
RIPK1 in Liver Parenchymal Cells Limits Murine Hepatitis during Acute CCl4-Induced Liver Injury

 ,
,  ,
, 
Abstract
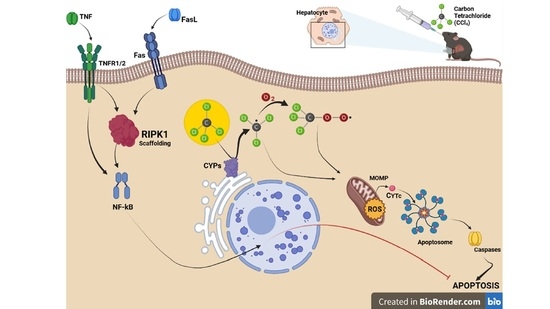
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. Scaffolding Properties of RIPK1 in Liver Parenchymal Cell Limit Apoptosis Occurrence in Acute CCl4-Induced Hepatitis
2.2. Ripk1 Deficiency in Liver Parenchymal Cells Potentiates Oxidative Stress, Inflammation and Immune Infiltration in CCl4-Induced Hepatitis
2.3. Neutralization of TNF-α Further Potentiates CCl4-Induced Hepatotoxicity in Ripk1fl/fl and Ripk1LPC-KO Mice
2.4. Neutralization of FasL Partially Increased CCl4-Induced Hepatotoxicity in Ripk1LPC-KO
3. Discussion
4. Materials and Methods
4.1. Animals, Treatment Protocols
4.2. Biochemical Studies
4.3. Histopathological Studies and Immunolocalization in Liver Tissues
4.4. RNA Isolation and qPCR Analysis
4.5. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Liang, D.; Guan, Y.; Zhu, J.; Wu, J.; Yu, X.; Qiu, K.; He, Z.; He, Q. Global research trends of drug-induced liver injury (DILI) in the past two decades: A bibliometric and visualized study. Ann. Palliat. Med. 2021, 10, 8651–8664. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.M. Etiologies of acute liver failure. Semin. Liver Dis. 2008, 28, 142–152. [Google Scholar] [CrossRef] [PubMed]
- Andrade, R.J.; Aithal, G.P.; Björnsson, E.S.; Kaplowitz, N.; Kullak-Ublick, G.A.; Larrey, D.; Karlsen, T.H. EASL Clinical Practice Guidelines: Drug-induced liver injury. J. Hepatol. 2019, 70, 1222–1261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McGill, M.R.; Jaeschke, H. Animal models of drug-induced liver injury. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 1031–1039. [Google Scholar] [CrossRef]
- Teschke, R. Liver Injury by Carbon Tetrachloride Intoxication in 16 Patients Treated with Forced Ventilation to Accelerate Toxin Removal via the Lungs: A Clinical Report. Toxics 2018, 6, 25. [Google Scholar] [CrossRef] [Green Version]
- Meaden, C.W.; Procopio, G.; Calello, D.P.; Nelson, L.S.; Ruck, B.; Gupta, A.; Jacob, J.E. Carbon tetrachloride poisoning from an antique fire extinguisher. Am. J. Emerg. Med. 2020, 38, 2139–2141. [Google Scholar] [CrossRef]
- Boll, M.; Weber, L.W.; Becker, E.; Stampfl, A. Mechanism of carbon tetrachloride-induced hepatotoxicity. Hepatocellular damage by reactive carbon tetrachloride metabolites. Z. Nat. C J. Biosci. 2001, 56, 649–659. [Google Scholar] [CrossRef]
- Weber, L.W.; Boll, M.; Stampfl, A. Hepatotoxicity and mechanism of action of haloalkanes: Carbon tetrachloride as a toxicological model. Crit. Rev. Toxicol. 2003, 33, 105–136. [Google Scholar] [CrossRef]
- Dai, C.; Xiao, X.; Li, D.; Tun, S.; Wang, Y.; Velkov, T.; Tang, S. Chloroquine ameliorates carbon tetrachloride-induced acute liver injury in mice via the concomitant inhibition of inflammation and induction of apoptosis. Cell Death Dis. 2018, 9, 1164. [Google Scholar] [CrossRef]
- Andrade, R.J.; Chalasani, N.; Björnsson, E.S.; Suzuki, A.; Kullak-Ublick, G.A.; Watkins, P.B.; Devarbhavi, H.; Merz, M.; Lucena, M.I.; Kaplowitz, N.; et al. Drug-induced liver injury. Nat. Rev. Dis. Primers 2019, 5, 58. [Google Scholar] [CrossRef] [Green Version]
- Green, D.R.; Galluzzi, L.; Kroemer, G. Cell biology. Metabolic control of cell death. Science 2014, 345, 1250256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shojaie, L.; Iorga, A.; Dara, L. Cell Death in Liver Diseases: A Review. Int. J. Mol. Sci. 2020, 21, 9682. [Google Scholar] [CrossRef] [PubMed]
- Jaeschke, H.; Ramachandran, A. Acetaminophen-induced apoptosis: Facts versus fiction. J. Clin. Transl. Res. 2020, 6, 36–47. [Google Scholar] [PubMed]
- Fink, S.L.; Cookson, B.T. Apoptosis, pyroptosis, and necrosis: Mechanistic description of dead and dying eukaryotic cells. Infect. Immun. 2005, 73, 1907–1916. [Google Scholar] [CrossRef] [Green Version]
- Gough, P.; Myles, I.A. Tumor Necrosis Factor Receptors: Pleiotropic Signaling Complexes and Their Differential Effects. Front. Immunol. 2020, 11, 585880. [Google Scholar] [CrossRef]
- Ofengeim, D.; Yuan, J. Regulation of RIP1 kinase signalling at the crossroads of inflammation and cell death. Nat. Rev. Mol. Cell Biol. 2013, 14, 727–736. [Google Scholar] [CrossRef]
- Farooq, M.; Filliol, A.; Simoes Eugénio, M.; Piquet-Pellorce, C.; Dion, S.; Raguenes-Nicol, C.; Jan, A.; Dimanche-Boitrel, M.T.; Le Seyec, J.; Samson, M. Depletion of RIPK1 in hepatocytes exacerbates liver damage in fulminant viral hepatitis. Cell Death Dis. 2019, 10, 12. [Google Scholar] [CrossRef]
- Filliol, A.; Piquet-Pellorce, C.; Le Seyec, J.; Farooq, M.; Genet, V.; Lucas-Clerc, C.; Bertin, J.; Gough, P.J.; Dimanche-Boitrel, M.T.; Vandenabeele, P.; et al. RIPK1 protects from TNF-α-mediated liver damage during hepatitis. Cell Death Dis. 2016, 7, e2462. [Google Scholar] [CrossRef] [Green Version]
- Filliol, A.; Piquet-Pellorce, C.; Raguénès-Nicol, C.; Dion, S.; Farooq, M.; Lucas-Clerc, C.; Vandenabeele, P.; Bertrand, M.J.M.; Le Seyec, J.; Samson, M. RIPK1 protects hepatocytes from Kupffer cells-mediated TNF-induced apoptosis in mouse models of PAMP-induced hepatitis. J. Hepatol. 2017, 66, 1205–1213. [Google Scholar] [CrossRef]
- Dong, Y.; Liu, Y.; Kou, X.; Jing, Y.; Sun, K.; Sheng, D.; Yu, G.; Yu, D.; Zhao, Q.; Zhao, X.; et al. The protective or damaging effect of Tumor necrosis factor-α in acute liver injury is concentration-dependent. Cell Biosci. 2016, 6, 8. [Google Scholar] [CrossRef] [Green Version]
- Simeonova, P.P.; Gallucci, R.M.; Hulderman, T.; Wilson, R.; Kommineni, C.; Rao, M.; Luster, M.I. The role of tumor necrosis factor-alpha in liver toxicity, inflammation, and fibrosis induced by carbon tetrachloride. Toxicol. Appl. Pharmacol. 2001, 177, 112–120. [Google Scholar] [CrossRef] [PubMed]
- Van, T.M.; Polykratis, A.; Straub, B.K.; Kondylis, V.; Papadopoulou, N.; Pasparakis, M. Kinase-independent functions of RIPK1 regulate hepatocyte survival and liver carcinogenesis. J. Clin. Investig. 2017, 127, 2662–2677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kondylis, V.; Pasparakis, M. RIP Kinases in Liver Cell Death, Inflammation and Cancer. Trends Mol. Med. 2019, 25, 47–63. [Google Scholar] [CrossRef] [PubMed]
- Farooq, M.; Simoes Eugénio, M.; Piquet-Pellorce, C.; Dion, S.; Raguenes-Nicol, C.; Santamaria, K.; Kara-Ali, G.H.; Larcher, T.; Dimanche-Boitrel, M.-T.; Samson, M.; et al. Receptor-interacting protein kinase-1 ablation in liver parenchymal cells promotes liver fibrosis in murine NASH without affecting other symptoms. J. Mol. Med. 2022, 100, 1027–1038. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.S.; Kang, J.W.; Lee, S.M. Melatonin attenuates carbon tetrachloride-induced liver fibrosis via inhibition of necroptosis. Transl. Res. J. Lab. Clin. Med. 2015, 166, 292–303. [Google Scholar] [CrossRef] [PubMed]
- Scholten, D.; Trebicka, J.; Liedtke, C.; Weiskirchen, R. The carbon tetrachloride model in mice. Lab. Anim. 2015, 49, 4–11. [Google Scholar] [CrossRef]
- Hameed, H.; Farooq, M.; Piquet-Pellorce, C.; Hamon, A.; Samson, M.; Le Seyec, J. Questioning the RIPK1 kinase activity involvement in acetaminophen-induced hepatotoxicity in mouse. Free Radic. Biol. Med. 2022, 178, 243–245. [Google Scholar] [CrossRef]
- Morio, L.A.; Chiu, H.; Sprowles, K.A.; Zhou, P.; Heck, D.E.; Gordon, M.K.; Laskin, D.L. Distinct roles of tumor necrosis factor-alpha and nitric oxide in acute liver injury induced by carbon tetrachloride in mice. Toxicol. Appl. Pharmacol. 2001, 172, 44–51. [Google Scholar] [CrossRef]
- Guicciardi, M.E.; Gores, G.J. Fasl and fas in liver homeostasis and hepatic injuries. In Fas Signaling; Springer: Boston, MA, USA, 2006; pp. 103–117. [Google Scholar]
- Pinkoski, M.J.; Brunner, T.; Green, D.R.; Lin, T. Fas and Fas ligand in gut and liver. Am. J. Physiol. Gastrointest. Liver Physiol. 2000, 278, G354–G366. [Google Scholar] [CrossRef] [Green Version]
- Filliol, A.; Farooq, M.; Piquet-Pellorce, C.; Genet, V.; Dimanche-Boitrel, M.T.; Vandenabeele, P.; Bertrand, M.J.M.; Samson, M.; Le Seyec, J. RIPK1 protects hepatocytes from death in Fas-induced hepatitis. Sci. Rep. 2017, 7, 9205. [Google Scholar] [CrossRef] [Green Version]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hameed, H.; Farooq, M.; Vuillier, C.; Piquet-Pellorce, C.; Hamon, A.; Dimanche-Boitrel, M.-T.; Samson, M.; Le Seyec, J. RIPK1 in Liver Parenchymal Cells Limits Murine Hepatitis during Acute CCl4-Induced Liver Injury. Int. J. Mol. Sci. 2022, 23, 7367. https://doi.org/10.3390/ijms23137367
Hameed H, Farooq M, Vuillier C, Piquet-Pellorce C, Hamon A, Dimanche-Boitrel M-T, Samson M, Le Seyec J. RIPK1 in Liver Parenchymal Cells Limits Murine Hepatitis during Acute CCl4-Induced Liver Injury. International Journal of Molecular Sciences. 2022; 23(13):7367. https://doi.org/10.3390/ijms23137367
Chicago/Turabian StyleHameed, Huma, Muhammad Farooq, Céline Vuillier, Claire Piquet-Pellorce, Annaïg Hamon, Marie-Thérèse Dimanche-Boitrel, Michel Samson, and Jacques Le Seyec. 2022. "RIPK1 in Liver Parenchymal Cells Limits Murine Hepatitis during Acute CCl4-Induced Liver Injury" International Journal of Molecular Sciences 23, no. 13: 7367. https://doi.org/10.3390/ijms23137367
APA StyleHameed, H., Farooq, M., Vuillier, C., Piquet-Pellorce, C., Hamon, A., Dimanche-Boitrel, M. -T., Samson, M., & Le Seyec, J. (2022). RIPK1 in Liver Parenchymal Cells Limits Murine Hepatitis during Acute CCl4-Induced Liver Injury. International Journal of Molecular Sciences, 23(13), 7367. https://doi.org/10.3390/ijms23137367