
Genome Assembly and Analysis of the Flavonoid and Phenylpropanoid Biosynthetic Pathways in Fingerroot Ginger (Boesenbergia rotunda)

 , ,
, ,  , and
, and
Abstract
:1. Introduction
2. Results
2.1. Chromosomes and Location of rDNA Sites
2.2. Genome Assembly
2.3. Annotation of the B. rotunda Genome
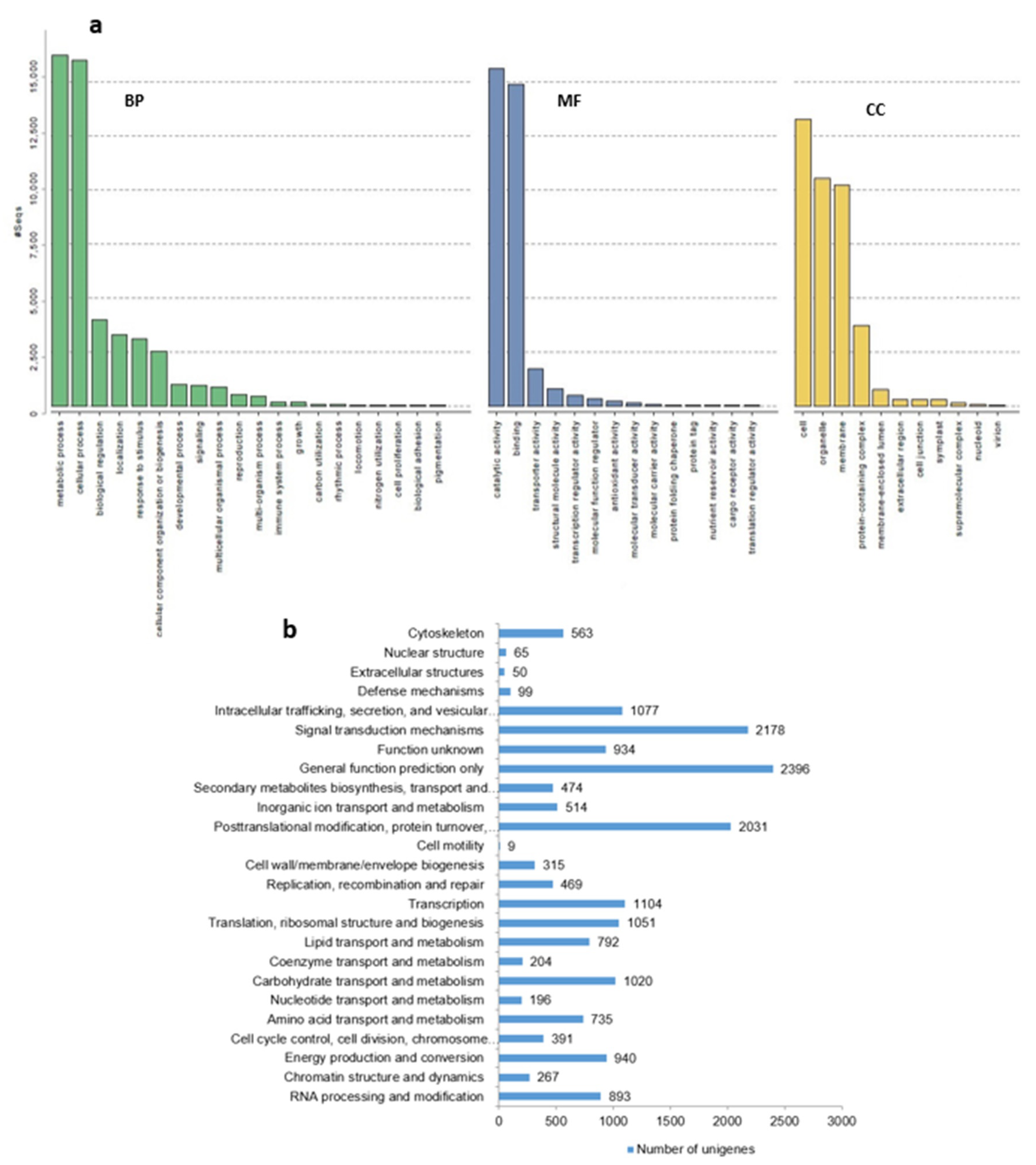
2.4. Functional Classification by Gene Ontology
2.5. Phylogenetic Orthology Inference of B. rotunda Genes
Gene Family Expansion and Contraction
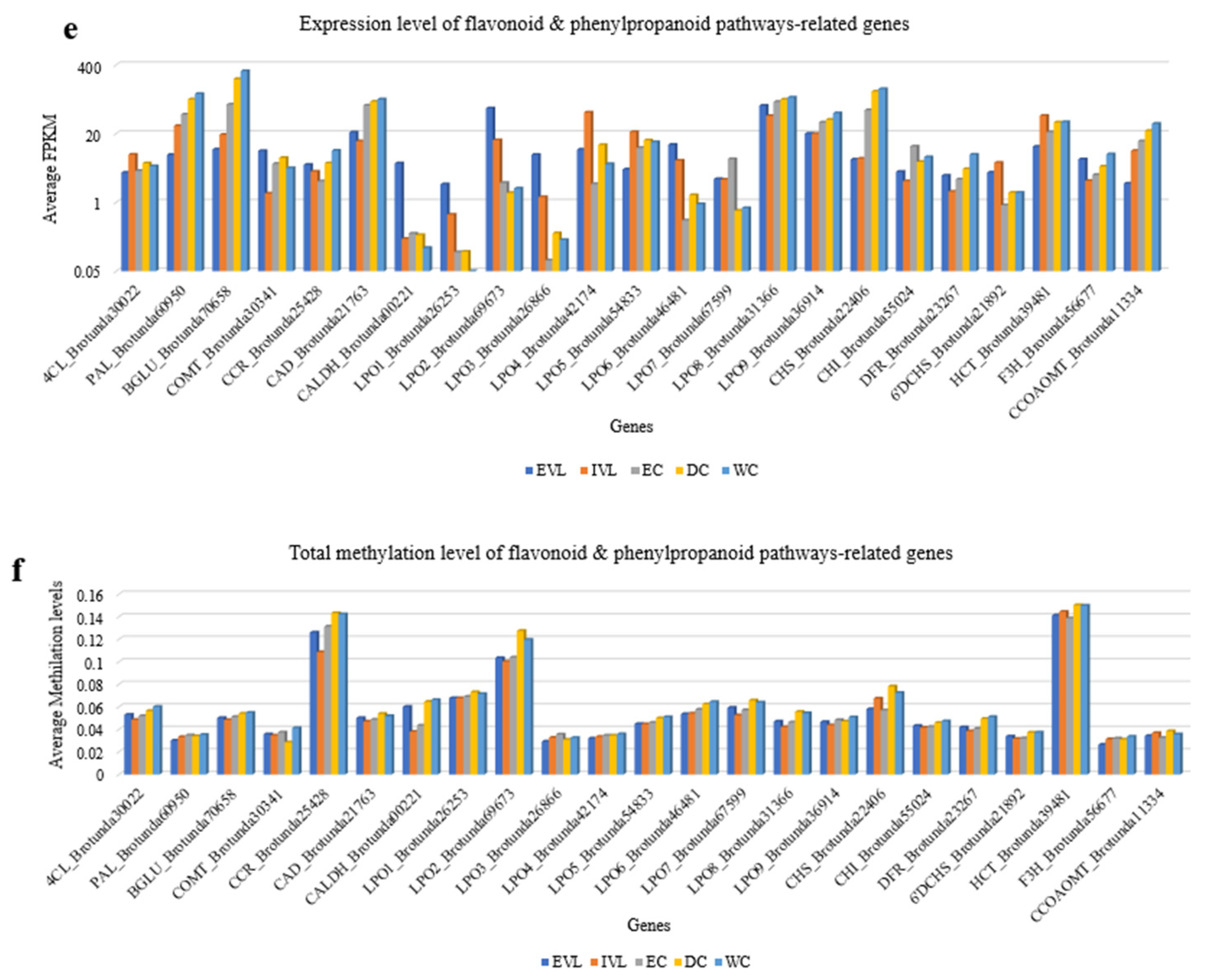
2.6. Transcriptome Changes of B. rotunda Unigenes Related to Flavonoid and Phenylpropanoid Biosynthesis Pathways
2.7. DNA Methylation Analysis Using Bisulfite Sequencing
2.8. Correlation between Gene Expression Levels and DNA Methylation Levels of Genes Related to Methylation, Somatic Embryogenesis and Secondary Metabolite Pathway
3. Discussion
4. Materials and Methods
4.1. Ethics
4.2. Plant Materials and Samples Preparation
4.3. DNA Extraction and Sequencing for Genome and Bisulfite Sequence (BS-Seq) Analysis
4.4. RNA Extraction and Sequencing for Transcriptome (RNA-Seq) Analysis
4.5. Determination of Chromosome Number and Location of 45S and 5S rDNA Sites on Metaphase Chromosomes of B. rotunda (2n = 36) Using Fluorescent In Situ Hybridization (FISH)
4.6. Genome Size Estimation
4.7. Genome Assembly
4.8. Repeat Annotation
4.9. Gene Annotation
4.10. Non-Coding RNAs Annotation
4.11. Construction of Phylogenetic Trees
4.12. Gene Family Expansion and Contraction Analysis
4.13. DNA Methylation Analysis Using Bisulfite Sequencing (BS-Seq)
4.14. De Novo Transcriptome Assembly of B. rotunda and Functional Annotation
4.15. Estimation of Transcript Abundance and Differential Expression
4.16. Mining of Simple Sequence Repeats (SSRs) from B. rotunda Transcriptome and Genome Assembly
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Benedict, J.C.; Smith, S.Y.; Specht, C.D.; Collinson, M.E.; Leong-Škorničková, J.; Parkinson, D.Y.; Marone, F. Species diversity driven by morphological and ecological disparity: A case study of comparative seed morphology and anatomy across a large monocot order. AoB Plants 2016, 8, plw063. [Google Scholar] [CrossRef] [PubMed]
- Christenhusz, M.J.; Byng, J.W. The number of known plants species in the world and its annual increase. Phytotaxa 2016, 261, 201–217. [Google Scholar] [CrossRef] [Green Version]
- Baker, J.G. The Flora of British India. In Scitamineae; Hooker, J.D., Ed.; Reeve & Co.: London, UK, 1890; Volume 6. [Google Scholar]
- Burkill, I.H. A dictionary of the economic products of the Malay Peninsula. In A Dictionary of the Economic Products of the Malay Peninsula; Ministry of Agriculture and Co-Operatives: Kuala Lumpur, Malaysia, 1966; Volume 2. [Google Scholar]
- Larsen, K. A preliminary checklist of the Zingiberaceae of Thailand. Thai For. Bull. 1996, 24, 35–49. [Google Scholar]
- Larsen, K.; Ibrahim, H.; Khaw, S.; Saw, L. Gingers of Peninsular Malaysia and Singapore; Natural History Borneo: Kota Kinabalu, Malaysia, 1999. [Google Scholar]
- Rachkeeree, A.; Kantadoung, K.; Suksathan, R.; Puangpradab, R.; Page, P.A.; Sommano, S.R. Nutritional compositions and phytochemical properties of the edible flowers from selected Zingiberaceae found in Thailand. Front. Nutr. 2018, 5, 3. [Google Scholar] [CrossRef] [Green Version]
- Jing, L.J.; Mohamed, M.; Rahmat, A.; Bakar, M.F.A. Phytochemicals, antioxidant properties and anticancer investigations of the different parts of several gingers species (Boesenbergia rotunda, Boesenbergia pulchella var attenuata and Boesenbergia armeniaca). J. Med. Plant Res. 2010, 4, 027–032. [Google Scholar]
- Larsen, K.; Lock, J.; Maas, H.; Maas, P. Zingiberaceae. In Flowering Plants Monocotyledons; Springer: Berlin/Heidelberg, Germany, 1998; pp. 474–495. [Google Scholar]
- Mood, J.; Veldkamp, J.; Dey, S.; Prince, L. Nomenclatural changes in Zingiberaceae: Caulokaempferia is a superfluous name for Monolophus and Jirawongsea is reduced to Boesenbergia. Bull. Singap. 2014, 66, 215–231. [Google Scholar]
- Eng-Chong, T.; Yean-Kee, L.; Chin-Fei, C.; Choon-Han, H.; Sher-Ming, W.; Li-Ping, C.T.; Gen-Teck, F.; Khalid, N.; Abd Rahman, N.; Karsani, S.A. Boesenbergia rotunda: From ethnomedicine to drug discovery. Evid. Based Complement. Altern. Med. 2012, 2012, 25. [Google Scholar] [CrossRef] [Green Version]
- Jirakiattikul, Y.; Rithichai, P.; Prachai, R.; Itharat, A. Elicitation enhancement of bioactive compound accumulation and antioxidant activity in shoot cultures of Boesenbergia rotunda L. Agric. Nat. Resour 2021, 55, 456–463. [Google Scholar]
- Shen, N.; Wang, T.; Gan, Q.; Liu, S.; Wang, L.; Biao, J. Plant flavonoids: Classification, distribution, biosynthesis, and antioxidant activity. Food Chem. 2022, 383, 132531. [Google Scholar] [CrossRef]
- Trakoontivakorn, G.; Nakahara, K.; Shinmoto, H.; Takenaka, M.; Onishi-Kameyama, M.; Ono, H.; Yoshida, M.; Nagata, T.; Tsushida, T. Structural analysis of a novel antimutagenic compound, 4-hydroxypanduratin A, and the antimutagenic activity of flavonoids in a Thai spice, fingerroot (Boesenbergia pandurata Schult.) against mutagenic heterocyclic amines. J. Agric. Food Chem. 2001, 49, 3046–3050. [Google Scholar] [CrossRef]
- Jaipetch, T.; Kanghae, S.; Pancharoen, O.; Patrick, V.; Reutrakul, V.; Tuntiwachwuttikul, P.; White, A. Constituents of Boesenbergia pandurata (syn. Kaempferia pandurata): Isolation, crystal structure and synthesis of (±)-Boesenbergin A. Aust. J. Chem. 1982, 35, 351–361. [Google Scholar]
- Tan, S.K. Flavanoids from Boesenbergia rotunda (L.) Mansf: Chemistry, Bioactivity and Accumulation; Universiti Malaya: Kuala Lumpur, Malaysia, 2005. [Google Scholar]
- Tan, B.C.; Tan, S.K.; Wong, S.M.; Ata, N.; Rahman, N.A.; Khalid, N. Distribution of flavonoids and cyclohexenyl chalcone derivatives in conventional propagated and in vitro-derived field-grown Boesenbergia rotunda (L.) Mansf. Evid. Based Complement. Altern. Med. 2015, 2015, 451870. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yusuf, N.A.; M Annuar, M.S.; Khalid, N. Existence of bioactive flavonoids in rhizomes and plant cell cultures of ‘Boesenbergia rotund’(L.) Mansf. Kulturpfl. Aust. J. Crop Sci. 2013, 7, 730. [Google Scholar]
- Rosdianto, A.M.; Puspitasari, I.M.; Lesmana, R.; Levita, J. Determination of Quercetin and Flavonol Synthase in Boesenbergia rotunda Rhizome. Pak. J. Biol. Sci. 2020, 23, 264–270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ching, A.Y.L.; Wah, T.S.; Sukari, M.A.; Cheng Lian, G.E.; Rahmani, M.; Khalid, K. Characterization of flavonoid derivatives from Boesenbergia rotunda (L.). Malays. J. Anal. Sci. 2007, 11, 154–159. [Google Scholar]
- Tuchinda, P.; Reutrakul, V.; Claeson, P.; Pongprayoon, U.; Sematong, T.; Santisuk, T.; Taylor, W.C. Anti-inflammatory cyclohexenyl chalcone derivatives in Boesenbergia pandurata. Phytochemistry 2002, 59, 169–173. [Google Scholar] [CrossRef]
- Rosdianto, A.M.; Puspitasari, I.M.; Lesmana, R.; Levita, J.J.J. Bioactive compounds of Boesenbergia sp. and their anti-inflammatory mechanism: A review. J. Appl. Pharm. Sci. 2020, 10, 116–126. [Google Scholar]
- Break, M.K.B.; Chiang, M.; Wiart, C.; Chin, C.-F.; Khoo, A.S.B.; Khoo, T.-J. Cytotoxic Activity of Boesenbergia rotunda Extracts against Nasopharyngeal Carcinoma Cells (HK1). Cardamonin, a Boesenbergia rotunda Constituent, Inhibits Growth and Migration of HK1 Cells by Inducing Caspase-Dependent Apoptosis and G2/M–Phase Arrest. Nutr. Cancer 2021, 73, 473–483. [Google Scholar] [CrossRef]
- Kirana, C.; Jones, G.P.; Record, I.R.; McIntosh, G.H. Anticancer properties of panduratin A isolated from Boesenbergia pandurata (Zingiberaceae). J. Nat. Med. 2007, 61, 131–137. [Google Scholar] [CrossRef]
- Liu, Q.; Cao, Y.; Zhou, P.; Gui, S.; Wu, X.; Xia, Y.; Tu, J. Panduratin A inhibits cell proliferation by inducing G0/G1 phase cell cycle arrest and induces apoptosis in breast cancer cells. Biomol. Ther. 2018, 26, 328. [Google Scholar] [CrossRef]
- Tanigaki, R.; Takahashi, R.; Nguyen, M.T.T.; Nguyen, N.T.; Do, T.V.N.; Nguyen, H.X.; Kataoka, T. 4-Hydroxypanduratin A and isopanduratin A inhibit tumor necrosis factor α-stimulated gene expression and the nuclear factor κB-dependent signaling pathway in human lung adenocarcinoma A549 cells. Biol. Pharm. Bull. 2019, 42, 26–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Win, N.N.; Awale, S.; Esumi, H.; Tezuka, Y.; Kadota, S. Panduratins D—I, novel secondary metabolites from rhizomes of Boesenbergia pandurata. Chem. Pharm. Bull. 2008, 56, 491–496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheenpracha, S.; Karalai, C.; Ponglimanont, C.; Subhadhirasakul, S.; Tewtrakul, S.J.B. Anti-HIV-1 protease activity of compounds from Boesenbergia pandurata. Bioorg. Med. Chem. 2006, 14, 1710–1714. [Google Scholar] [CrossRef] [PubMed]
- Chee, C.F.; Abdullah, I.; Buckle, M.J.; Rahman, N.A. An efficient synthesis of (±)-panduratin A and (±)-isopanduratin A, inhibitors of dengue-2 viral activity. Tetrahedron Lett. 2010, 51, 495–498. [Google Scholar] [CrossRef]
- Kiat, T.S.; Pippen, R.; Yusof, R.; Ibrahim, H.; Khalid, N.; Rahman, N.A. Inhibitory activity of cyclohexenyl chalcone derivatives and flavonoids of fingerroot, Boesenbergia rotunda (L.), towards dengue-2 virus NS3 protease. Bioorg. Med. Chem. Lett. 2006, 16, 3337–3340. [Google Scholar] [CrossRef]
- Kanjanasirirat, P.; Suksatu, A.; Manopwisedjaroen, S.; Munyoo, B.; Tuchinda, P.; Jearawuttanakul, K.; Seemakhan, S.; Charoensutthivarakul, S.; Wongtrakoongate, P.; Rangkasenee, N. High-content screening of Thai medicinal plants reveals Boesenbergia rotunda extract and its component Panduratin A as anti-SARS-CoV-2 agents. Sci. Rep. 2020, 10, 19963. [Google Scholar] [CrossRef]
- Hwang, J.-K.; Chung, J.-Y.; Baek, N.-I.; Park, J.-H. Isopanduratin A from Kaempferia pandurata as an active antibacterial agent against cariogenic Streptococcus mutans. Int. J. Antimicrob. Agents 2004, 23, 377–381. [Google Scholar] [CrossRef]
- Mazlan, R.R.; Zakaria, M.; Rukayadi, Y. Antimicrobial activity of fingerroot [Boesenbergia rotunda (L.) Mansf. A.] Extract against Streptococcus mutans and Streptococcus sobrinus. J. Pure. Appl. Microbiol. 2016, 10, 1755–1762. [Google Scholar]
- Bhamarapravati, S.; Juthapruth, S.; Mahachai, W.; Mahady, G. Antibacterial activity of Boesenbergia rotunda (L.) Mansf. and Myristica fragrans Houtt. against Helicobacter pylori. Songklanakarin J. Sci. Technol. 2006, 28, 157–163. [Google Scholar]
- Pattaratanawadee, E.; Rachtanapun, C.; Wanchaitanawong, P.; Mahakarnchanakul, W. Antimicrobial activity of spice extracts against pathogenic and spoilage microorganisms. Kasetsart J. Nat. Sci. 2006, 40, 159–165. [Google Scholar]
- Hwang, J.K.; Kim, S.Y.; Kim, M.B. Composition Comprising Panduratin or Fingerroot (Boesenbergia pandurata) Extract for Treating, Preventing, or Ameliorating Bone Loss Disease. Patent Application No. WO2017150934A1, 3 March 2017. [Google Scholar]
- Adhikari, D.; Gong, D.-S.; Oh, S.H.; Sung, E.H.; Lee, S.O.; Kim, D.-W.; Oak, M.-H.; Kim, H.J.J.P. Vasorelaxant Effect of Boesenbergia rotunda and Its Active Ingredients on an Isolated Coronary Artery. Plants 2020, 9, 1688. [Google Scholar] [CrossRef] [PubMed]
- Tan, S.; Pippen, R.; Yusof, R.; Ibrahim, H.; Rahman, N.; Khalid, N. Simple one-medium formulation regeneration of fingerroot [Boesenbergia rotunda (L.) Mansf. Kulturpfl.] via somatic embryogenesis. Vitr. Cell. Dev. Biol. Plant 2005, 41, 757–761. [Google Scholar] [CrossRef]
- Wong, S.M.; Salim, N.; Harikrishna, J.A.; Khalid, N. Highly efficient plant regeneration via somatic embryogenesis from cell suspension cultures of Boesenbergia rotunda. Vitr. Cell. Dev. Biol.-Plant 2013, 49, 665–673. [Google Scholar] [CrossRef]
- Yusuf, N.A.; Annuar, M.S.; Khalid, N. Rapid micropropagation of Boesenbergia rotunda (L.) Mansf. Kulturpfl. (a valuable medicinal plant) from shoot bud explants. Afr. J. Biotechnol. 2011, 10, 1194–1199. [Google Scholar]
- Md-Mustafa, N.D.; Khalid, N.; Gao, H.; Peng, Z.; Alimin, M.F.; Bujang, N.; Ming, W.S.; Mohd-Yusuf, Y.; Harikrishna, J.A.; Othman, R.Y. Transcriptome profiling shows gene regulation patterns in a flavonoid pathway in response to exogenous phenylalanine in Boesenbergia rotunda cell culture. BMC Genom. 2014, 15, 984. [Google Scholar] [CrossRef] [Green Version]
- Wu, M.; Li, Q.; Hu, Z.; Li, X.; Chen, S. The complete Amomum kravanh chloroplast genome sequence and phylogenetic analysis of the commelinids. Molecules 2017, 22, 1875. [Google Scholar] [CrossRef] [Green Version]
- Ng, T.L.M.; Karim, R.; Tan, Y.S.; Teh, H.F.; Danial, A.D.; Ho, L.S.; Khalid, N.; Appleton, D.R.; Harikrishna, J.A. Amino acid and secondary metabolite production in embryogenic and non-embryogenic callus of Fingerroot ginger (Boesenbergia rotunda). PLoS ONE 2016, 11, e0156714. [Google Scholar] [CrossRef] [Green Version]
- Chen, D.-x.; Pan, Y.; Wang, Y.; Cui, Y.-Z.; Zhang, Y.-J.; Mo, R.-y.; Wu, X.-l.; Tan, J.; Zhang, J.; Guo, L.-a. The chromosome-level reference genome of Coptis chinensis provides insights into genomic evolution and berberine biosynthesis. Hort. Res. 2021, 8, 121. [Google Scholar] [CrossRef]
- Xia, Z.; Huang, D.; Zhang, S.; Wang, W.; Ma, F.; Wu, B.; Xu, Y.; Xu, B.; Chen, D.; Zou, M. Chromosome-scale genome assembly provides insights into the evolution and flavor synthesis of passion fruit (Passiflora edulis Sims). Hort. Res. 2021, 8, 14. [Google Scholar] [CrossRef]
- Xu, X.; Yuan, H.; Yu, X.; Huang, S.; Sun, Y.; Zhang, T.; Liu, Q.; Tong, H.; Zhang, Y.; Wang, Y. The chromosome-level Stevia genome provides insights into steviol glycoside biosynthesis. Hort. Res. 2021, 8, 189. [Google Scholar] [CrossRef]
- Chakraborty, A.; Mahajan, S.; Jaiswal, S.K.; Sharma, V.K. Genome sequencing of turmeric provides evolutionary insights into its medicinal properties. Commun. Biol. 2021, 4, 1193. [Google Scholar] [CrossRef] [PubMed]
- Droc, G.; Lariviere, D.; Guignon, V.; Yahiaoui, N.; This, D.; Garsmeur, O.; Dereeper, A.; Hamelin, C.; Argout, X.; Dufayard, J.F. The banana genome hub. Database 2013, 2013, bat035. [Google Scholar] [CrossRef] [PubMed]
- Rijzaani, H.; Bayer, P.E.; Rouard, M.; Doležel, J.; Batley, J.; Edwards, D. The pangenome of banana highlights differences between genera and genomes. Plant Genome 2021, 15, e20100. [Google Scholar] [CrossRef] [PubMed]
- Luo, M.-C.; Gu, Y.Q.; Puiu, D.; Wang, H.; Twardziok, S.O.; Deal, K.R.; Huo, N.; Zhu, T.; Wang, L.; Wang, Y. Genome sequence of the progenitor of the wheat D genome Aegilops tauschii. Nature 2017, 551, 498–502. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Ong-Abdullah, M.; Low, E.-T.L.; Manaf, M.A.A.; Rosli, R.; Nookiah, R.; Ooi, L.C.-L.; Ooi, S.E.; Chan, K.-L.; Halim, M.A. Oil palm genome sequence reveals divergence of interfertile species in Old and New worlds. Nature 2013, 500, 335–339. [Google Scholar] [CrossRef] [Green Version]
- Schnable, P.S.; Ware, D.; Fulton, R.S.; Stein, J.C.; Wei, F.; Pasternak, S.; Liang, C.; Zhang, J.; Fulton, L.; Graves, T.A. The B73 maize genome: Complexity, diversity, and dynamics. Science 2009, 326, 1112–1115. [Google Scholar] [CrossRef] [Green Version]
- Wei, C.; Yang, H.; Wang, S.; Zhao, J.; Liu, C.; Gao, L.; Xia, E.; Lu, Y.; Tai, Y.; She, G. Draft genome sequence of Camellia sinensis var. sinensis provides insights into the evolution of the tea genome and tea quality. Proc. Natl. Acad. Sci. USA 2018, 115, E4151–E4158. [Google Scholar] [CrossRef] [Green Version]
- Xia, E.-H.; Tong, W.; Wu, Q.; Wei, S.; Zhao, J.; Zhang, Z.-Z.; Wei, C.-L.; Wan, X.-C. Tea plant genomics: Achievements, challenges and perspectives. Hort. Res. 2020, 7, 7. [Google Scholar] [CrossRef] [Green Version]
- Jayakodi, M.; Choi, B.-S.; Lee, S.-C.; Kim, N.-H.; Park, J.Y.; Jang, W.; Lakshmanan, M.; Mohan, S.V.; Lee, D.-Y.; Yang, T.-J. Ginseng genome database: An open-access platform for genomics of Panax ginseng. BMC Plant Biol. 2018, 18, 62. [Google Scholar] [CrossRef]
- Michael, T.P.; VanBuren, R. Building near-complete plant genomes. Curr. Opin. Plant Biol. 2020, 54, 26–33. [Google Scholar] [CrossRef]
- Wang, Z.; Rouard, M.; Biswas, M.K.; Droc, G.; Cui, D.; Roux, N.; Baurens, F.-C.; Ge, X.-J.; Schwarzacher, T.; Heslop-Harrison, P.J. A chromosome-level reference genome of Ensete glaucum gives insight into diversity and chromosomal and repetitive sequence evolution in the Musaceae. GigaScience 2022, 11, giac027. [Google Scholar] [CrossRef] [PubMed]
- Tan, E.C.; Karsani, S.A.; Foo, G.T.; Wong, S.M.; Rahman, N.A.; Khalid, N.; Othman, S.; Yusof, R. Proteomic analysis of cell suspension cultures of Boesenbergia rotunda induced by phenylalanine: Identification of proteins involved in flavonoid and phenylpropanoid biosynthesis pathways. Plant Cell Tissue Organ Cult. (PCTOC) 2012, 111, 219–229. [Google Scholar] [CrossRef]
- Bennetzen, J.L.; Wang, H. The contributions of transposable elements to the structure, function, and evolution of plant genomes. Annu. Rev. Plant Biol. 2014, 65, 505–530. [Google Scholar] [CrossRef]
- Liu, N.; Niu, Y.; Zhang, G.; Feng, Z.; Bo, Y.; Lian, J.; Wang, B.; Gong, Y. Genome sequencing and population resequencing provide insights into the genetic basis of domestication and diversity of vegetable soybean. Hort. Res. 2022, 9, uhab052. [Google Scholar] [CrossRef] [PubMed]
- Simão, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Emms, D.M.; Kelly, S. OrthoFinder: Phylogenetic orthology inference for comparative genomics. Genome Biol. 2019, 20, 238. [Google Scholar] [CrossRef] [Green Version]
- Emms, D.; Kelly, S. STAG: Species tree inference from all genes. BioRxiv 2018, 267914. [Google Scholar] [CrossRef]
- Emms, D.M.; Kelly, S. STRIDE: Species tree root inference from gene duplication events. Mol. Biol. Evol. 2017, 34, 3267–3278. [Google Scholar] [CrossRef]
- Guardo, M.D.; Moretto, M.; Moser, M.; Catalano, C.; Troggio, M.; Deng, Z.; Cestaro, A.; Caruso, M.; Distefano, G.; Malfa, S.L. The haplotype-resolved reference genome of lemon (Citrus limon L. Burm f.). Tree Genet. Genom. 2021, 17, 46. [Google Scholar] [CrossRef]
- Baek, S.; Choi, K.; Kim, G.-B.; Yu, H.-J.; Cho, A.; Jang, H.; Kim, C.; Kim, H.-J.; Chang, K.S.; Kim, J.-H. Draft genome sequence of wild Prunus yedoensis reveals massive inter-specific hybridization between sympatric flowering cherries. Genome Biol. 2018, 19, 127. [Google Scholar] [CrossRef]
- Chin, C.-S.; Peluso, P.; Sedlazeck, F.J.; Nattestad, M.; Concepcion, G.T.; Clum, A.; Dunn, C.; O’Malley, R.; Figueroa-Balderas, R.; Morales-Cruz, A. Phased diploid genome assembly with single-molecule real-time sequencing. Nat. Methods 2016, 13, 1050–1054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chia, Y.C.; Teh, S.-H.; Mohamed, Z. Isolation and characterization of Chalcone Isomerase (CHI) Gene from Boesenbergia rotunda. S. Afr. J. Bot. 2020, 130, 475–482. [Google Scholar] [CrossRef]
- Liew, Y.J.M.; Lee, Y.K.; Khalid, N.; Abd Rahman, N.; Tan, B.C. Enhancing flavonoid production by promiscuous activity of prenyltransferase, BrPT2 from Boesenbergia rotunda. PeerJ 2020, 8, e9094. [Google Scholar] [CrossRef]
- Wu, W.; Yang, Y.-L.; He, W.-M.; Rouard, M.; Li, W.-M.; Xu, M.; Roux, N.; Ge, X.-J. Whole genome sequencing of a banana wild relative Musa itinerans provides insights into lineage-specific diversification of the Musa genus. Sci. Rep. 2016, 6, 31586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’hont, A.; Denoeud, F.; Aury, J.-M.; Baurens, F.-C.; Carreel, F.; Garsmeur, O.; Noel, B.; Bocs, S.; Droc, G.; Rouard, M. The banana (Musa acuminata) genome and the evolution of monocotyledonous plants. Nature 2012, 488, 213–217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, S.-P.; Jia, K.H.; Liu, H.; Zhang, R.G.; Li, Z.C.; Zhou, S.S.; Shi, T.L.; Ma, A.C.; Yu, C.W.; Gao, C. Haplotype-resolved genome assembly and allele-specific gene expression in cultivated ginger. Hort. Res. 2021, 8, 188. [Google Scholar] [CrossRef]
- Macas, J.; Novak, P.; Pellicer, J.; Čížková, J.; Koblížková, A.; Neumann, P.; Fukova, I.; Doležel, J.; Kelly, L.J.; Leitch, I.J. In depth characterization of repetitive DNA in 23 plant genomes reveals sources of genome size variation in the legume tribe Fabeae. PLoS ONE 2015, 10, e0143424. [Google Scholar] [CrossRef]
- McCann, J.; Macas, J.; Novák, P.; Stuessy, T.F.; Villaseñor, J.L.; Weiss-Schneeweiss, H. Differential genome size and repetitive DNA evolution in diploid species of Melampodium sect. Melampodium (Asteraceae). Front. Plant Sci. 2020, 11, 362. [Google Scholar] [CrossRef]
- Li, H.-L.; Wu, L.; Dong, Z.; Jiang, Y.; Jiang, S.; Xing, H.; Li, Q.; Liu, G.; Tian, S.; Wu, Z. Haplotype-resolved genome of diploid ginger (Zingiber officinale) and its unique gingerol biosynthetic pathway. Hort. Res. 2021, 8, 189. [Google Scholar] [CrossRef]
- Bennetzen, J.L. The structure and evolution of angiosperm nuclear genomes. Curr. Opin. Plant Biol. 1998, 1, 103–108. [Google Scholar] [CrossRef]
- Alonso, C.; Pérez, R.; Bazaga, P.; Herrera, C.M. Global DNA cytosine methylation as an evolving trait: Phylogenetic signal and correlated evolution with genome size in angiosperms. Front. Genet. 2015, 6, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fedoroff, N.V. Transposable elements, epigenetics, and genome evolution. Science 2012, 338, 758–767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rabinowicz, P.D.; Citek, R.; Budiman, M.A.; Nunberg, A.; Bedell, J.A.; Lakey, N.; O’Shaughnessy, A.L.; Nascimento, L.U.; McCombie, W.R.; Martienssen, R.A. Differential methylation of genes and repeats in land plants. Genome Res. 2005, 15, 1431–1440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rabinowicz, P.D.; Palmer, L.E.; May, B.P.; Hemann, M.T.; Lowe, S.W.; McCombie, W.R.; Martienssen, R.A. Genes and transposons are differentially methylated in plants, but not in mammals. Genome Res. 2003, 13, 2658–2664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaughn, M.W.; Tanurdžić, M.; Lippman, Z.; Jiang, H.; Carrasquillo, R.; Rabinowicz, P.D.; Dedhia, N.; McCombie, W.R.; Agier, N.; Bulski, A. Epigenetic natural variation in Arabidopsis thaliana. PLoS Biol. 2007, 5, e174. [Google Scholar] [CrossRef]
- Crisp, P.A.; Marand, A.P.; Noshay, J.M.; Zhou, P.; Lu, Z.; Schmitz, R.J.; Springer, N.M. Stable unmethylated DNA demarcates expressed genes and their cis-regulatory space in plant genomes. Proc. Natl. Acad. Sci. USA 2020, 117, 23991–24000. [Google Scholar] [CrossRef]
- Aya, K.; Kobayashi, M.; Tanaka, J.; Ohyanagi, H.; Suzuki, T.; Yano, K.; Takano, T.; Yano, K.; Matsuoka, M.J.P.; Physiology, C. De novo transcriptome assembly of a fern, Lygodium japonicum, and a web resource database, Ljtrans DB. Plant Cell Physiol. 2015, 56, e5. [Google Scholar] [CrossRef] [Green Version]
- Karim, R.; Tan, Y.S.; Singh, P.; Khalid, N.; Harikrishna, J.A. Expression and DNA methylation of SERK, BBM, LEC2 and WUS genes in in vitro cultures of Boesenbergia rotunda (L.) Mansf. Physiol. Mol. Biol. Plants 2018, 24, 741–751. [Google Scholar] [CrossRef]
- Karim, R.; Tan, Y.S.; Singh, P.; Nuruzzaman, M.; Khalid, N.; Harikrishna, J.A. Expression and DNA methylation of MET1, CMT3 and DRM2 during in vitro culture of Boesenbergia rotunda (L.) Mansf. Philipp. Agric. Sci. 2018, 101, 261–270. [Google Scholar]
- Biswas, M.K.; Bagchi, M.; Nath, U.K.; Biswas, D.; Natarajan, S.; Jesse, D.M.I.; Park, J.-I.; Nou, I.-S. Transcriptome wide SSR discovery cross-taxa transferability and development of marker database for studying genetic diversity population structure of Lilium species. Sci. Rep. 2020, 10, 18621. [Google Scholar] [CrossRef]
- Biswas, M.K.; Darbar, J.N.; Borrell, J.S.; Bagchi, M.; Biswas, D.; Nuraga, G.W.; Demissew, S.; Wilkin, P.; Schwarzacher, T.; Heslop-Harrison, J. The landscape of microsatellites in the enset (Ensete ventricosum) genome and web-based marker resource development. Sci. Rep. 2020, 10, 15312. [Google Scholar] [CrossRef] [PubMed]
- Devi, K.D.; Punyarani, K.; Singh, N.S.; Devi, H.S. An efficient protocol for total DNA extraction from the members of order Zingiberales-suitable for diverse PCR based downstream applications. SpringerPlus 2013, 2, 669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, M.; Chung, C.Y.-L.; Li, M.-W.; Wong, F.-L.; Wang, X.; Liu, A.; Wang, Z.; Leung, A.K.-Y.; Wong, T.-H.; Tong, S.-W. A reference-grade wild soybean genome. Nat. Commun. 2019, 10, 1216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kiefer, E.; Heller, W.; Ernst, D. A simple and efficient protocol for isolation of functional RNA from plant tissues rich in secondary metabolites. Plant Mol. Biol. Rep. 2000, 18, 33–39. [Google Scholar] [CrossRef]
- Schwarzacher, T.; Heslop-Harrison, P. Practical In Situ Hybridization; BIOS Scientific Publishers Ltd.: Oxford, UK, 2000; p. 216. [Google Scholar]
- Gerlach, W.; Bedbrook, J. Cloning and characterization of ribosomal RNA genes from wheat and barley. Nucleic Acids Res. 1979, 7, 1869–1885. [Google Scholar] [CrossRef]
- Yan, L.; Wang, X.; Liu, H.; Tian, Y.; Lian, J.; Yang, R.; Hao, S.; Wang, X.; Yang, S.; Li, Q. The genome of Dendrobium officinale illuminates the biology of the important traditional Chinese orchid herb. Mol. Plant 2015, 8, 922–934. [Google Scholar] [CrossRef] [Green Version]
- Lin, E.; Zhuang, H.; Yu, J.; Liu, X.; Huang, H.; Zhu, M.; Tong, Z. Genome survey of Chinese fir (Cunninghamia lanceolata): Identification of genomic SSRs and demonstration of their utility in genetic diversity analysis. Sci. Rep. 2020, 10, 4698. [Google Scholar] [CrossRef]
- Marçais, G.; Kingsford, C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 2011, 27, 764–770. [Google Scholar] [CrossRef] [Green Version]
- Ranallo-Benavidez, T.R.; Jaron, K.S.; Schatz, M.C. GenomeScope 2.0 and Smudgeplot for reference-free profiling of polyploid genomes. Nat. Commun. 2020, 11, 1432. [Google Scholar] [CrossRef] [Green Version]
- Pendleton, M.; Sebra, R.; Pang, A.W.C.; Ummat, A.; Franzen, O.; Rausch, T.; Stütz, A.M.; Stedman, W.; Anantharaman, T.; Hastie, A. Assembly and diploid architecture of an individual human genome via single-molecule technologies. Nat. Methods 2015, 12, 780. [Google Scholar] [CrossRef]
- Chin, C.-S.; Alexander, D.H.; Marks, P.; Klammer, A.A.; Drake, J.; Heiner, C.; Clum, A.; Copeland, A.; Huddleston, J.; Eichler, E.E. Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data. Nat. Methods 2013, 10, 563. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Wu, S.; Li, A.; Ruan, J. SMARTdenovo: A de novo assembler using long noisy reads. Gigabyte 2021, 2021, 1–9. [Google Scholar] [CrossRef]
- Walker, B.J.; Abeel, T.; Shea, T.; Priest, M.; Abouelliel, A.; Sakthikumar, S.; Cuomo, C.A.; Zeng, Q.; Wortman, J.; Young, S.K. Pilon: An integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS ONE 2014, 9, e112963. [Google Scholar] [CrossRef]
- Boetzer, M.; Henkel, C.V.; Jansen, H.J.; Butler, D.; Pirovano, W. Scaffolding pre-assembled contigs using SSPACE. Bioinformatics 2011, 27, 578–579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- English, A.C.; Richards, S.; Han, Y.; Wang, M.; Vee, V.; Qu, J.; Qin, X.; Muzny, D.M.; Reid, J.G.; Worley, K.C. Mind the gap: Upgrading genomes with Pacific Biosciences RS long-read sequencing technology. PLoS ONE 2012, 7, e47768. [Google Scholar]
- Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv 2013, arXiv:1303.3997. [Google Scholar]
- Benson, G. Tandem repeats finder: A program to analyze DNA sequences. Nucleic Acids Res. 1999, 27, 573–580. [Google Scholar] [CrossRef] [Green Version]
- Bao, W.; Kojima, K.K.; Kohany, O. Repbase Update, a database of repetitive elements in eukaryotic genomes. Mob. DNA 2015, 6, 11. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzberg, S.L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 2019, 37, 907–915. [Google Scholar] [CrossRef]
- Stanke, M.; Diekhans, M.; Baertsch, R.; Haussler, D. Using native and syntenically mapped cDNA alignments to improve de novo gene finding. Bioinformatics 2008, 24, 637–644. [Google Scholar] [CrossRef] [Green Version]
- Stanke, M.; Keller, O.; Gunduz, I.; Hayes, A.; Waack, S.; Morgenstern, B. AUGUSTUS: Ab initio prediction of alternative transcripts. Nucleic Acids Res. 2006, 34 (Suppl. S2), W435–W439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burge, C.; Karlin, S. Prediction of complete gene structures in human genomic DNA. J. Mol. Biol. 1997, 268, 78–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korf, I. Gene finding in novel genomes. BMC Bioinform. 2004, 5, 59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cantarel, B.L.; Korf, I.; Robb, S.M.; Parra, G.; Ross, E.; Moore, B.; Holt, C.; Alvarado, A.S.; Yandell, M. MAKER: An easy-to-use annotation pipeline designed for emerging model organism genomes. Genome Res. 2008, 18, 188–196. [Google Scholar] [CrossRef] [Green Version]
- Deng, Y.; Li, J.; Wu, S.; Zhu, Y.; Chen, Y.; He, F.; Chen, Y.; Deng, L.Y.; LI, J.; WU, S. Integrated nr database in protein annotation system and its localization. Comput. Eng. 2006, 32, 71–74. [Google Scholar]
- Jones, P.; Binns, D.; Chang, H.-Y.; Fraser, M.; Li, W.; McAnulla, C.; McWilliam, H.; Maslen, J.; Mitchell, A.; Nuka, G. InterProScan 5: Genome-scale protein function classification. Bioinformatics 2014, 30, 1236–1240. [Google Scholar] [CrossRef] [Green Version]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T. Gene ontology: Tool for the unification of biology. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [Green Version]
- Kanehisa, M. The KEGG database. In ‘In Silico’ Simulation of Biological Processes; Bock, G., Goode, J.A., Eds.; Novartis Foundation Symposium; John Wiley & Sons Ltd.: London, UK, 2002; Volume 247, pp. 91–100. [Google Scholar]
- Bairoch, A.; Apweiler, R. The SWISS-PROT protein sequence database and its supplement TrEMBL in 2000. Nucleic Acids Res. 2000, 28, 45–48. [Google Scholar] [CrossRef]
- Tatusov, R.L.; Fedorova, N.D.; Jackson, J.D.; Jacobs, A.R.; Kiryutin, B.; Koonin, E.V.; Krylov, D.M.; Mazumder, R.; Mekhedov, S.L.; Nikolskaya, A.N. The COG database: An updated version includes eukaryotes. BMC Bioinform. 2003, 4, 41. [Google Scholar] [CrossRef] [Green Version]
- Lowe, T.M.; Eddy, S.R. tRNAscan-SE: A program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997, 25, 955–964. [Google Scholar] [CrossRef]
- Kalvari, I.; Argasinska, J.; Quinones-Olvera, N.; Nawrocki, E.P.; Rivas, E.; Eddy, S.R.; Bateman, A.; Finn, R.D.; Petrov, A.I. Rfam 13.0: Shifting to a genome-centric resource for non-coding RNA families. Nucleic Acids Res. 2018, 46, D335–D342. [Google Scholar] [CrossRef] [PubMed]
- Nawrocki, E.P.; Eddy, S.R. Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics 2013, 29, 2933–2935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Conway, J.R.; Lex, A.; Gehlenborg, N. UpSetR: An R package for the visualization of intersecting sets and their properties. Bioinformatics 2017, 33, 2938–2940. [Google Scholar] [CrossRef] [Green Version]
- Mendes, F.K.; Vanderpool, D.; Fulton, B.; Hahn, M.W. CAFE 5 models variation in evolutionary rates among gene families. Bioinformatics 2021, 36, 5516–5518. [Google Scholar] [CrossRef] [PubMed]
- Britton, T.; Anderson, C.L.; Jacquet, D.; Lundqvist, S.; Bremer, K. Estimating divergence times in large phylogenetic trees. Syst. Biol. 2007, 56, 741–752. [Google Scholar] [CrossRef] [Green Version]
- Jeremy, S.; Dan, R.; Kerrie, B.; Susan, L.; Miranda, H.-S.; Kathleen, L.; Hope, T.; Jane, G.; Neil, M.; Naxin, H. Genome sequencing and analysis of the model grass Brachypodium distachyon. Nature 2010, 463, 7282. [Google Scholar]
- Vandepoele, K.; Saeys, Y.; Simillion, C.; Raes, J.; Van de Peer, Y. The automatic detection of homologous regions (ADHoRe) and its application to microcolinearity between Arabidopsis and rice. Genome Res. 2002, 12, 1792–1801. [Google Scholar] [CrossRef] [Green Version]
- Krueger, F. Trim Galore: A Wrapper Tool around Cutadapt and FastQC to Consistently Apply Quality and Adapter Trimming to FastQ Files. Available online: https://github.com/FelixKrueger/TrimGalore (accessed on 1 June 2020).
- Krueger, F.; Andrews, S.R. Bismark: A flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics 2011, 27, 1571–1572. [Google Scholar] [CrossRef]
- Song, Q.; Decato, B.; Hong, E.E.; Zhou, M.; Fang, F.; Qu, J.; Garvin, T.; Kessler, M.; Zhou, J.; Smith, A.D. A reference methylome database and analysis pipeline to facilitate integrative and comparative epigenomics. PLoS ONE 2013, 8, e81148. [Google Scholar] [CrossRef] [Green Version]
- Schulz, M.H.; Zerbino, D.R.; Vingron, M.; Birney, E. Oases: Robust de novo RNA-seq assembly across the dynamic range of expression levels. Bioinformatics 2012, 28, 1086–1092. [Google Scholar] [CrossRef] [PubMed]
- Simpson, J.T.; Wong, K.; Jackman, S.D.; Schein, J.E.; Jones, S.J.; Birol, I. ABySS: A parallel assembler for short read sequence data. Genome Res. 2009, 19, 1117–1123. [Google Scholar] [CrossRef] [Green Version]
- Xie, Y.; Wu, G.; Tang, J.; Luo, R.; Patterson, J.; Liu, S.; Huang, W.; He, G.; Gu, S.; Li, S. SOAPdenovo-Trans: De novo transcriptome assembly with short RNA-Seq reads. Bioinformatics 2014, 30, 1660–1666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henschel, R.; Lieber, M.; Wu, L.-S.; Nista, P.M.; Haas, B.J.; LeDuc, R.D. Trinity RNA-Seq Assembler Performance Optimization. In Proceedings of the XSEDE ‘12 1st Conference of the Extreme Science and Engineering Discovery Environment: Bridging from the Extreme to the Campus and Beyond, Chicago, IL, USA, 16–20 July 2012; ACM: Chicago, IL, USA, 2012; p. 45. [Google Scholar]
- Li, W.; Godzik, A. Cd-hit: A fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 2006, 22, 1658–1659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pertea, G.; Huang, X.; Liang, F.; Antonescu, V.; Sultana, R.; Karamycheva, S.; Lee, Y.; White, J.; Cheung, F.; Parvizi, B. TIGR Gene Indices clustering tools (TGICL): A software system for fast clustering of large EST datasets. Bioinformatics 2003, 19, 651–652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conesa, A.; Götz, S.; García-Gómez, J.M.; Terol, J.; Talón, M.; Robles, M. Blast2GO: A universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 2005, 21, 3674–3676. [Google Scholar] [CrossRef] [Green Version]
- Götz, S.; García-Gómez, J.M.; Terol, J.; Williams, T.D.; Nagaraj, S.H.; Nueda, M.J.; Robles, M.; Talón, M.; Dopazo, J.; Conesa, A. High-throughput functional annotation and data mining with the Blast2GO suite. Nucleic Acids Res. 2008, 36, 3420–3435. [Google Scholar] [CrossRef]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [Green Version]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [Green Version]
- Biswas, M.; Nath, U.; Howlader, J.; Bagchi, M.; Natarajan, S.; Kayum, M.A.; Kim, H.-T.; Park, J.-I.; Kang, J.-G.; Nou, I.-S. Exploration and exploitation of novel SSR markers for candidate transcription factor genes in Lilium species. Genes 2018, 9, 97. [Google Scholar] [CrossRef] [Green Version]
- Untergasser, A.; Cutcutache, I.; Koressaar, T.; Ye, J.; Faircloth, B.C.; Remm, M.; Rozen, S.G. Primer3—New capabilities and interfaces. Nucleic Acids Res. 2012, 40, e115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schuler, G.D. Sequence mapping by electronic PCR. Genome Res. 1997, 7, 541–550. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Scaffolds | Contigs | |||||
|---|---|---|---|---|---|---|
| No. | Size (bp) | No. | Size (bp) | Gaps | ||
| With gaps | Without Gaps | |||||
| Total Number | 10,627 | 27,491 | 16,864 | |||
| Min | - | 5830 | 5830 | - | 5198 | 25 |
| Median | - | 136,187 | 131,005 | - | 55,047 | 2415 |
| Mean | - | 220,901 | 213,350 | - | 82,473 | 4758 |
| Max | - | 2,848,924 | 2,758,809 | - | 1,033,476 | 38,914 |
| Total size | - | 2,347,517,452 | 2,267,274,222 | - | 2,267,274,222 | 80,243,230 |
| N50 | - | 394,682 | 379,106 | - | 123,867 | 11,038 |
| N90 | - | 107,821 | 103,307 | - | 37,045 | 2551 |
| N95 | - | 69,101 | 66,089 | - | 27,170 | 1540 |
| GC content (%) | 40.1 | |||||
| Species | Read Length (bp) | Data | Sequence Depth (X) | Mapped (%) | Properly Paired (%) | Singletons (%) | Reference Total Length (Gb) | Reads Covered Length (Gb) | Coverage (%) |
|---|---|---|---|---|---|---|---|---|---|
| B. rotunda | 250_250 | 260 (Gb) | 104 | 95.24 | 84.47 | 0.20 | 2.35 | 2.25 | 96 |
| Features | SOAP-denovo (K25) | Oases (K21) | TransAbyss (K25) | Trinity (K25) |
|---|---|---|---|---|
| N50 size (bp) | 410 | 1019 | 495 | 487 |
| N50 No. | 22,910 | 14,286 | 28,234 | 36,730 |
| Contig number | 78,492 | 72,085 | 111,327 | 158,465 |
| Transcript’s size (bp) | 30,869,274 | 51,258,323 | 50,358,442 | 70,949,809 |
| Avg. transcript length (bp) | 393 | 711 | 452 | 448 |
| Min. contig length(bp) | 200 | 200 | 200 | 200 |
| Max. contig length (bp) | 15,760 | 12,523 | 33,886 | 13,325 |
| Assessment assembly after merged assembly of non-redundant contigs from different k-mers via TGICL | ||||
| N50 size (bp) | 572 | 1013 | 607 | 536 |
| N50 no. | 18,528 | 17,329 | 28,419 | 26,034 |
| Contig number | 78,963 | 95,847 | 132,572 | 115,096 |
| Transcriptome size (bp) | 38,503,434 | 66,535,881 | 67,286,353 | 54,131,258 |
| Average length (bp) | 488 | 694 | 508 | 470 |
| Min. contig length(bp) | 200 | 200 | 200 | 200 |
| Max. contig length (bp) | 43,900 | 88,053 | 100,968 | 19,761 |
| Type | Repeat Size (bp) | % of Genome | |
|---|---|---|---|
| TRF | 162,927,183 | 6.94 | |
| RepeatMasker (RepBase TEs) | DNA | 23,107,771 | 0.98 |
| LINE | 7,269,664 | 0.31 | |
| LTR | 308,993,979 | 13.16 | |
| SINE | 50,226 | 0.00 | |
| Other | 1305 | 0.00 | |
| Unknown | 0.00 | 0.00 | |
| Total | 339,001,341 | 14.44 | |
| RepeatProteinMask (TE proteins) | DNA | 30,071,545 | 1.28 |
| LINE | 15,711,684 | 0.67 | |
| LTR | 449,069,450 | 19.13 | |
| SINE | 0.00 | 0 | |
| Other | 0.00 | 0 | |
| Unknown | 0.00 | 0 | |
| Total | 494,297,946 | 21.05 | |
| De novo | DNA | 49,253,612 | 2.10 |
| LINE | 9,765,795 | 0.42 | |
| LTR | 1,524,782,230 | 64.95 | |
| SINE | 789,305 | 0.03 | |
| Other | 0 | 0.00 | |
| Satellite | 8,247,215 | 0.351316 | |
| Simple_repeat | 6,742,687 | 0.287226 | |
| Unknown | 2,202,787 | 0.09 | |
| Total | 1,591,591,610 | 67.80 | |
| Combined TEs | DNA | 77,273,965 | 3.29 |
| LINE | 23,221,220 | 0.99 | |
| LTR | 1,576,612,191 | 67.16 | |
| SINE | 832,585 | 0.04 | |
| Other | 1305 | 0.00 | |
| Unknown | 2,202,787 | 0.09 | |
| Total | 1,653,717,174 | 70.45 | |
| Total | 1,702,210,889 | 72.51 |
| Item | Genome-Wide | % | Transcriptome-Wide | % |
|---|---|---|---|---|
| Total number of sequences examined | 10,627 | 95,847 | ||
| Total size of examined sequences (bp) | 2,347,517,452 | 66,535,881 | ||
| Total number of identified microsatellites | 238,441 | 4579 | ||
| Number of microsatellites containing sequences | 10,381 | 4032 | ||
| Sequences contain more than one microsatellite | 9803 | 384 | ||
| Microsatellites in compound formation | 4309 | 27 | ||
| Microsatellite’s density (per Mbp) | 102 | 69 | ||
| Class I microsatellites | 82,414 | 35.20 | 949 | 20.85 |
| Class II microsatellites | 151,718 | 64.80 | 3603 | 79.15 |
| AT rich microsatellites | 176,052 | 75.19 | 2778 | 61.03 |
| GC rich microsatellites | 43,155 | 18.43 | 1275 | 28.01 |
| AT/GC balance microsatellites | 14,925 | 6.37 | 499 | 10.96 |
| Mono-nucleotide repeats | 68,961 | 28.92 | 1137 | 24.83 |
| Di-nucleotide repeats | 61,439 | 25.77 | 574 | 12.54 |
| Tri-nucleotide repeats | 84,932 | 35.62 | 2366 | 51.67 |
| Tera-nucleotide repeats | 5330 | 2.24 | 148 | 3.23 |
| Penta-nucleotide repeats | 9917 | 4.16 | 185 | 4.04 |
| Hexa-nucleotide repeats | 7862 | 3.30 | 169 | 3.69 |
| Primer modelling was successful | 223,678 | 93.81 | 3348 | 73.12 |
| Primer modelling failed | 14,763 | 6.60 | 1231 | 36.77 |
| Non redundant primer | 132,792 | 59.37 | 1888 | 56.39 |
| No. of Primer Mapped on M. acuminata genome | 100 | 0.075 | 30 | 1.59 |
| No. of Primer Mapped on M. balbisiana genome | 105 | 0.079 | 25 | 1.32 |
| No. of Primer Mapped on M. Itinerans genome | 102 | 0.077 | 32 | 1.69 |
| No. of Primer Mapped on Ensete ventricosum genome | 121 | 0.091 | 27 | 1.43 |
| No. of primer tested | 6 | 100 | 8 | 100 |
| No. of primer amplified | 6 | 100 | 8 | 100 |
| Database | Number | Percent (%) |
|---|---|---|
| NR | 71,072 | 97.22 |
| InterPro | 69,525 | 95.11 |
| GO | 45,256 | 61.91 |
| KEGG | 59,649 | 81.60 |
| Swissprot | 57,622 | 78.82 |
| COG | 24,851 | 33.99 |
| TrEMBL | 70,990 | 97.11 |
| Total annotated | 73,102 | 97.81 |
| Unannotated | 1602 | 2.19 |
| Type | Copy | Average Length (bp) | Total Length (bp) | % of Genome |
|---|---|---|---|---|
| miRNA | 213 | 119 | 25,384 | 0.001081 |
| tRNA | 2727 | 75 | 205,538 | 0.008756 |
| rRNA | 486 | 232 | 112,876 | 0.004808 |
| 18S | 105 | 666 | 69,922 | 0.002979 |
| 28S | 147 | 119 | 17,441 | 0.000743 |
| 5.8S | 40 | 148 | 5931 | 0.000253 |
| 5S | 194 | 101 | 19,582 | 0.000834 |
| snRNA | 2136 | 154 | 329,909 | 0.014054 |
| CD-box | 600 | 105 | 62,771 | 0.002674 |
| HACA-box | 53 | 134 | 7091 | 0.000302 |
| splicing | 1483 | 175 | 260,047 | 0.011078 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Taheri, S.; Teo, C.H.; Heslop-Harrison, J.S.; Schwarzacher, T.; Tan, Y.S.; Wee, W.Y.; Khalid, N.; Biswas, M.K.; Mutha, N.V.R.; Mohd-Yusuf, Y.; et al. Genome Assembly and Analysis of the Flavonoid and Phenylpropanoid Biosynthetic Pathways in Fingerroot Ginger (Boesenbergia rotunda). Int. J. Mol. Sci. 2022, 23, 7269. https://doi.org/10.3390/ijms23137269
Taheri S, Teo CH, Heslop-Harrison JS, Schwarzacher T, Tan YS, Wee WY, Khalid N, Biswas MK, Mutha NVR, Mohd-Yusuf Y, et al. Genome Assembly and Analysis of the Flavonoid and Phenylpropanoid Biosynthetic Pathways in Fingerroot Ginger (Boesenbergia rotunda). International Journal of Molecular Sciences. 2022; 23(13):7269. https://doi.org/10.3390/ijms23137269
Chicago/Turabian StyleTaheri, Sima, Chee How Teo, John S. Heslop-Harrison, Trude Schwarzacher, Yew Seong Tan, Wei Yee Wee, Norzulaani Khalid, Manosh Kumar Biswas, Naresh V. R. Mutha, Yusmin Mohd-Yusuf, and et al. 2022. "Genome Assembly and Analysis of the Flavonoid and Phenylpropanoid Biosynthetic Pathways in Fingerroot Ginger (Boesenbergia rotunda)" International Journal of Molecular Sciences 23, no. 13: 7269. https://doi.org/10.3390/ijms23137269
APA StyleTaheri, S., Teo, C. H., Heslop-Harrison, J. S., Schwarzacher, T., Tan, Y. S., Wee, W. Y., Khalid, N., Biswas, M. K., Mutha, N. V. R., Mohd-Yusuf, Y., Gan, H. M., & Harikrishna, J. A. (2022). Genome Assembly and Analysis of the Flavonoid and Phenylpropanoid Biosynthetic Pathways in Fingerroot Ginger (Boesenbergia rotunda). International Journal of Molecular Sciences, 23(13), 7269. https://doi.org/10.3390/ijms23137269