
Roles of Fatty Acids in Microglial Polarization: Evidence from In Vitro and In Vivo Studies on Neurodegenerative Diseases



Abstract
:1. Introduction
1.1. Neuroinflammation and Neurodegeneration
1.2. Microglia: A Double-Edged Sword
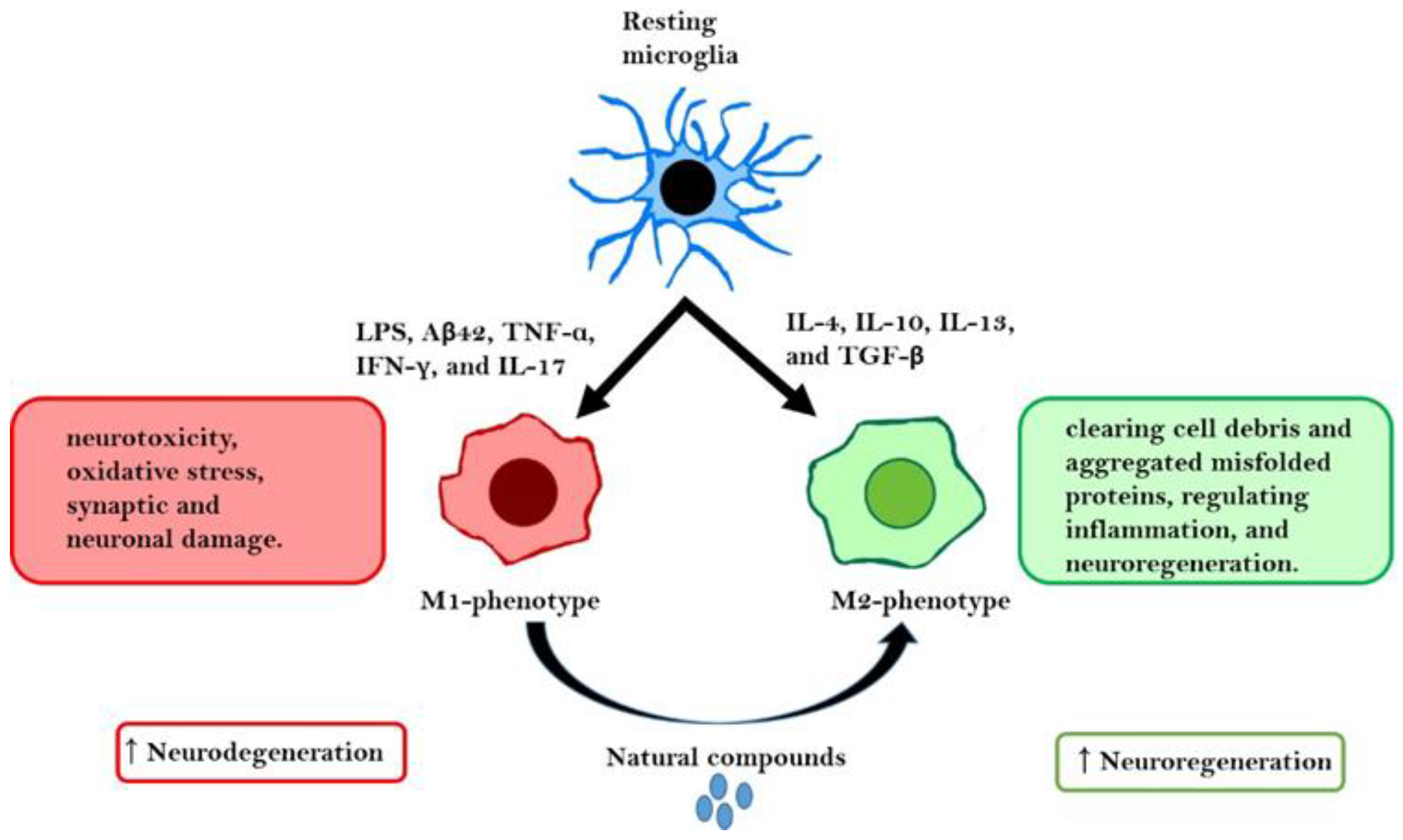
1.3. Microglial Polarization as an Inflammation/Anti-Inflammation Switch
1.4. Fatty Acids and Their Bioactive Potential
2. Literature Search Strategy
3. Role of Fatty Acids in Microglial Polarization in NDs
3.1. Role of Fatty Acids in Microglial Polarization in Neuroinflammation

{kind=link}
{kind=link}
| Compound | Cell/Animal | Treatment | Findings | Ref |
|---|---|---|---|---|
| DHA | Primary microglial cultures | DHA (0.1, 1, 10, or 20 μM) + LPS (10 ng/mL) or IFN-γ (200 U/mL) for 24 h. | ↓NO, ↓iNOS, ↓TNFα, ↓IL-6, ↓Arg1, ↓IL-10, ↓PGE2, ↓MAPK, ↑PPARγ nuclear translocation, and ↑NPC survival and differentiation. | [67] |
| ω-3 PUFAs | C57BL6/J and CX3CR1-eGFP | Sunflower oil (6% fat; rich in LA, ω-3 PUFA-deficient diet) or a mixture of different oils containing ALA (ω-3 PUFA diet). | ↓ω-3 PUFAs, ↑IL-1β, ↑IL-6, ↑COX-2, ↓CD36 ↓CD206, ↓microglial motility, ↑Erg-1, ↓c-Jun, and ↓BDNF. | [75] |
| DHA +PhA | BV-2 cells | DHA containing 50 or 500 ppm of PhA for 1 h + LPS (0.1 µg/mL) or H2O2 (0.8 mM) for 24 h. | ↑Cell viability, ↓LDH, ↓caspase-3, ↓O2−, ↓COX-2, ↓IL-6, ↓iNOS, ↓CD11b, ↑Arg1, ↓GtPx, ↓GtRd, ↓SOD-1, and ↑BDNF. | [76] |
| DHA | Primary neuron/glia, neuron, and microglial cultures (SD rats) | DHA (50 μM) for 3 h + JEV (MOI = 5) for 1 h + DHA (50 μM) for 3 h. JEV (MOI = 5) for 1 h + DHA (50 μM) for 48 h | ↓Nitrite, ↓IL-1β, ↓TNFα, ↓PGE2, ↓ROS, ↓neuronal death, ↓phagocytic microglia, ↓LDH efflux, ↓JEV RNA, ↓NS1, ↓viral particle production, ↓CD68, ↓iNOS, ↓COX-2, ↓IRF5, ↓IRF8, ↓P2X4R, ↓P2X7R, ↓P2Y12R, ↑CD163, ↑CD206, ↑Arg1, ↑Nrf2, ↑HO-1, ↑miR-124, ↓NF-κB, ↓AP-1, ↓CREB, ↓TLR4, ↓TLR7, ↓MyD88, ↓IRF1, ↓NLRP3, ↓ASC, and ↓phosphorylation (TAK1, TBK1, IRF3, ERK, JNK, p38, Akt, cPLA2, Jak1, Jak2, Tyk2, Src, Stat1, and Stat2). Pretreatment > post-treatment. | [79] |
| DHA | C57Bl/6J mice | Fish hydrolysate (DHA; 143 µg) or DHA (10 mg) for 18 days + LPS (125 µg/kg bodyweight) for 2 h. | ↓TLR4, ↓IL-1β, ↓TNFα, ↓IL-6, ↓CCL-2, ↓CD68, ↓CD11b, ↓CD86, ↓SOCS3, ↓NGF, and ↓COX-2. | [85] |
| Syn and EPEA | SIM-A9 cells, C57BL/6 mice | Syn or EPEA (10 µM) for 1 h + LPS (1 µg/mL) for 24 h. Syn or EPEA (100 mg/mL) + LPS (750 mg/kg bodyweight), injection/day for 7 days. | (Syn): ↓IL-1β, (Syn and EPEA): ↓TNFα, ↓IL-6, and ↑IL-10. (Syn): ↓IL-1β, ↓TNFα, ↑IL-10, ↓CD86, ↓Iba1, and ↑BDNF (Syn and EPEA): ↑IL-4, ↓MHC-II, ↑CD206, ↑Arg1, ↓GFAP, ↓S100β, and ↑LTP. | [87] |
| DPA | BV-2, SH-SY5Y cells | BV-2 cells; DPA (0, 12.5, 25, 50, or 100 µM) for 24 h + LPS (100 ng/mL) for 24 h; supernatant → SH-SY5Y cells | ↑Cell viability, ↓NO, ↓Iba-1, ↓CD11b, ↑Arg1, ↑CD206, ↑IL-10, ↓IL-1β, ↓TNF-α, and ↓IL-1R1. | [88] |
3.2. Role of Fatty Acids in Microglial Polarization in Spinal Cord Injury (SCI)
3.3. Role of Fatty Acids in Microglial Polarization in Alzheimer’s Disease (AD)
| Model | Compound | Cell/Animal | Treatment | Findings | Ref. |
|---|---|---|---|---|---|
| SCI | DHA | Wistar rats PMCs | DHA (45 mg/kg bodyweight), 3 weeks post-SCI. DHA (1 μM) for 2 or 4 days. | ↑Activation and proliferation of microglial cells, ↓CD86, and ↑CD163. ↑SOD and ↑microglial cell proliferation. | [91] |
| DHA | SD rats C57BL6 mice BMDMs (C57BL/6 mice) PC12 cells BV-2 cells PMCs (Wistar rats) | DHA (500 nmol/kg bodyweight), 30 min post-SCI, 7 and 28 days, 35 days for C57BL/6 mice. LPS (100 ng/mL) or IFN-γ (20 ng/ mL) or IL-4 (20 ng/mL) for 30 min + DHA (1 or 3 μM) for 24 h. UV-stressed PC12 + DHA-treated BV-2 cells (DHA; 3 μM for 24 h) DHA (1 and 3 μM) for 24 h | ↑Locomotor recovery, ↑neuronal survival, ↑ED1, ↑CD16/32, ↑Arg1, ↑miR-124, and ↓myelin phagocytosis. ↓M1 (CD16/32) ↓Phagocytic activity of microglia, ↓myelin phagocytosis, and ↑miR-124 ↑miR-124 | [92] | |
| AD | DHA and EPA | CHME3 cells | DHA or EPA (0.05, 0.1, 0.5, or 1 μM) + Aβ42 (1 μg/mL) for 2, 6, or 24 h. | ↑Aβ42-Phagocytosis, ↑BDNF, ↓TNF-α, ↓CD40, ↓CD86, and ↑CD206. | [109] |
| DHA and EPA | MG6 cells BV-2 cells | DHA or EPA (200 μM each), DHA + EPA (100 μM) for 30 min + LPS (100 ng/mL) for 24 h. | ↓TNF-α, ↓IL-6, ↑SIRT1, ↓NF-κB, ↑NAMPT, ↑NAD+, and ↑autophagy. | [66] | |
| EPA | C57BL/6 mice | Ethyl (E)-EPA (0.8%; 8–10 g per mouse/day) for 42 days. On day 28, treat with 2 μg Aβ. | ↑Spatial learning and memory, ↓CCL-2, ↑Arg1, ↓IL-1β, ↓TNF-α, ↓IL-6, ↑BDNF, ↑NGF, and ↓neural death. | [115] |
3.4. Role of Fatty Acids in Microglial Polarization in MS
3.5. Role of Fatty Acids in Microglial Polarization in Cerebral Ischemia
| Model | Compound | Cell/Animal Type | Treatment | Findings | Ref. |
|---|---|---|---|---|---|
| MS | DHA and EPA | PMCs C57/BL6 mice | DHA or EPA (5–80 mM) for 24 h + LPS (2.5 ng/mL) for 24 h or DHA or EPA (20 mM) for 24 h + myelin (1, 5, or 10 mg/mL) w/o IFN-γ (5 ng/mL) for 24 h. Cuprizone (0.2%) with low ω-3 PUFA (0.3%) or with high ω-3 PUFA (DHA 1 EPA, 15 g/kg bodyweight) for 5 weeks. | ↓NO, ↓TNF-α, ↑myelin phagocytosis, ↓IL-6, ↓CCL5, ↓TNF-α, ↓CD14, ↓CCL2, ↓IL-23a, ↓CCL4, ↓IL-1α, ↓iNOS, ↑CCL22, ↑CCL17, ↑Arg1, ↑IL-5, ↑Chi3l1, ↑Gata3, ↑CD206, ↑CCL11, ↑CCR2, ↑TGFβ, and ↑CCL1. ↓Demyelination, ↓neurological deficits, ↓CD16, ↓iNOS, ↑CD206, ↑YM1/2, and ↑Arg1. | [129] |
| nFAG | C57BL/6J mice | nFAG (10 mg/daily) on day 16 post-immunization with MOG. | Delays the onset of EAE, ↓TNF-α, ↓IL-1β, ↓IL-6, ↓IL-8, ↓ICAM-1, ↓GFAP, ↓iNOS, ↓RGC degeneration, and ↓RGC loss. | [130] | |
| SFAs and USFAs | C57BL/6J mice | 9 MOG-treated mice (20% SFAs + 17.5% USFAs) for 16 days. | ↓CXCL-10, ↓CXCL-11, ↓IL-12, ↓IL-23, ↑CCL-2, ↑CCL-22, ↑CD163, ↑Arg1, ↑IL-10, ↓NF-κB, ↓optic nerve damage, and ↑PhNR amplitude | [132] | |
| ALA and VA | N9 microglial cells Oli-Neu cells CGNs | LPS (100 ng/mL) + VPA or ALA or diamide 1 or ethanolamide 2 (0.5, 1.5, or 10 μM) and VPA + ALA (1:1) Each compound (1–10 μM) for 24 h. Each compound was pretreated (5, 10, or 25 μM) for 6 h + co-treatment with glutamate (100 μM)/glycine (10 μM) + 24 h. | ↓iNOS ↑OPC differentiation, ↓Olig2, and ↑CNPase ↑Neuroprotection | [138] | |
| KO diet (rich in ω-3 PUFAs) | C57BL/6 mice | KO diet containing 0.2% CPZ for 5 weeks. | ↓Motor abnormalities and cognitive deficit, ↑GSH-Px, ↑SOD, ↑GSH, ↓MDA, ↑myelin sheet recovery, ↓NG2+ OPCs, ↓CD68+ cells, ↓Iba1+ cells, ↓CD16, ↓iNOS, ↑Arg1, ↑CD206, ↓HDAC3, ↓p-STAT3, and ↓NF-κB. | [140] | |
| Cerebral Ischemia | ω-3 PUFAs | Wistar Rats, primary astroglial and neuronal cultures, BV-2 microglia | MCAO + DHA (140 mg/kg bodyweight/day), EPA (220 mg/kg bodyweight/day) for 24 h. Hypoxia for 1 h (neuronal cells) or 3 h (astroglia, BV-2 cells) w/o emulsion. | ↓Ischemic damage, ↑behavioral performance, ↑GAP-43, ↑Tau mRNA, ↓HIF1a, ↓IL-1β, ↓TNF-α, ↓NLRP3, ↓Arg1. ↓TNF-α, ↓iNOS, ↓CCL5, ↓COX-2, ↓Bax, and ↑Bcl-2 | [152] |
| DHA and FO | C57BL/6J mice | MCAO for 2 h + DHA (10 mg/kg bodyweight/day) for 14 days w/o FO supplementation and 5 days post-MCAO | ↑Long-term histological and functional outcomes, ↓white matter injury, ↑BrdU, ↑APC, ↑MBP, ↓SMI-32, ↓CD16/32+/Iba1+ cells, and ↑CD206+/Iba1+ cells | [156] | |
| DHA | C57BL/6 mice | tMCAO for 1 h + DHA (10 mg/kg bodyweight) for 3 days. | ↓Brain infarct, ↓neurological deficit, ↓infiltration (macrophages, neutrophils, T and B lymphocytes), ↑CD206+ CD16-, ↑CD206+Iba1+, ↑CD206+CD16−, ↑IL-10, ↑Arg1, ↑TGFβ, ↓CCL1, ↓CCL2, ↓CCL3, ↓CCL17, ↓CXCL1, ↓CXCL2, ↓CXCL10, ↓CXCL12, ↓CXCL13, ↓C5/5a, ↓IL-1α, ↓IL-1rα, ↓IL-27, ↓IFNγ, ↓TNFα, ↓GCSF, ↓TIMP1, and ↓TREM1. | [160] | |
| FO | Wistar rats | Phospholipid emulsion; EPA (70 mg/kg bodyweight) and DHA (80 mg/kg bodyweight) daily for 21 days post-MCAO. | ↑HPS, ↓iNOS, ↑Arg1, ↓granulopoiesis, and ↓myeloperoxidase positivity | [161] | |
| DHA | Human and I/R mouse brain tissue C57/BL6 mice Mouse peritoneal macrophages and brain microglia | Assessed for NALP3 and M1 and M2 cells. tMCAO + DHA (5 mg/kg bodyweight) after tMCAO. DHA (1 µmol/L) on days 2 and 10. After 24 h, treated with IFN-γ (100 U/mL), LPS (100 ng/mL), and IL-4 (100 U/mL) for 72 h. | ↑NALP3+ cells on day 4, but decreased on day 6, ↑iNOS, and ↑Ym-1 ↓I/R injury, ↓iNOS, ↓IL-1β, ↓IL-23 ↑Arg1, ↑Ym-1, ↑IL-10, and ↓NALP3 ↓NALP3 and ↓IL-1β | [162] |
3.6. Role of Fatty Acids in Microglial Polarization in Traumatic Brain Injury (TBI)
3.7. Role of Fatty Acids in Microglial Polarization in Depression
| Model | Compound | Cell/Animal Type | Treatment | Findings | Ref. |
|---|---|---|---|---|---|
| TBI | DHA | SD rats | TBI for 5 min + DHA (16 mg/kg bodyweight/daily in DMSO) for 3, 7, or 21 days. | ↓CD16/32+, ↑CD206+, ↓ER stress, ↓NF-κB, ↓CHOP+ neurons, ↓LAMP-1, and ↓TNF-α. | [172] |
| ALA | Pregnant C57BL6/N mice | Flaxseed oil (3.1% ALA) for 4 months + TBI. | ↑Brain DHA, ↓DPA/DHA, ↓IL-1β, ↓TNFα, ↓IL-6, ↓CCL2, ↑CD206, ↓GFAP, and ↓motor and cognitive abilities | [174] | |
| DHA | SD rats | CCI for 30 min + DHA (100–150 mg/kg bodyweight/day) | ↓Nitrate/nitrite, ↓microgliosis, ↓CD68, ↓MHC-II, ↑CD206, ↓cell number, ↓CD86, ↓CD32, ↓IL-1 β, ↓TNF-α, ↓IL-10, ↓TGFβ, and ↑novel object recognition | [83] | |
| ω-3 PUFA | SD rats | TBI for 30 min + ω-3 PUFA (2 mL/kg bodyweight/day) for 7 days. | ↑Neurological functions, ↓brain water content, ↓neuronal apoptosis, ↓CD16+, ↑CD206+, ↓IL-1β, ↓TNF-α, ↓IL-6, ↑IL-10, ↓HMGB1/NF-κB, and ↑SIRT1 | [179] | |
| Depression | Soybean oil | Heterozygous transgenic Fat-1 mice | Soybean oil (10%) + 3 µg/µL LPS for 24 h. | ↓CD11b, ↓IL-1β, ↓TNF-α, ↓IL-17, ↑IL-4, ↑IL-10, ↑TGF-β1, ↑BDNF, ↑TrkB, ↓p75, ↓NO, and ↓iNOS. | [182] |
| DHA and EPA | SD rats | OVX surgery +1.5 g refined fish oil/kg bodyweight (approximate EPA and DHA contents were 340 and 240 mg/g, respectively) for 10 weeks. | ↑Anti-anxiety, ↓apoptotic cells, ↓Iba-1, ↓IL-1β, ↓IL-6, ↓TNF-α, ↓CD86, ↓iNOS, ↑IL-10, ↑IL-4, ↑CD206, ↑Arg1, ↓NF-κB, and ↑IκB. | [183] |
4. Conclusions
5. Limitations and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gitler, A.D.; Dhillon, P.; Shorter, J. Neurodegenerative disease: Models, mechanisms, and a new hope. Dis. Models Mech. 2017, 10, 499–502. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.S.; Joh, T.H. Microglia, major player in the brain inflammation: Their roles in the pathogenesis of Parkinson’s disease. Exp. Mol. Med. 2006, 38, 333–347. [Google Scholar] [CrossRef] [Green Version]
- Saijo, K.; Crotti, A.; Glass, C.K. Regulation of microglia activation and deactivation by nuclear receptors. Glia 2013, 61, 104–111. [Google Scholar] [CrossRef]
- González-Scarano, F.; Baltuch, G. Microglia as mediators of inflammatory and degenerative diseases. Annu. Rev. Neurosci. 1999, 22, 219–240. [Google Scholar] [CrossRef]
- Liu, B.; Hong, J.S. Role of microglia in inflammation-mediated neurodegenerative diseases: Mechanisms and strategies for therapeutic intervention. J. Pharmacol. Exp. Ther. 2003, 304, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Kwon, H.S.; Koh, S.H. Neuroinflammation in neurodegenerative disorders: The roles of microglia and astrocytes. Transl. Neurodegener. 2020, 9, 42. [Google Scholar] [CrossRef]
- Lawson, L.J.; Perry, V.H.; Dri, P.; Gordon, S. Heterogeneity in the distribution and morphology of microglia in the normal adult mouse brain. Neuroscience 1990, 39, 151–170. [Google Scholar] [CrossRef]
- Kierdorf, K.; Erny, D.; Goldmann, T.; Sander, V.; Schulz, C.; Perdiguero, E.G.; Wieghofer, P.; Heinrich, A.; Riemke, P.; Hölscher, C.; et al. Microglia emerge from erythromyeloid precursors via Pu.1- and Irf8-dependent pathways. Nat. Neurosci. 2013, 16, 273–280. [Google Scholar] [CrossRef]
- Arcuri, C.; Mecca, C.; Bianchi, R.; Giambanco, I.; Donato, R. The Pathophysiological Role of Microglia in Dynamic Surveillance, Phagocytosis and Structural Remodeling of the Developing CNS. Front. Mol. Neurosci. 2017, 10, 191. [Google Scholar] [CrossRef] [Green Version]
- Perry, V.H.; Nicoll, J.A.; Holmes, C. Microglia in neurodegenerative disease. Nat. Rev. Neurol. 2010, 6, 193–201. [Google Scholar] [CrossRef]
- Xu, L.; He, D.; Bai, Y. Microglia-Mediated Inflammation and Neurodegenerative Disease. Mol. Neurobiol. 2016, 53, 6709–6715. [Google Scholar] [CrossRef] [PubMed]
- Zrzavy, T.; Hametner, S.; Wimmer, I.; Butovsky, O.; Weiner, H.L.; Lassmann, H. Loss of ‘homeostatic’ microglia and patterns of their activation in active multiple sclerosis. Brain 2017, 140, 1900–1913. [Google Scholar] [CrossRef] [PubMed]
- Block, M.L.; Zecca, L.; Hong, J.S. Microglia-mediated neurotoxicity: Uncovering the molecular mechanisms. Nat. Rev. Neurosci. 2007, 8, 57–69. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Le, W. Differential Roles of M1 and M2 Microglia in Neurodegenerative Diseases. Mol. Neurobiol. 2016, 53, 1181–1194. [Google Scholar] [CrossRef] [PubMed]
- Martinez, F.O.; Gordon, S. The M1 and M2 paradigm of macrophage activation: Time for reassessment. F1000Prime Rep. 2014, 6, 13. [Google Scholar] [CrossRef] [Green Version]
- Ransohoff, R.M. A polarizing question: Do M1 and M2 microglia exist? Nat. Neurosci. 2016, 19, 987–991. [Google Scholar] [CrossRef]
- Cherry, J.D.; Olschowka, J.A.; O’Banion, M.K. Neuroinflammation and M2 microglia: The good, the bad, and the inflamed. J. Neuroinflamm. 2014, 11, 98. [Google Scholar] [CrossRef] [Green Version]
- Davis, M.J.; Tsang, T.M.; Qiu, Y.; Dayrit, J.K.; Freij, J.B.; Huffnagle, G.B.; Olszewski, M.A. Macrophage M1/M2 polarization dynamically adapts to changes in cytokine microenvironments in Cryptococcus neoformans infection. mBio 2013, 4, e00264-00213. [Google Scholar] [CrossRef] [Green Version]
- Franco, R.; Fernandez-Suarez, D. Alternatively activated microglia and macrophages in the central nervous system. Prog. Neurobiol. 2015, 131, 65–86. [Google Scholar] [CrossRef]
- Zhang, L.; Zhang, J.; You, Z. Switching of the Microglial Activation Phenotype Is a Possible Treatment for Depression Disorder. Front. Cell. Neurosci. 2018, 12, 306. [Google Scholar] [CrossRef]
- Wang, Y.; Zhao, M.; Liu, S.; Guo, J.; Lu, Y.; Cheng, J.; Liu, J. Macrophage-derived extracellular vesicles: Diverse mediators of pathology and therapeutics in multiple diseases. Cell Death Dis. 2020, 11, 924. [Google Scholar] [CrossRef]
- Chen, W.W.; Zhang, X.; Huang, W.J. Role of neuroinflammation in neurodegenerative diseases (Review). Mol. Med. Rep. 2016, 13, 3391–3396. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.M.; Yang, S.; Huang, S.S.; Tang, B.S.; Guo, J.F. Microglial Activation in the Pathogenesis of Huntington’s Disease. Front. Aging Neurosci. 2017, 9, 193. [Google Scholar] [CrossRef]
- Su, F.; Bai, F.; Zhou, H.; Zhang, Z. Reprint of: Microglial toll-like receptors and Alzheimer’s disease. Brain Behav. Immun. 2016, 55, 166–178. [Google Scholar] [CrossRef]
- Zhang, Q.-S.; Heng, Y.; Yuan, Y.-H.; Chen, N.-H. Pathological α-synuclein exacerbates the progression of Parkinson’s disease through microglial activation. Toxicol. Lett. 2017, 265, 30–37. [Google Scholar] [CrossRef]
- Wang, L.X.; Zhang, S.X.; Wu, H.J.; Rong, X.L.; Guo, J. M2b macrophage polarization and its roles in diseases. J. Leukoc. Biol. 2019, 106, 345–358. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Zhang, L.; Zhang, H.; Sun, X.; Liu, D.; Zhang, J.; Zhang, Y.; Cheng, L.; Santos, H.A.; Cui, W. Programmable immune activating electrospun fibers for skin regeneration. Bioact. Mater. 2021, 6, 3218–3230. [Google Scholar] [CrossRef]
- Jordão, M.J.C.; Sankowski, R.; Brendecke, S.M.; Sagar; Locatelli, G.; Tai, Y.H.; Tay, T.L.; Schramm, E.; Armbruster, S.; Hagemeyer, N.; et al. Single-cell profiling identifies myeloid cell subsets with distinct fates during neuroinflammation. Science 2019, 363, eaat7554. [Google Scholar] [CrossRef]
- Matcovitch-Natan, O.; Winter, D.R.; Giladi, A.; Vargas Aguilar, S.; Spinrad, A.; Sarrazin, S.; Ben-Yehuda, H.; David, E.; Zelada González, F.; Perrin, P.; et al. Microglia development follows a stepwise program to regulate brain homeostasis. Science 2016, 353, aad8670. [Google Scholar] [CrossRef]
- Mathys, H.; Penney, J.; Tsai, L.H. A Developmental Switch in Microglial HDAC Function. Immunity 2018, 48, 476–478. [Google Scholar] [CrossRef] [Green Version]
- Masuda, T.; Sankowski, R.; Staszewski, O.; Böttcher, C.; Amann, L.; Sagar; Scheiwe, C.; Nessler, S.; Kunz, P.; van Loo, G.; et al. Spatial and temporal heterogeneity of mouse and human microglia at single-cell resolution. Nature 2019, 566, 388–392. [Google Scholar] [CrossRef]
- Sankowski, R.; Böttcher, C.; Masuda, T.; Geirsdottir, L.; Sagar; Sindram, E.; Seredenina, T.; Muhs, A.; Scheiwe, C.; Shah, M.J.; et al. Mapping microglia states in the human brain through the integration of high-dimensional techniques. Nat. Neurosci. 2019, 22, 2098–2110. [Google Scholar] [CrossRef]
- Masuda, T.; Sankowski, R.; Staszewski, O.; Prinz, M. Microglia Heterogeneity in the Single-Cell Era. Cell Rep. 2020, 30, 1271–1281. [Google Scholar] [CrossRef]
- Shukla, A.K.; McIntyre, L.L.; Marsh, S.E.; Schneider, C.A.; Hoover, E.M.; Walsh, C.M.; Lodoen, M.B.; Blurton-Jones, M.; Inlay, M.A. CD11a expression distinguishes infiltrating myeloid cells from plaque-associated microglia in Alzheimer’s disease. Glia 2019, 67, 844–856. [Google Scholar] [CrossRef]
- Yin, Z.; Raj, D.; Saiepour, N.; Van Dam, D.; Brouwer, N.; Holtman, I.R.; Eggen, B.J.L.; Möller, T.; Tamm, J.A.; Abdourahman, A.; et al. Immune hyperreactivity of Aβ plaque-associated microglia in Alzheimer’s disease. Neurobiol. Aging 2017, 55, 115–122. [Google Scholar] [CrossRef]
- Grubman, A.; Choo, X.Y.; Chew, G.; Ouyang, J.F.; Sun, G.; Croft, N.P.; Rossello, F.J.; Simmons, R.; Buckberry, S.; Landin, D.V.; et al. Transcriptional signature in microglia associated with Aβ plaque phagocytosis. Nat. Commun. 2021, 12, 3015. [Google Scholar] [CrossRef]
- Hammond, T.R.; Dufort, C.; Dissing-Olesen, L.; Giera, S.; Young, A.; Wysoker, A.; Walker, A.J.; Gergits, F.; Segel, M.; Nemesh, J.; et al. Single-Cell RNA Sequencing of Microglia throughout the Mouse Lifespan and in the Injured Brain Reveals Complex Cell-State Changes. Immunity 2019, 50, 253–271.e256. [Google Scholar] [CrossRef] [Green Version]
- Krasemann, S.; Madore, C.; Cialic, R.; Baufeld, C.; Calcagno, N.; El Fatimy, R.; Beckers, L.; O’Loughlin, E.; Xu, Y.; Fanek, Z.; et al. The TREM2-APOE Pathway Drives the Transcriptional Phenotype of Dysfunctional Microglia in Neurodegenerative Diseases. Immunity 2017, 47, 566–581.e569. [Google Scholar] [CrossRef] [Green Version]
- Sala Frigerio, C.; Wolfs, L.; Fattorelli, N.; Thrupp, N.; Voytyuk, I.; Schmidt, I.; Mancuso, R.; Chen, W.T.; Woodbury, M.E.; Srivastava, G.; et al. The Major Risk Factors for Alzheimer’s Disease: Age, Sex, and Genes Modulate the Microglia Response to Aβ Plaques. Cell Rep. 2019, 27, 1293–1306.e1296. [Google Scholar] [CrossRef] [Green Version]
- Safaiyan, S.; Besson-Girard, S.; Kaya, T.; Cantuti-Castelvetri, L.; Liu, L.; Ji, H.; Schifferer, M.; Gouna, G.; Usifo, F.; Kannaiyan, N.; et al. White matter aging drives microglial diversity. Neuron 2021, 109, 1100–1117.e1110. [Google Scholar] [CrossRef]
- Li, Q.; Cheng, Z.; Zhou, L.; Darmanis, S.; Neff, N.F.; Okamoto, J.; Gulati, G.; Bennett, M.L.; Sun, L.O.; Clarke, L.E.; et al. Developmental Heterogeneity of Microglia and Brain Myeloid Cells Revealed by Deep Single-Cell RNA Sequencing. Neuron 2019, 101, 207–223.e210. [Google Scholar] [CrossRef] [Green Version]
- Marschallinger, J.; Iram, T.; Zardeneta, M.; Lee, S.E.; Lehallier, B.; Haney, M.S.; Pluvinage, J.V.; Mathur, V.; Hahn, O.; Morgens, D.W.; et al. Lipid-droplet-accumulating microglia represent a dysfunctional and proinflammatory state in the aging brain. Nat. Neurosci. 2020, 23, 194–208. [Google Scholar] [CrossRef]
- Keren-Shaul, H.; Spinrad, A.; Weiner, A.; Matcovitch-Natan, O.; Dvir-Szternfeld, R.; Ulland, T.K.; David, E.; Baruch, K.; Lara-Astaiso, D.; Toth, B.; et al. A Unique Microglia Type Associated with Restricting Development of Alzheimer’s Disease. Cell 2017, 169, 1276–1290.e1217. [Google Scholar] [CrossRef]
- Hagemeyer, N.; Hanft, K.M.; Akriditou, M.A.; Unger, N.; Park, E.S.; Stanley, E.R.; Staszewski, O.; Dimou, L.; Prinz, M. Microglia contribute to normal myelinogenesis and to oligodendrocyte progenitor maintenance during adulthood. Acta Neuropathol. 2017, 134, 441–458. [Google Scholar] [CrossRef] [Green Version]
- Wlodarczyk, A.; Holtman, I.R.; Krueger, M.; Yogev, N.; Bruttger, J.; Khorooshi, R.; Benmamar-Badel, A.; de Boer-Bergsma, J.J.; Martin, N.A.; Karram, K.; et al. A novel microglial subset plays a key role in myelinogenesis in developing brain. EMBO J. 2017, 36, 3292–3308. [Google Scholar] [CrossRef]
- Staszewski, O.; Hagemeyer, N. Unique microglia expression profile in developing white matter. BMC Res. Notes 2019, 12, 367. [Google Scholar] [CrossRef]
- Czapski, G.A.; Strosznajder, J.B. Glutamate and GABA in Microglia-Neuron Cross-Talk in Alzheimer’s Disease. Int. J. Mol. Sci. 2021, 22, 11677. [Google Scholar] [CrossRef]
- Tsay, H.J.; Liu, H.K.; Kuo, Y.H.; Chiu, C.S.; Liang, C.C.; Chung, C.W.; Chen, C.C.; Chen, Y.P.; Shiao, Y.J. EK100 and Antrodin C Improve Brain Amyloid Pathology in APP/PS1 Transgenic Mice by Promoting Microglial and Perivascular Clearance Pathways. Int. J. Mol. Sci. 2021, 22, 413. [Google Scholar] [CrossRef]
- Soriano, S.; Curry, K.; Wang, Q.; Chow, E.; Treangen, T.J.; Villapol, S. Fecal Microbiota Transplantation Derived from Alzheimer’s Disease Mice Worsens Brain Trauma Outcomes in Wild-Type Controls. Int. J. Mol. Sci. 2022, 23, 4476. [Google Scholar] [CrossRef]
- Calder, P.C. Fatty acids and inflammation: The cutting edge between food and pharma. Eur. J. Pharmacol. 2011, 668 (Suppl. 1), S50–S58. [Google Scholar] [CrossRef]
- Satari, B.; Karimi, K. Mucoralean fungi for sustainable production of bioethanol and biologically active molecules. Appl. Microbiol. Biotechnol. 2018, 102, 1097–1117. [Google Scholar] [CrossRef] [PubMed]
- Shahidi, F.; Ambigaipalan, P. Omega-3 Polyunsaturated Fatty Acids and Their Health Benefits. Annu. Rev. Food Sci. Technol. 2018, 9, 345–381. [Google Scholar] [CrossRef] [PubMed]
- Kumar, N.G.; Contaifer, D.; Madurantakam, P.; Carbone, S.; Price, E.T.; Van Tassell, B.; Brophy, D.F.; Wijesinghe, D.S. Dietary Bioactive Fatty Acids as Modulators of Immune Function: Implications on Human Health. Nutrients 2019, 11, 2974. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sokoła-Wysoczańska, E.; Wysoczański, T.; Wagner, J.; Czyż, K.; Bodkowski, R.; Lochyński, S.; Patkowska-Sokoła, B. Polyunsaturated Fatty Acids and Their Potential Therapeutic Role in Cardiovascular System Disorders—A Review. Nutrients 2018, 10, 1561. [Google Scholar] [CrossRef] [Green Version]
- Calder, P.C. Marine omega-3 fatty acids and inflammatory processes: Effects, mechanisms and clinical relevance. Biochim. Biophys. Acta 2015, 1851, 469–484. [Google Scholar] [CrossRef]
- Calder, P.C. Omega-3 fatty acids and inflammatory processes: From molecules to man. Biochem. Soc. Trans. 2017, 45, 1105–1115. [Google Scholar] [CrossRef] [Green Version]
- Braverman, N.E.; Moser, A.B. Functions of plasmalogen lipids in health and disease. Biochim. Biophys. Acta 2012, 1822, 1442–1452. [Google Scholar] [CrossRef] [Green Version]
- Nadjar, A.; Leyrolle, Q.; Joffre, C.; Laye, S. Bioactive lipids as new class of microglial modulators: When nutrition meets neuroimunology. Prog Neuropsychopharm. Biol Psychiatry 2017, 79, 19–26. [Google Scholar] [CrossRef]
- Bazinet, R.P.; Layé, S. Polyunsaturated fatty acids and their metabolites in brain function and disease. Nat. Rev. Neurosci. 2014, 15, 771–785. [Google Scholar] [CrossRef]
- Simopoulos, A.P. Evolutionary aspects of diet: The omega-6/omega-3 ratio and the brain. Mol. Neurobiol. 2011, 44, 203–215. [Google Scholar] [CrossRef]
- Weiser, M.J.; Butt, C.M.; Mohajeri, M.H. Docosahexaenoic Acid and Cognition throughout the Lifespan. Nutrients 2016, 8, 99. [Google Scholar] [CrossRef]
- Layé, S.; Nadjar, A.; Joffre, C.; Bazinet, R.P. Anti-Inflammatory Effects of Omega-3 Fatty Acids in the Brain: Physiological Mechanisms and Relevance to Pharmacology. Pharmacol. Rev. 2018, 70, 12–38. [Google Scholar] [CrossRef] [PubMed]
- Rey, C.; Nadjar, A.; Joffre, F.; Amadieu, C.; Aubert, A.; Vaysse, C.; Pallet, V.; Layé, S.; Joffre, C. Maternal n-3 polyunsaturated fatty acid dietary supply modulates microglia lipid content in the offspring. Prostaglandins Leukot Essent Fat. Acids 2018, 133, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Madore, C.; Leyrolle, Q.; Morel, L.; Rossitto, M.; Greenhalgh, A.D.; Delpech, J.C.; Martinat, M.; Bosch-Bouju, C.; Bourel, J.; Rani, B.; et al. Essential omega-3 fatty acids tune microglial phagocytosis of synaptic elements in the mouse developing brain. Nat. Commun. 2020, 11, 6133. [Google Scholar] [CrossRef] [PubMed]
- Rey, C.; Nadjar, A.; Buaud, B.; Vaysse, C.; Aubert, A.; Pallet, V.; Layé, S.; Joffre, C. Resolvin D1 and E1 promote resolution of inflammation in microglial cells in vitro. Brain Behav. Immun. 2016, 55, 249–259. [Google Scholar] [CrossRef] [PubMed]
- Inoue, T.; Tanaka, M.; Masuda, S.; Ohue-Kitano, R.; Yamakage, H.; Muranaka, K.; Wada, H.; Kusakabe, T.; Shimatsu, A.; Hasegawa, K. Omega-3 polyunsaturated fatty acids suppress the inflammatory responses of lipopolysaccharide-stimulated mouse microglia by activating SIRT1 pathways. Biochim. Biophys. Acta (BBA) Mol. Cell Biol. Lipids 2017, 1862, 552–560. [Google Scholar] [CrossRef] [PubMed]
- Antonietta Ajmone-Cat, M.; Lavinia Salvatori, M.; De Simone, R.; Mancini, M.; Biagioni, S.; Bernardo, A.; Cacci, E.; Minghetti, L. Docosahexaenoic acid modulates inflammatory and antineurogenic functions of activated microglial cells. J. Neurosci. Res. 2012, 90, 575–587. [Google Scholar] [CrossRef]
- De Smedt-Peyrusse, V.; Sargueil, F.; Moranis, A.; Harizi, H.; Mongrand, S.; Layé, S. Docosahexaenoic acid prevents lipopolysaccharide-induced cytokine production in microglial cells by inhibiting lipopolysaccharide receptor presentation but not its membrane subdomain localization. J. Neurochem. 2008, 105, 296–307. [Google Scholar] [CrossRef]
- Ghosh, S.; Castillo, E.; Frias, E.S.; Swanson, R.A. Bioenergetic regulation of microglia. Glia 2018, 66, 1200–1212. [Google Scholar] [CrossRef]
- Longo, V.D.; Mattson, M.P. Fasting: Molecular mechanisms and clinical applications. Cell Metab. 2014, 19, 181–192. [Google Scholar] [CrossRef] [Green Version]
- Fu, S.P.; Wang, J.F.; Xue, W.J.; Liu, H.M.; Liu, B.R.; Zeng, Y.L.; Li, S.N.; Huang, B.X.; Lv, Q.K.; Wang, W.; et al. Anti-inflammatory effects of BHBA in both in vivo and in vitro Parkinson’s disease models are mediated by GPR109A-dependent mechanisms. J. Neuroinflamm. 2015, 12, 9. [Google Scholar] [CrossRef] [Green Version]
- Heppner, F.L.; Ransohoff, R.M.; Becher, B. Immune attack: The role of inflammation in Alzheimer disease. Nat. Rev. Neurosci. 2015, 16, 358–372. [Google Scholar] [CrossRef] [PubMed]
- Breunig, J.J.; Guillot-Sestier, M.V.; Town, T. Brain injury, neuroinflammation and Alzheimer’s disease. Front. Aging Neurosci. 2013, 5, 26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quero, L.; Hanser, E.; Manigold, T.; Tiaden, A.N.; Kyburz, D. TLR2 stimulation impairs anti-inflammatory activity of M2-like macrophages, generating a chimeric M1/M2 phenotype. Arthritis Res. Ther. 2017, 19, 245. [Google Scholar] [CrossRef] [Green Version]
- Madore, C.; Nadjar, A.; Delpech, J.-C.; Séré, A.; Aubert, A.; Portal, C.; Joffre, C.; Layé, S. Nutritional n-3 PUFAs deficiency during perinatal periods alters brain innate immune system and neuronal plasticity-associated genes. Brain Behav. Immun. 2014, 41, 22–31. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Roso, M.B.; Olivares-Álvaro, E.; Quintela, J.C.; Ballesteros, S.; Espinosa-Parrilla, J.F.; Ruiz-Roso, B.; Lahera, V.; de Las Heras, N.; Martín-Fernández, B. Effects of Low Phytanic Acid-Concentrated DHA on Activated Microglial Cells: Comparison with a Standard Phytanic Acid-Concentrated DHA. Neuromol. Med. 2018, 20, 328–342. [Google Scholar] [CrossRef]
- Gibson, R.A.; Neumann, M.A.; Lien, E.L.; Boyd, K.A.; Tu, W.C. Docosahexaenoic acid synthesis from alpha-linolenic acid is inhibited by diets high in polyunsaturated fatty acids. Prostaglandins Leukot Essent Fat. Acids 2013, 88, 139–146. [Google Scholar] [CrossRef]
- Lauritzen, L.; Hansen, H.S.; Jørgensen, M.H.; Michaelsen, K.F. The essentiality of long chain n-3 fatty acids in relation to development and function of the brain and retina. Prog. Lipid. Res. 2001, 40, 1–94. [Google Scholar] [CrossRef]
- Chang, C.Y.; Wu, C.C.; Wang, J.D.; Li, J.R.; Wang, Y.Y.; Lin, S.Y.; Chen, W.Y.; Liao, S.L.; Chen, C.J. DHA attenuated Japanese Encephalitis virus infection-induced neuroinflammation and neuronal cell death in cultured rat Neuron/glia. Brain Behav. Immun. 2021, 93, 194–205. [Google Scholar] [CrossRef]
- Hsieh, J.T.; St John, A.L. Japanese encephalitis virus and its mechanisms of neuroinvasion. PLoS Pathog. 2020, 16, e1008260. [Google Scholar] [CrossRef]
- Yakass, M.B.; Franco, D.; Quaye, O. Suppressors of Cytokine Signaling and Protein Inhibitors of Activated Signal Transducer and Activator of Transcriptions As Therapeutic Targets in Flavivirus Infections. J. Interferon Cytokine Res. 2020, 40, 1–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kao, T.K.; Ou, Y.C.; Lin, S.Y.; Pan, H.C.; Song, P.J.; Raung, S.L.; Lai, C.Y.; Liao, S.L.; Lu, H.C.; Chen, C.J. Luteolin inhibits cytokine expression in endotoxin/cytokine-stimulated microglia. J. Nutr. Biochem. 2011, 22, 612–624. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Chen, C.; Fan, S.; Wu, S.; Yang, F.; Fang, Z.; Fu, H.; Li, Y. Omega-3 polyunsaturated fatty acid attenuates the inflammatory response by modulating microglia polarization through SIRT1-mediated deacetylation of the HMGB1/NF-κB pathway following experimental traumatic brain injury. J. Neuroinflamm. 2018, 15, 116. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Wang, X.; Ashraf, U.; Zheng, B.; Ye, J.; Zhou, D.; Zhang, H.; Song, Y.; Chen, H.; Zhao, S.; et al. Activation of neuronal N-methyl-D-aspartate receptor plays a pivotal role in Japanese encephalitis virus-induced neuronal cell damage. J. Neuroinflamm. 2018, 15, 238. [Google Scholar] [CrossRef]
- Chataigner, M.; Martin, M.; Lucas, C.; Pallet, V.; Layé, S.; Mehaignerie, A.; Bouvret, E.; Dinel, A.L.; Joffre, C. Fish Hydrolysate Supplementation Containing n-3 Long Chain Polyunsaturated Fatty Acids and Peptides Prevents LPS-Induced Neuroinflammation. Nutrients 2021, 13, 824. [Google Scholar] [CrossRef]
- Cochet, F.; Peri, F. The Role of Carbohydrates in the Lipopolysaccharide (LPS)/Toll-Like Receptor 4 (TLR4) Signalling. Int. J. Mol. Sci. 2017, 18, 2318. [Google Scholar] [CrossRef] [Green Version]
- Tyrtyshnaia, A.; Konovalova, S.; Bondar, A.; Ermolenko, E.; Sultanov, R.; Manzhulo, I. Anti-Inflammatory Activity of N-Docosahexaenoylethanolamine and N-Eicosapentaenoylethanolamine in a Mouse Model of Lipopolysaccharide-Induced Neuroinflammation. Int. J. Mol. Sci. 2021, 22, 728. [Google Scholar] [CrossRef]
- Liu, B.; Zhang, Y.; Yang, Z.; Liu, M.; Zhang, C.; Zhao, Y.; Song, C. ω-3 DPA Protected Neurons from Neuroinflammation by Balancing Microglia M1/M2 Polarizations through Inhibiting NF-κB/MAPK p38 Signaling and Activating Neuron-BDNF-PI3K/AKT Pathways. Mar. Drugs 2021, 19, 587. [Google Scholar] [CrossRef]
- Hulsebosch, C.E. Recent advances in pathophysiology and treatment of spinal cord injury. Adv. Physiol. Educ. 2002, 26, 238–255. [Google Scholar] [CrossRef]
- Amo-Aparicio, J.; Martínez-Muriana, A.; Sánchez-Fernández, A.; López-Vales, R. Neuroinflammation Quantification for Spinal Cord Injury. Curr. Protoc. Immunol. 2018, 123, e57. [Google Scholar] [CrossRef]
- Manzhulo, O.; Tyrtyshnaia, A.; Kipryushina, Y.; Dyuizen, I.; Manzhulo, I. Docosahexaenoic acid induces changes in microglia/macrophage polarization after spinal cord injury in rats. Acta Histochem. 2018, 120, 741–747. [Google Scholar] [CrossRef]
- Yip, P.K.; Bowes, A.L.; Hall, J.C.; Burguillos, M.A.; Ip, T.R.; Baskerville, T.; Liu, Z.-H.; Mohamed, M.A.; Getachew, F.; Lindsay, A.D. Docosahexaenoic acid reduces microglia phagocytic activity via miR-124 and induces neuroprotection in rodent models of spinal cord contusion injury. Hum. Mol. Genet. 2019, 28, 2427–2448. [Google Scholar] [CrossRef]
- Srivastava, S.; Ahmad, R.; Khare, S.K. Alzheimer’s disease and its treatment by different approaches: A review. Eur. J. Med. Chem. 2021, 216, 113320. [Google Scholar] [CrossRef]
- Battaglia, S.; Thayer, J.F. Functional interplay between central and autonomic nervous systems in human fear conditioning. Trends Neurosci. 2022, 45, 504–506. [Google Scholar] [CrossRef]
- Battaglia, S.; Orsolini, S.; Borgomaneri, S.; Barbieri, R.; Diciotti, S.; di Pellegrino, G. Characterizing cardiac autonomic dynamics of fear learning in humans. Psychophysiology 2022, e14122. [Google Scholar] [CrossRef]
- Tanaka, M.; Toldi, J.; Vécsei, L. Exploring the etiological links behind neurodegenerative diseases: Inflammatory cytokines and bioactive kynurenines. Int. J. Mol. Sci. 2020, 21, 2431. [Google Scholar] [CrossRef] [Green Version]
- Török, N.; Tanaka, M.; Vécsei, L. Searching for Peripheral Biomarkers in Neurodegenerative Diseases: The Tryptophan-Kynurenine Metabolic Pathway. Int. J. Mol. Sci. 2020, 21, 9338. [Google Scholar] [CrossRef]
- González de San Román, E.; Llorente-Ovejero, A.; Martínez-Gardeazabal, J.; Moreno-Rodríguez, M.; Giménez-Llort, L.; Manuel, I.; Rodríguez-Puertas, R. Modulation of Neurolipid Signaling and Specific Lipid Species in the Triple Transgenic Mouse Model of Alzheimer’s Disease. Int. J. Mol. Sci. 2021, 22, 12256. [Google Scholar] [CrossRef]
- Akiyama, H.; Barger, S.; Barnum, S.; Bradt, B.; Bauer, J.; Cole, G.M.; Cooper, N.R.; Eikelenboom, P.; Emmerling, M.; Fiebich, B.L.; et al. Inflammation and Alzheimer’s disease. Neurobiol. Aging 2000, 21, 383–421. [Google Scholar] [CrossRef]
- Stewart, C.R.; Stuart, L.M.; Wilkinson, K.; van Gils, J.M.; Deng, J.; Halle, A.; Rayner, K.J.; Boyer, L.; Zhong, R.; Frazier, W.A.; et al. CD36 ligands promote sterile inflammation through assembly of a Toll-like receptor 4 and 6 heterodimer. Nat. Immunol. 2010, 11, 155–161. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.C.; Zheng, M.H.; Du, Y.L.; Wang, L.; Kuang, F.; Qin, H.Y.; Zhang, B.F.; Han, H. N9 microglial cells polarized by LPS and IL4 show differential responses to secondary environmental stimuli. Cell Immunol. 2012, 278, 84–90. [Google Scholar] [CrossRef] [PubMed]
- Wirz, K.T.; Bossers, K.; Stargardt, A.; Kamphuis, W.; Swaab, D.F.; Hol, E.M.; Verhaagen, J. Cortical beta amyloid protein triggers an immune response, but no synaptic changes in the APPswe/PS1dE9 Alzheimer’s disease mouse model. Neurobiol. Aging 2013, 34, 1328–1342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Solito, E.; Sastre, M. Microglia function in Alzheimer’s disease. Front. Pharmacol. 2012, 3, 14. [Google Scholar] [CrossRef] [Green Version]
- Jiang, T.; Zhang, Y.D.; Chen, Q.; Gao, Q.; Zhu, X.C.; Zhou, J.S.; Shi, J.Q.; Lu, H.; Tan, L.; Yu, J.T. TREM2 modifies microglial phenotype and provides neuroprotection in P301S tau transgenic mice. Neuropharmacology 2016, 105, 196–206. [Google Scholar] [CrossRef] [PubMed]
- McDonald, D.R.; Brunden, K.R.; Landreth, G.E. Amyloid fibrils activate tyrosine kinase-dependent signaling and superoxide production in microglia. J. Neurosci. 1997, 17, 2284–2294. [Google Scholar] [CrossRef] [PubMed]
- Lue, L.F.; Kuo, Y.M.; Beach, T.; Walker, D.G. Microglia activation and anti-inflammatory regulation in Alzheimer’s disease. Mol. Neurobiol. 2010, 41, 115–128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Battaglia, S.; Garofalo, S.; di Pellegrino, G. Context-dependent extinction of threat memories: Influences of healthy aging. Sci. Rep. 2018, 8, 12592. [Google Scholar] [CrossRef] [PubMed]
- Díaz, M.; Mesa-Herrera, F.; Marín, R. DHA and Its Elaborated Modulation of Antioxidant Defenses of the Brain: Implications in Aging and AD Neurodegeneration. Antioxidants 2021, 10, 907. [Google Scholar] [CrossRef]
- Hjorth, E.; Zhu, M.; Toro, V.C.; Vedin, I.; Palmblad, J.; Cederholm, T.; Freund-Levi, Y.; Faxen-Irving, G.; Wahlund, L.O.; Basun, H.; et al. Omega-3 fatty acids enhance phagocytosis of Alzheimer’s disease-related amyloid-β42 by human microglia and decrease inflammatory markers. J. Alzheimer’s Dis. 2013, 35, 697–713. [Google Scholar] [CrossRef] [Green Version]
- Galloway, D.A.; Phillips, A.E.M.; Owen, D.R.J.; Moore, C.S. Phagocytosis in the Brain: Homeostasis and Disease. Front. Immunol. 2019, 10, 790. [Google Scholar] [CrossRef] [Green Version]
- Jiao, S.S.; Shen, L.L.; Zhu, C.; Bu, X.L.; Liu, Y.H.; Liu, C.H.; Yao, X.Q.; Zhang, L.L.; Zhou, H.D.; Walker, D.G.; et al. Brain-derived neurotrophic factor protects against tau-related neurodegeneration of Alzheimer’s disease. Transl. Psychiatry 2016, 6, e907. [Google Scholar] [CrossRef] [PubMed]
- Ng, T.K.S.; Ho, C.S.H.; Tam, W.W.S.; Kua, E.H.; Ho, R.C. Decreased Serum Brain-Derived Neurotrophic Factor (BDNF) Levels in Patients with Alzheimer’s Disease (AD): A Systematic Review and Meta-Analysis. Int. J. Mol. Sci. 2019, 20, 257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Velagapudi, R.; El-Bakoush, A.; Lepiarz, I.; Ogunrinade, F.; Olajide, O.A. AMPK and SIRT1 activation contribute to inhibition of neuroinflammation by thymoquinone in BV2 microglia. Mol. Cell. Biochem. 2017, 435, 149–162. [Google Scholar] [CrossRef]
- Chen, J.; Zhou, Y.; Mueller-Steiner, S.; Chen, L.F.; Kwon, H.; Yi, S.; Mucke, L.; Gan, L. SIRT1 protects against microglia-dependent amyloid-beta toxicity through inhibiting NF-kappaB signaling. J. Biol. Chem. 2005, 280, 40364–40374. [Google Scholar] [CrossRef] [Green Version]
- Dong, Y.; Long, S.; Gu, Y.; Liu, W. Eicosapentaenoic acid attenuates Aβ-induced neurotoxicity by decreasing neuroinflammation through regulation of microglial polarization. Neuro. Endocrinol. Lett. 2021, 42, 91–98. [Google Scholar] [PubMed]
- Joly-Amado, A.; Hunter, J.; Quadri, Z.; Zamudio, F.; Rocha-Rangel, P.V.; Chan, D.; Kesarwani, A.; Nash, K.; Lee, D.C.; Morgan, D.; et al. CCL2 Overexpression in the Brain Promotes Glial Activation and Accelerates Tau Pathology in a Mouse Model of Tauopathy. Front. Immunol. 2020, 11, 997. [Google Scholar] [CrossRef] [PubMed]
- Ju, L.; Zeng, H.; Chen, Y.; Wu, Y.; Wang, B.; Xu, Q. Dual polarization of microglia isolated from mixed glial cell cultures. J. Neurosci. Res. 2015, 93, 1345–1352. [Google Scholar] [CrossRef]
- Ghosh, M.; Xu, Y.; Pearse, D.D. Cyclic AMP is a key regulator of M1 to M2a phenotypic conversion of microglia in the presence of Th2 cytokines. J. Neuroinflamm. 2016, 13, 9. [Google Scholar] [CrossRef] [Green Version]
- Walton, C.; King, R.; Rechtman, L.; Kaye, W.; Leray, E.; Marrie, R.A.; Robertson, N.; La Rocca, N.; Uitdehaag, B.; van der Mei, I.; et al. Rising prevalence of multiple sclerosis worldwide: Insights from the Atlas of MS, third edition. Mult. Scler. J. 2020, 26, 1816–1821. [Google Scholar] [CrossRef]
- Sospedra, M.; Martin, R. Immunology of multiple sclerosis. Annu. Rev. Immunol. 2005, 23, 683–747. [Google Scholar] [CrossRef] [Green Version]
- Steinman, L. Immunology of relapse and remission in multiple sclerosis. Annu. Rev. Immunol. 2014, 32, 257–281. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.J.; Chen, W.W.; Zhang, X. Multiple sclerosis: Pathology, diagnosis and treatments. Exp. Ther. Med. 2017, 13, 3163–3166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frischer, J.M.; Weigand, S.D.; Guo, Y.; Kale, N.; Parisi, J.E.; Pirko, I.; Mandrekar, J.; Bramow, S.; Metz, I.; Brück, W.; et al. Clinical and pathological insights into the dynamic nature of the white matter multiple sclerosis plaque. Ann. Neurol. 2015, 78, 710–721. [Google Scholar] [CrossRef] [PubMed]
- Giovannoni, G.; Popescu, V.; Wuerfel, J.; Hellwig, K.; Iacobaeus, E.; Jensen, M.B.; García-Domínguez, J.M.; Sousa, L.; De Rossi, N.; Hupperts, R.; et al. Smouldering multiple sclerosis: The ‘real MS’. Ther. Adv. Neurol. Disord. 2022, 15, 17562864211066751. [Google Scholar] [CrossRef]
- Tanaka, M.; Vécsei, L. Editorial of Special Issue Crosstalk between Depression, Anxiety, and Dementia: Comorbidity in Behavioral Neurology and Neuropsychiatry. Biomedicines 2021, 9, 517. [Google Scholar] [CrossRef]
- Battaglia, S.; Fabius, J.H.; Moravkova, K.; Fracasso, A.; Borgomaneri, S. The Neurobiological Correlates of Gaze Perception in Healthy Individuals and Neurologic Patients. Biomedicines 2022, 10, 627. [Google Scholar] [CrossRef]
- Ellena, G.; Battaglia, S.; Làdavas, E. The spatial effect of fearful faces in the autonomic response. Exp. Brain Res. 2020, 238, 2009–2018. [Google Scholar] [CrossRef]
- Luo, C.; Jian, C.; Liao, Y.; Huang, Q.; Wu, Y.; Liu, X.; Zou, D.; Wu, Y. The role of microglia in multiple sclerosis. Neuropsychiatr. Dis. Treat. 2017, 13, 1661–1667. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.; Zhang, H.; Pu, H.; Wang, G.; Li, W.; Leak, R.K.; Chen, J.; Liou, A.K.; Hu, X. n-3 PUFA supplementation benefits microglial responses to myelin pathology. Sci. Rep. 2014, 4, 7458. [Google Scholar] [CrossRef]
- Dal Monte, M.; Cammalleri, M.; Locri, F.; Amato, R.; Marsili, S.; Rusciano, D.; Bagnoli, P. Fatty Acids Dietary Supplements Exert Anti-Inflammatory Action and Limit Ganglion Cell Degeneration in the Retina of the EAE Mouse Model of Multiple Sclerosis. Nutrients 2018, 10, 325. [Google Scholar] [CrossRef] [Green Version]
- Horstmann, L.; Schmid, H.; Heinen, A.P.; Kurschus, F.C.; Dick, H.B.; Joachim, S.C. Inflammatory demyelination induces glia alterations and ganglion cell loss in the retina of an experimental autoimmune encephalomyelitis model. J. Neuroinflamm. 2013, 10, 120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Locri, F.; Cammalleri, M.; Pini, A.; Dal Monte, M.; Rusciano, D.; Bagnoli, P. Further Evidence on Efficacy of Diet Supplementation with Fatty Acids in Ocular Pathologies: Insights from the EAE Model of Optic Neuritis. Nutrients 2018, 10, 1447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilsey, L.J.; Fortune, B. Electroretinography in glaucoma diagnosis. Curr. Opin. Ophthalmol. 2016, 27, 118–124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hossain, D.M.; Panda, A.K.; Manna, A.; Mohanty, S.; Bhattacharjee, P.; Bhattacharyya, S.; Saha, T.; Chakraborty, S.; Kar, R.K.; Das, T.; et al. Retracted: FoxP3 acts as a cotranscription factor with STAT3 in tumor-induced regulatory T cells. Immunity 2013, 39, 1057–1069. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tugal, D.; Liao, X.; Jain, M.K. Transcriptional control of macrophage polarization. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 1135–1144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.C. NF-κB signaling in inflammation. Signal Transduct. Target Ther. 2017, 2, 17023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ponath, G.; Park, C.; Pitt, D. The Role of Astrocytes in Multiple Sclerosis. Front. Immunol. 2018, 9, 217. [Google Scholar] [CrossRef]
- Rossi, M.; Petralla, S.; Protti, M.; Baiula, M.; Kobrlova, T.; Soukup, O.; Spampinato, S.M.; Mercolini, L.; Monti, B.; Bolognesi, M.L. α-Linolenic Acid-Valproic Acid Conjugates: Toward Single-Molecule Polypharmacology for Multiple Sclerosis. ACS Med. Chem. Lett. 2020, 11, 2406–2413. [Google Scholar] [CrossRef]
- Skaper, S.D. Oligodendrocyte precursor cells as a therapeutic target for demyelinating diseases. Prog. Brain Res. 2019, 245, 119–144. [Google Scholar] [CrossRef]
- Zhang, N.; Jin, L.; Liu, C.; Zhang, R.; Siebert, H.-C.; Li, Y.; Loers, G.; Petridis, A.K.; Xia, Z.; Dong, H. An antarctic krill oil-based diet elicits neuroprotective effects by inhibiting oxidative stress and rebalancing the M1/M2 microglia phenotype in a cuprizone model for demyelination. J. Funct. Foods 2021, 76, 104309. [Google Scholar] [CrossRef]
- Xia, M.; Zhao, Q.; Zhang, H.; Chen, Y.; Yuan, Z.; Xu, Y.; Zhang, M. Proteomic Analysis of HDAC3 Selective Inhibitor in the Regulation of Inflammatory Response of Primary Microglia. Neural Plast. 2017, 2017, 6237351. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Liu, C.; Zhang, R.; Jin, L.; Yin, X.; Zheng, X.; Siebert, H.C.; Li, Y.; Wang, Z.; Loers, G.; et al. Amelioration of clinical course and demyelination in the cuprizone mouse model in relation to ketogenic diet. Food Funct. 2020, 11, 5647–5663. [Google Scholar] [CrossRef] [PubMed]
- Haberland, M.; Montgomery, R.L.; Olson, E.N. The many roles of histone deacetylases in development and physiology: Implications for disease and therapy. Nat. Rev. Genet. 2009, 10, 32–42. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Barozzi, I.; Termanini, A.; Prosperini, E.; Recchiuti, A.; Dalli, J.; Mietton, F.; Matteoli, G.; Hiebert, S.; Natoli, G. Requirement for the histone deacetylase Hdac3 for the inflammatory gene expression program in macrophages. Proc. Natl. Acad. Sci. USA 2012, 109, E2865–E2874. [Google Scholar] [CrossRef] [Green Version]
- Frisullo, G.; Angelucci, F.; Caggiula, M.; Nociti, V.; Iorio, R.; Patanella, A.K.; Sancricca, C.; Mirabella, M.; Tonali, P.A.; Batocchi, A.P. pSTAT1, pSTAT3, and T-bet expression in peripheral blood mononuclear cells from relapsing-remitting multiple sclerosis patients correlates with disease activity. J. Neurosci. Res. 2006, 84, 1027–1036. [Google Scholar] [CrossRef]
- Jakkula, E.; Leppä, V.; Sulonen, A.M.; Varilo, T.; Kallio, S.; Kemppinen, A.; Purcell, S.; Koivisto, K.; Tienari, P.; Sumelahti, M.L.; et al. Genome-wide association study in a high-risk isolate for multiple sclerosis reveals associated variants in STAT3 gene. Am. J. Hum. Genet. 2010, 86, 285–291. [Google Scholar] [CrossRef] [Green Version]
- Bai, B.; Yan, Z.; Hao, Y.; Zhang, Z.; Li, G.; Dekker, J.; Qiu, C. A randomised controlled multimodal intervention trial in patients with ischaemic stroke in Shandong, China: Design and rationale. Lancet 2017, 390, S13. [Google Scholar] [CrossRef] [Green Version]
- Ekdahl, C.T.; Claasen, J.H.; Bonde, S.; Kokaia, Z.; Lindvall, O. Inflammation is detrimental for neurogenesis in adult brain. Proc. Natl. Acad. Sci. USA 2003, 100, 13632–13637. [Google Scholar] [CrossRef] [Green Version]
- Xiong, X.Y.; Liu, L.; Yang, Q.W. Functions and mechanisms of microglia/macrophages in neuroinflammation and neurogenesis after stroke. Prog. Neurobiol. 2016, 142, 23–44. [Google Scholar] [CrossRef]
- Zhu, H.; Hu, S.; Li, Y.; Sun, Y.; Xiong, X.; Hu, X.; Chen, J.; Qiu, S. Interleukins and ischemic stroke. Front. Immunol. 2022, 188, 828447. [Google Scholar] [CrossRef]
- Virani, S.S.; Alonso, A.; Aparicio, H.J.; Benjamin, E.J.; Bittencourt, M.S.; Callaway, C.W.; Carson, A.P.; Chamberlain, A.M.; Cheng, S.; Delling, F.N.; et al. Heart Disease and Stroke Statistics-2021 Update: A Report From the American Heart Association. Circulation 2021, 143, e254–e743. [Google Scholar] [CrossRef] [PubMed]
- Zendedel, A.; Habib, P.; Dang, J.; Lammerding, L.; Hoffmann, S.; Beyer, C.; Slowik, A. Omega-3 polyunsaturated fatty acids ameliorate neuroinflammation and mitigate ischemic stroke damage through interactions with astrocytes and microglia. J. Neuroimmunol. 2015, 278, 200–211. [Google Scholar] [CrossRef] [PubMed]
- Kaur, C.; Sivakumar, V.; Zhang, Y.; Ling, E.A. Hypoxia-induced astrocytic reaction and increased vascular permeability in the rat cerebellum. Glia 2006, 54, 826–839. [Google Scholar] [CrossRef] [PubMed]
- Lin, B. Encephalopathy: A vicious cascade following forebrain ischemia and hypoxia. Cent. Nerv. Syst. Agents Med. Chem. 2013, 13, 57–70. [Google Scholar] [CrossRef]
- Li, Y.; Han, F.; Shi, Y. Increased neuronal apoptosis in medial prefrontal cortex is accompanied with changes of Bcl-2 and Bax in a rat model of post-traumatic stress disorder. J. Mol. Neurosci. 2013, 51, 127–137. [Google Scholar] [CrossRef]
- Jiang, X.; Pu, H.; Hu, X.; Wei, Z.; Hong, D.; Zhang, W.; Gao, Y.; Chen, J.; Shi, Y. A Post-stroke Therapeutic Regimen with Omega-3 Polyunsaturated Fatty Acids that Promotes White Matter Integrity and Beneficial Microglial Responses after Cerebral Ischemia. Transl. Stroke Res. 2016, 7, 548–561. [Google Scholar] [CrossRef]
- Franklin, R.J.; Ffrench-Constant, C. Remyelination in the CNS: From biology to therapy. Nat. Rev. Neurosci. 2008, 9, 839–855. [Google Scholar] [CrossRef]
- Han, L.; Cai, W.; Mao, L.; Liu, J.; Li, P.; Leak, R.K.; Xu, Y.; Hu, X.; Chen, J. Rosiglitazone Promotes White Matter Integrity and Long-Term Functional Recovery After Focal Cerebral Ischemia. Stroke 2015, 46, 2628–2636. [Google Scholar] [CrossRef] [Green Version]
- Fancy, S.P.; Harrington, E.P.; Baranzini, S.E.; Silbereis, J.C.; Shiow, L.R.; Yuen, T.J.; Huang, E.J.; Lomvardas, S.; Rowitch, D.H. Parallel states of pathological Wnt signaling in neonatal brain injury and colon cancer. Nat. Neurosci. 2014, 17, 506–512. [Google Scholar] [CrossRef] [Green Version]
- Cai, W.; Liu, S.; Hu, M.; Sun, X.; Qiu, W.; Zheng, S.; Hu, X.; Lu, Z. Post-stroke DHA Treatment Protects Against Acute Ischemic Brain Injury by Skewing Macrophage Polarity Toward the M2 Phenotype. Transl. Stroke Res. 2018, 9, 669–680. [Google Scholar] [CrossRef]
- Horváth, E.; Huțanu, A.; Orădan, A.; Chiriac, L.; Muntean, D.L.; Nagy, E.-E.; Dobreanu, M. N-3 polyunsaturated fatty acids induce granulopoiesis and early monocyte polarization in the bone marrow of a tMCAO rat model. Rev. Romana Med. Lab. 2019, 27, 51–61. [Google Scholar] [CrossRef] [Green Version]
- Horváth, E.; Orădan, A.; Chiriac, L.; Dobreanu, M.; Nagy, E.-E.; Voidăzan, S.; Berei, R.; Muntean, D.L.; Huțanu, A. Fish-oil preconditioning up-regulates expression of splenic ARG1positive M2 type macrophages and the ARG1/INOS2 ratio after experimental induced transient cerebral ischemia. Farmacia 2019, 67, 820–829. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Huang, Y.; Gao, T.; Zhang, T.; Hao, Y.; Hu, Q.; Han, Y.; Xu, Z.; Wang, D.; Zhang, J. DHA Plays a Protective Role in Cerebral Ischemia-Reperfusion Injury by Affecting Macrophage/Microglia Type Polarization. 2021. Available online: https://ssrn.com/abstract=3937113 (accessed on 6 October 2021).
- Fleminger, S.; Oliver, D.L.; Lovestone, S.; Rabe-Hesketh, S.; Giora, A. Head injury as a risk factor for Alzheimer’s disease: The evidence 10 years on; a partial replication. J. Neurol. Neurosurg. Psychiatry 2003, 74, 857–862. [Google Scholar] [CrossRef] [PubMed]
- Sivanandam, T.M.; Thakur, M.K. Traumatic brain injury: A risk factor for Alzheimer’s disease. Neurosci. Biobehav. Rev. 2012, 36, 1376–1381. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.J.; Yang, M.S.; Zhang, B.; Niu, F.; Dong, J.Q.; Liu, B.Y. Glucose metabolism: A link between traumatic brain injury and Alzheimer’s disease. Chin. J. Traumatol. 2021, 24, 5–10. [Google Scholar] [CrossRef]
- Jafari, S.; Etminan, M.; Aminzadeh, F.; Samii, A. Head injury and risk of Parkinson disease: A systematic review and meta-analysis. Mov. Disord. 2013, 28, 1222–1229. [Google Scholar] [CrossRef]
- Gardner, R.C.; Burke, J.F.; Nettiksimmons, J.; Goldman, S.; Tanner, C.M.; Yaffe, K. Traumatic brain injury in later life increases risk for Parkinson disease. Ann. Neurol. 2015, 77, 987–995. [Google Scholar] [CrossRef]
- Kang, J.H.; Lin, H.C. Increased risk of multiple sclerosis after traumatic brain injury: A nationwide population-based study. J. Neurotrauma 2012, 29, 90–95. [Google Scholar] [CrossRef] [Green Version]
- Gu, D.; Ou, S.; Tang, M.; Yin, Z.; Wang, Z.; Liu, G. Trauma and amyotrophic lateral sclerosis: A systematic review and meta-analysis. Amyotroph. Lateral Scler. Front. Degener. 2021, 22, 170–185. [Google Scholar] [CrossRef]
- Peterson, A.B.; Xu, L.; Daugherty, J.; Breiding, M.J. Surveillance Report of Traumatic Brain Injury-Related Emergency Department Visits, Hospitalizations, and Deaths, United States, 2014. 2019. Available online: https://stacks.cdc.gov/view/cdc/78062 (accessed on 3 June 2022).
- Harvey, L.D.; Yin, Y.; Attarwala, I.Y.; Begum, G.; Deng, J.; Yan, H.Q.; Dixon, C.E.; Sun, D. Administration of DHA Reduces Endoplasmic Reticulum Stress-Associated Inflammation and Alters Microglial or Macrophage Activation in Traumatic Brain Injury. ASN Neuro 2015, 7, 1759091415618969. [Google Scholar] [CrossRef] [Green Version]
- Hotamisligil, G.S. Endoplasmic reticulum stress and the inflammatory basis of metabolic disease. Cell 2010, 140, 900–917. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Desai, A.; Park, T.; Barnes, J.; Kevala, K.; Chen, H.; Kim, H.-Y. Reduced acute neuroinflammation and improved functional recovery after traumatic brain injury by α-linolenic acid supplementation in mice. J. Neuroinflamm. 2016, 13, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paudel, Y.N.; Shaikh, M.F.; Chakraborti, A.; Kumari, Y.; Aledo-Serrano, Á.; Aleksovska, K.; Alvim, M.K.M.; Othman, I. HMGB1: A Common Biomarker and Potential Target for TBI, Neuroinflammation, Epilepsy, and Cognitive Dysfunction. Front. Neurosci. 2018, 12, 628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Wu, S.; Chen, C.; Xie, B.; Fang, Z.; Hu, W.; Chen, J.; Fu, H.; He, H. Omega-3 polyunsaturated fatty acid supplementation attenuates microglial-induced inflammation by inhibiting the HMGB1/TLR4/NF-κB pathway following experimental traumatic brain injury. J. Neuroinflamm. 2017, 14, 143. [Google Scholar] [CrossRef] [PubMed]
- Lan, K.C.; Chao, S.C.; Wu, H.Y.; Chiang, C.L.; Wang, C.C.; Liu, S.H.; Weng, T.I. Salidroside ameliorates sepsis-induced acute lung injury and mortality via downregulating NF-κB and HMGB1 pathways through the upregulation of SIRT1. Sci. Rep. 2017, 7, 12026. [Google Scholar] [CrossRef] [Green Version]
- Godoy, J.A.; Zolezzi, J.M.; Braidy, N.; Inestrosa, N.C. Role of Sirt1 during the ageing process: Relevance to protection of synapses in the brain. Mol. Neurobiol. 2014, 50, 744–756. [Google Scholar] [CrossRef]
- Schober, M.E.; Requena, D.F.; Casper, T.C.; Velhorst, A.K.; Lolofie, A.; McFarlane, K.E.; Otto, T.E.; Terry, C.; Gensel, J.C. Docosahexaenoic acid decreased neuroinflammation in rat pups after controlled cortical impact. Exp. Neurol. 2019, 320, 112971. [Google Scholar] [CrossRef]
- Friedrich, M.J. Depression Is the Leading Cause of Disability Around the World. JAMA 2017, 317, 1517. [Google Scholar] [CrossRef]
- Baquero, M.; Martín, N. Depressive symptoms in neurodegenerative diseases. World J. Clin. Cases 2015, 3, 682–693. [Google Scholar] [CrossRef]
- Gu, M.; Li, Y.; Tang, H.; Zhang, C.; Li, W.; Zhang, Y.; Li, Y.; Zhao, Y.; Song, C. Endogenous Omega (n)-3 Fatty Acids in Fat-1 Mice Attenuated Depression-Like Behavior, Imbalance between Microglial M1 and M2 Phenotypes, and Dysfunction of Neurotrophins Induced by Lipopolysaccharide Administration. Nutrients 2018, 10, 1351. [Google Scholar] [CrossRef] [Green Version]
- Wu, B.; Song, Q.; Zhang, Y.; Wang, C.; Yang, M.; Zhang, J.; Han, W.; Jiang, P. Antidepressant activity of ω-3 polyunsaturated fatty acids in ovariectomized rats: Role of neuroinflammation and microglial polarization. Lipids Health Dis. 2020, 19, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Z.; Liu, B.; Yang, L.E.; Zhang, C. Platycodigenin as Potential Drug Candidate for Alzheimer’s Disease via Modulating Microglial Polarization and Neurite Regeneration. Molecules 2019, 24, 3207. [Google Scholar] [CrossRef] [PubMed] [Green Version]


| Pheno-Type | Sub-Type | Specific Cytokines | Chemokines | Specific Markers | Functions | Ref. |
|---|---|---|---|---|---|---|
| M1 | - | IL-1β, IL-6, IL-12, IL-23, and TNF-α | CCL4, CCL5, CCL8, CXCL2, CXCL4, CXCL9, and CXCL10 | CD86, CD16, CD32, CD68, iNOS, IL-1R, and MHC-II | Neurotoxicity, oxidative stress, neuronal, and synaptic damage | [21,23] |
| M2 | M2a | IL-4, IL-10, IL-13, and IL-1Ra | CCL24 and CCL22 | CD163, CD206, Arg1, Ym-1, FIZZ-1, and MHC-II | Tissue repair, phagocytosis, and encapsulation of parasites | [19,21,24,25] |
| M2b | IL-10, IL-1, and IL-6 | CCL1 | CD86 and MHC-II | Phagocytosis and regulation of inflammatory responses | [21,26] | |
| M2c | IL-10 and TGF-β | CCL16, CXCL13, and CCR5 | CD163, TLR1, and TLR8 | Immunoregulation and tissue healing | [21,27] | |
| M2d | IL-10, IL-12, TNF-α, and TGF-β | CXCL13, CCL16, and CCL18 | VEGF | Angiogenesis in tumor | [21] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sanjay; Park, M.; Lee, H.-J. Roles of Fatty Acids in Microglial Polarization: Evidence from In Vitro and In Vivo Studies on Neurodegenerative Diseases. Int. J. Mol. Sci. 2022, 23, 7300. https://doi.org/10.3390/ijms23137300
Sanjay, Park M, Lee H-J. Roles of Fatty Acids in Microglial Polarization: Evidence from In Vitro and In Vivo Studies on Neurodegenerative Diseases. International Journal of Molecular Sciences. 2022; 23(13):7300. https://doi.org/10.3390/ijms23137300
Chicago/Turabian StyleSanjay, Miey Park, and Hae-Jeung Lee. 2022. "Roles of Fatty Acids in Microglial Polarization: Evidence from In Vitro and In Vivo Studies on Neurodegenerative Diseases" International Journal of Molecular Sciences 23, no. 13: 7300. https://doi.org/10.3390/ijms23137300
APA StyleSanjay, Park, M., & Lee, H. -J. (2022). Roles of Fatty Acids in Microglial Polarization: Evidence from In Vitro and In Vivo Studies on Neurodegenerative Diseases. International Journal of Molecular Sciences, 23(13), 7300. https://doi.org/10.3390/ijms23137300