar] [CrossRef] [Green Version]
- Akiyama, H.; Barger, S.; Barnum, S.; Bradt, B.; Bauer, J.; Cole, G.M.; Cooper, N.R.; Eikelenboom, P.; Emmerling, M.; Berndt, L.; et al. Inflammation and Alzheimer’s disease. Neurobiol. Aging 2000, 21, 383–421. [Google Scholar] [CrossRef]
- Kinney, J.W.; Bemiller, M.; Murtishaw, S.; Leisgang, M.; Salazar, A.M.; Lamb, B.T. Inflammation as a central mechanism in Alzheimer’s disease. Alzheimer Dement. Transl. Res. Clin. Intervent. 2018, 4, 575–590. [Google Scholar] [CrossRef]
- Olabarria, M.; Noristani, H.N.; Verkhrstsky, A.; Rodriguez, J.J. Age-dependent decrease in glutamine synthetase expression in the hippocampal astroglia of the triple transgenic Alzheimer’s disease mouse model: Mechanism for deficient glutamatergic transmission? Mol. Neurodegen. 2011, 6, 55. [Google Scholar] [CrossRef] [Green Version]
- Heneka, M.T.; McManus, R.M.; Latz, E. Inflammasome signalling in brain function and neurodegenerative disease. Nat. Rev. Neurosci. 2018, 19, 610–621. [Google Scholar] [CrossRef] [PubMed]
- Terada, T.; Therriault, J.; Kang, M.S.P.; Savard, M.; Pascoal, T.A.; Lussier, F.; Tissot, C.; Wang, Y.-T.; Benedet, A.; Matsdaira, T.; et al. Mitochondrial complex I abnormalities is associated with tau and clinical symptoms in mild Alzheimer’s disease. Mol. Neurodegener. 2021, 16, 28. [Google Scholar] [CrossRef] [PubMed]
- Wisniewski, H.M.; Kozlowski, P.B. Evidence for blood-brain barrier changes in senile dementia of the Alzheimer type (SDAT). Ann. N. Y. Acad. Sci. 1982, 396, 119–129. [Google Scholar] [CrossRef]
- Ayton, S.; Portbury, S.; Kalinowski, P.; Agarwal, P.; Diouf, I.; Schneider, J.A.; Morris, M.-C.; Bush, A.I. Regional brain iron associated with deterioration in Alzheimer’s disease: A large cohort study and theoretical significance. Alzh. Dementia 2021, 17, 1244–1256. [Google Scholar] [CrossRef]
- Silvestri, L.; Camashella, C. A potential pathogenetic role of iron in Alzheimer’s disease. J. Cell Mol. Med. 2008, 12, 1548–1550. [Google Scholar] [CrossRef]
- Rogers, J.T.; Randall, J.D.; Cahill, C.M.; Eder, P.S.; Huang, X.; Gunshin, H.; Leiter, L.; McPhee, J.; Sarang, S.S.; Utsuki, T.; et al. An iron-responsive element type II in the 5′-untranslated region of the Alzheimer’s amyloid precursor protein transcript. J. Biol. Chem. 2002, 277, 45518–45528. [Google Scholar] [CrossRef] [Green Version]
- Connor, J.R.; Snyder, S.; Beard, J.L.; Fine, R.E.; Mufson, E.J. Regional distribution of iron and iron-regulatory proteins in the brain in aging and Alzheimer’s disease. J. Neurosci. Res. 1992, 31, 327–335. [Google Scholar] [CrossRef]
- Hensley, K. Neuroinflammation in Alzheimer’s disease: Mechanisms, pathological consequences, and potential for therapeutic intervention. J. Alzheimers Dis. 2010, 21, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Garg, G.; Singh, S.; Singh, A.K.; Rizvi, S.I. N-acetyl-l-cysteine attenuates oxidative damage and neurodegeneration in rat brain during aging. Can. J. Physiol. Pharmacol. 2018, 96, 1189–1196. [Google Scholar] [CrossRef]
- Monti, D.A.; Zabrecky, G.; Kremens, D.; Liang, T.W.; Wintering, N.A.; Bazzan, A.J.; Zhong, L.; Bowens, B.K.; Chervoneva, I.; Imtenzo, C.; et al. N-Acetyl Cysteine Is Associated With Dopaminergic Improvement in Parkinson’s Disease. Clin. Pharmacol. Ther. 2019, 106, 884–890. [Google Scholar] [CrossRef]
- Dexter, D.T.; Statton, S.A.; Whitmore, C.; Freinbichler, W.; Weinberger, P.; Tipton, K.F.; Della Corte, L.; Ward, R.J.; Crichton, R.R. Clinically available iron chelators induce neuroprotection in the 6-OHDA model of Parkinson’s disease after peripheral administration. J. Neural. Transm. 2011, 118, 223–231. [Google Scholar] [CrossRef] [PubMed]
- Devos, D.; Cabantchik, Z.I.; Moreau, C.; Danel, V.; Mahoney-Sanchez, L.; Bouchaoui, H.; Gouel, F.; Rolland, A.-S.; Duce, J.A.; Devedjian, J.-C. Fairpark-II, Fairals-II Conservative iron chelation for neurodegenerative diseases such as Parkinson’s disease and amyotrophic lateral sclerosis. J. Neural Transm. 2020, 127, 189–203. [Google Scholar] [CrossRef] [PubMed]
- Devos, D.; Moreau, C.; Devedjian, J.C.; Fluza, J.; Petrault, M.; Laloux, C.; Jommeaux, A.; Ryckewaert, G.; Garcon, G.; Rouaix, N.; et al. Targeting chelatable iron as a therapeutic modality in Parkinson’s disease. Antioxid. Redox. Signal. 2014, 21, 195–210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fairpark II Conservative Iron Chelation as a Disease-Modifying Strategy in Parkinson’s Disease: A Multicentric, Parallel-Group, Placebo-Controlled, Randomized Clinical Trial of Deferiprone. Periodic Reporting for Period 5. European Commission. Available online: https://cordis.europa.eu/project/id/633190/reporting (accessed on 20 April 2022).




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ward, R.J.; Dexter, D.T.; Crichton, R.R. Iron, Neuroinflammation and Neurodegeneration. Int. J. Mol. Sci. 2022, 23, 7267. https://doi.org/10.3390/ijms23137267
Ward RJ, Dexter DT, Crichton RR. Iron, Neuroinflammation and Neurodegeneration. International Journal of Molecular Sciences. 2022; 23(13):7267. https://doi.org/10.3390/ijms23137267
Chicago/Turabian StyleWard, Roberta J., David T. Dexter, and Robert R. Crichton. 2022. "Iron, Neuroinflammation and Neurodegeneration" International Journal of Molecular Sciences 23, no. 13: 7267. https://doi.org/10.3390/ijms23137267
APA StyleWard, R. J., Dexter, D. T., & Crichton, R. R. (2022). Iron, Neuroinflammation and Neurodegeneration. International Journal of Molecular Sciences, 23(13), 7267. https://doi.org/10.3390/ijms23137267