
The Role of Vitamin D in SARS-CoV-2 Infection and Acute Kidney Injury

 ,
,  ,
,  ,
,
Abstract
:1. Introduction
2. Antioxidant and Renoprotective Effect of Vitamin D in AKI Animal Models
3. Vitamin D and the Renin–Angiotensin–Aldosterone System (RAAS)
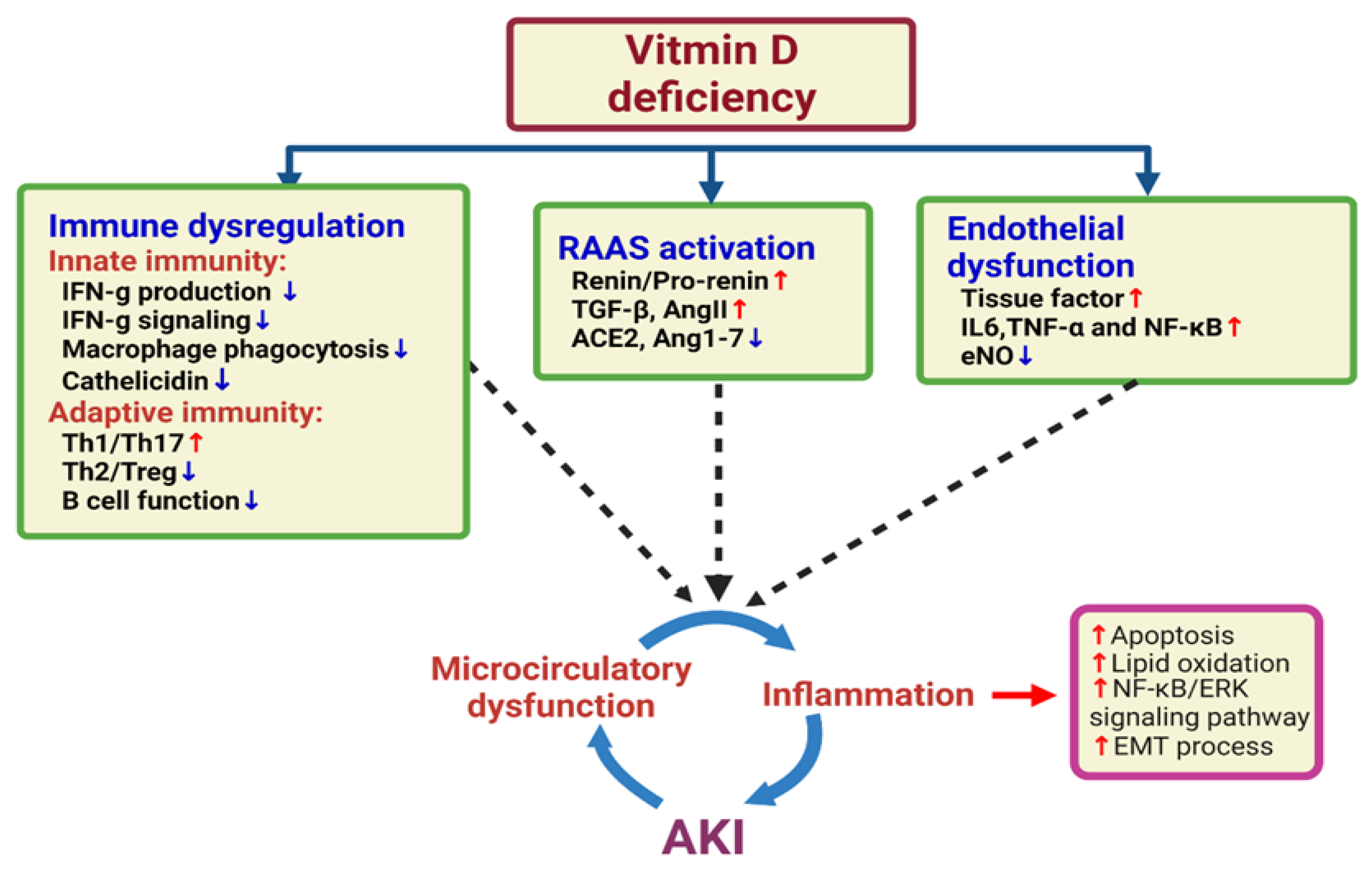
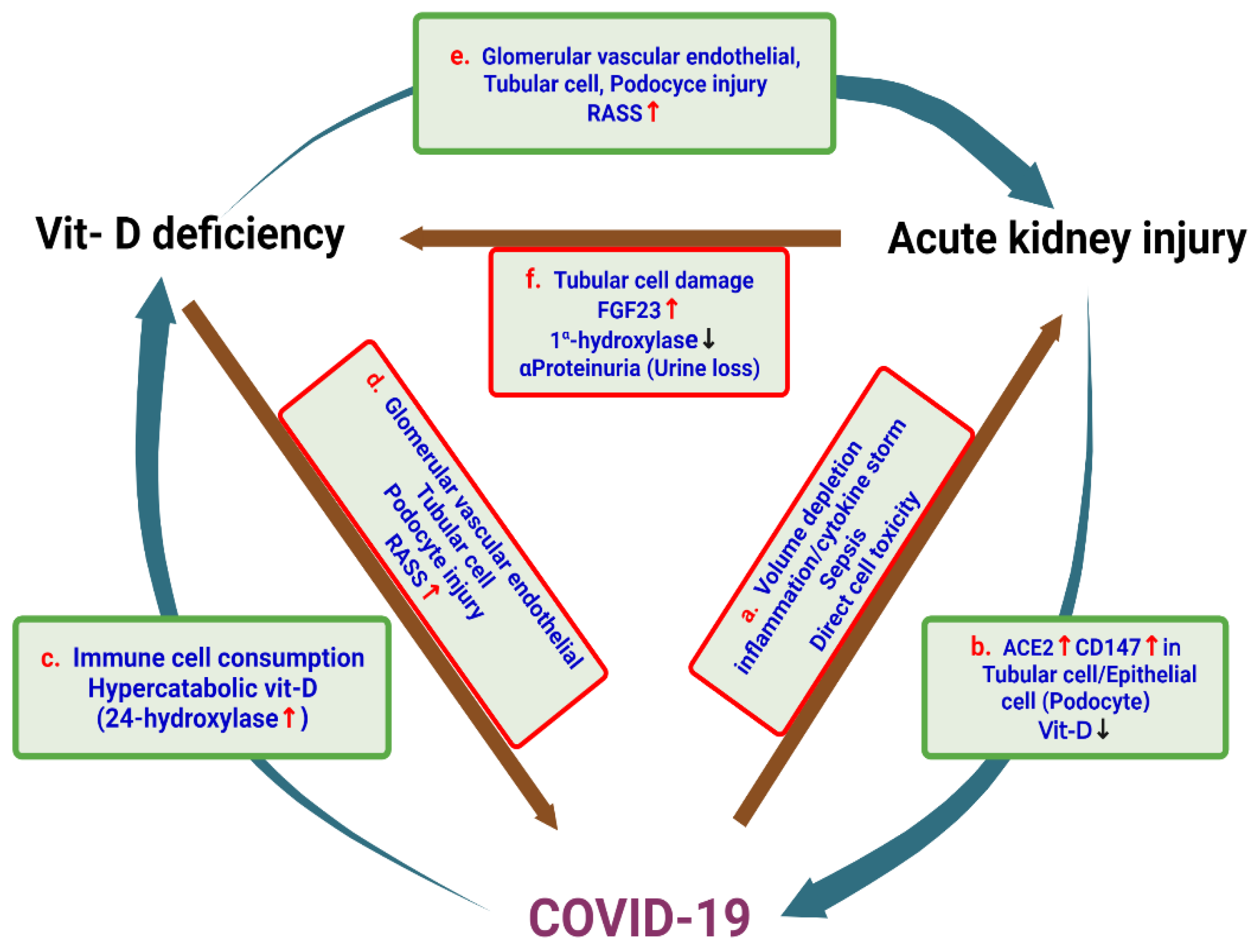
4. Vitamin D Deficiency and the Risk of AKI
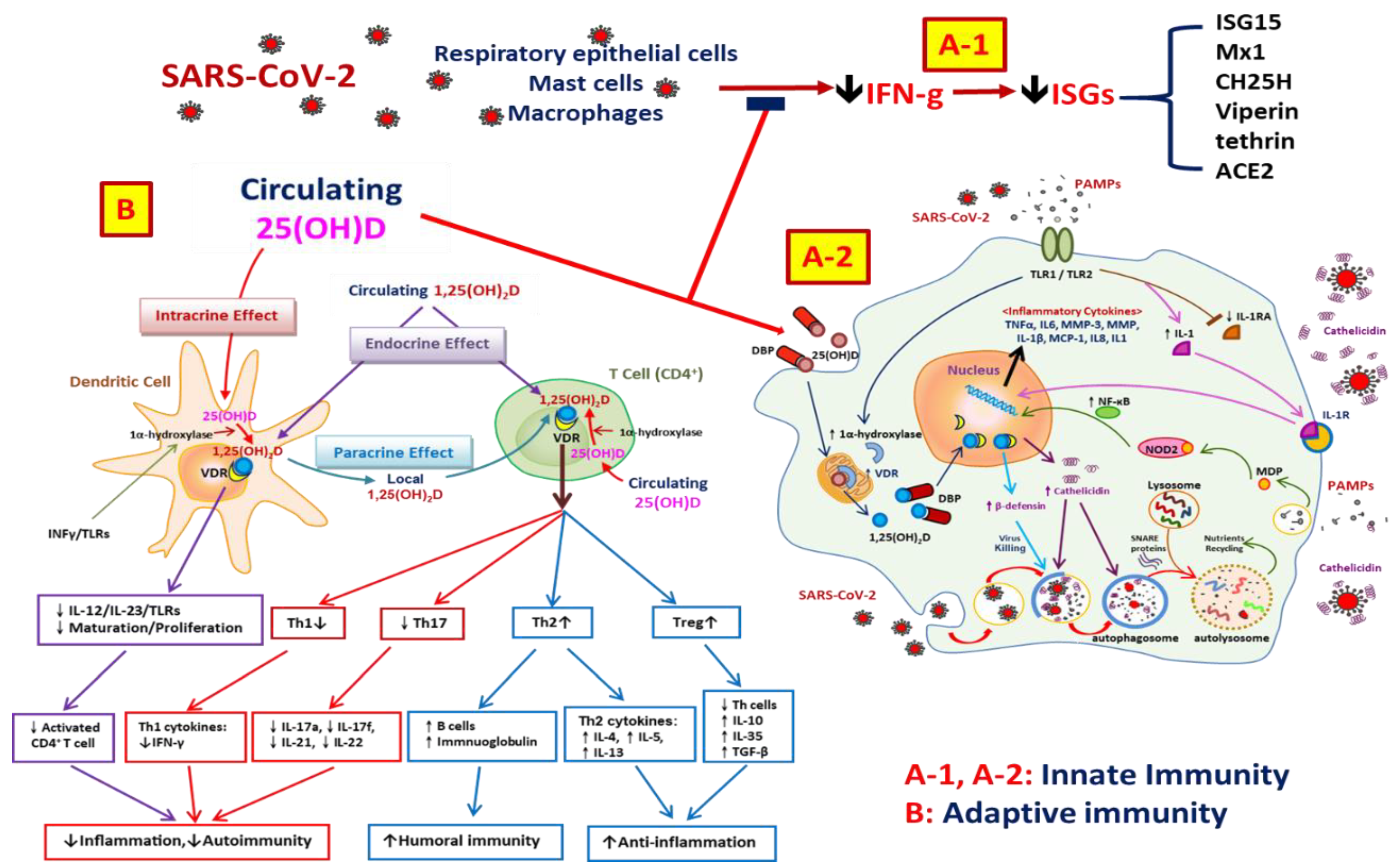
5. Vitamin D and the Immune System
6. Vitamin D and Endothelial Dysfunction
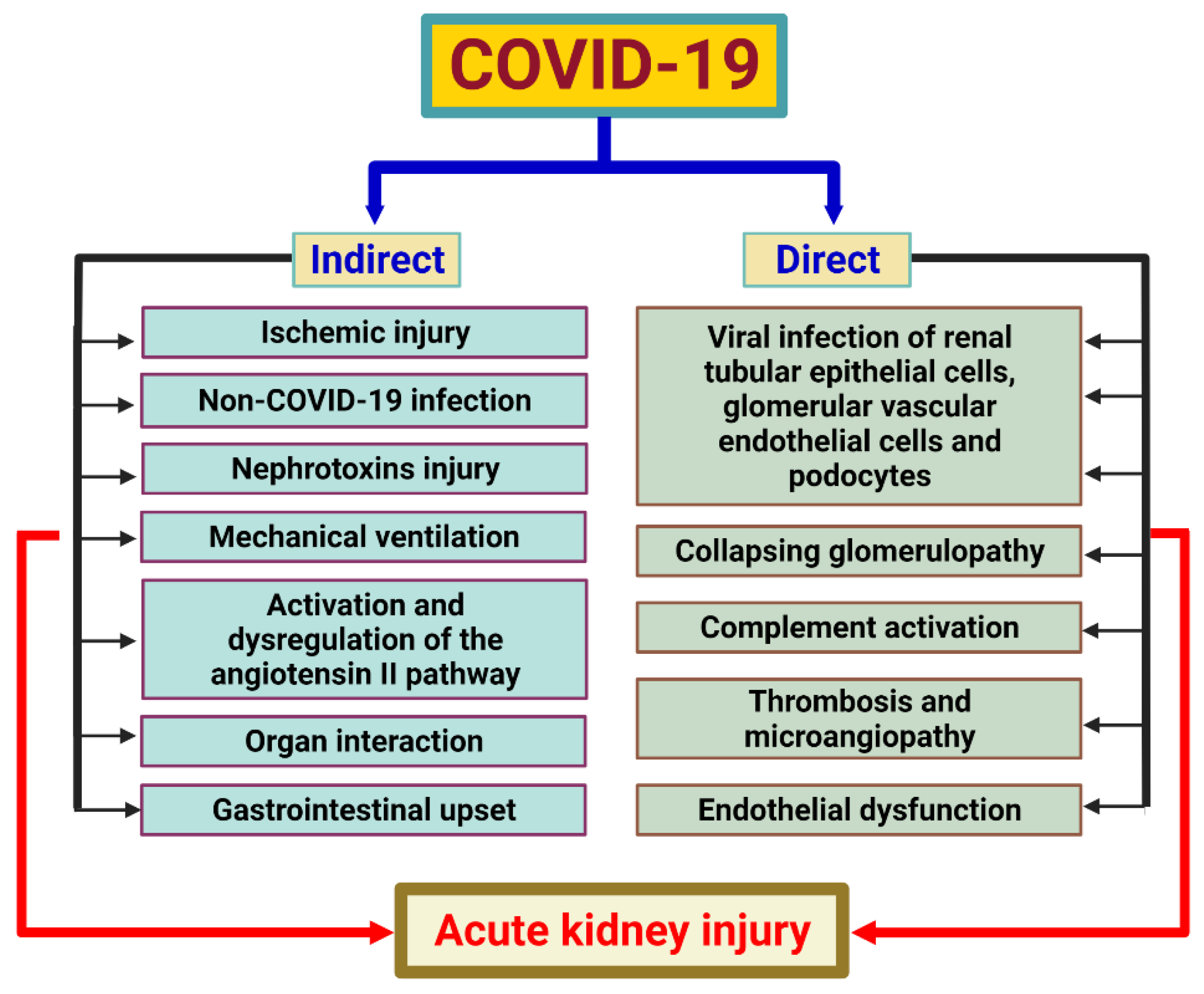
7. SARS-CoV-2 and Acute Kidney Injury
8. Anti-Inflammatory Effects of Vitamin D on SARS-CoV-2
9. Side Effects of Excess Vitamin D
10. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sung, C.C.; Liao, M.T.; Lu, K.C.; Wu, C.C. Role of vitamin D in insulin resistance. J. Biomed. Biotechnol. 2012, 2012, 634195. [Google Scholar] [CrossRef] [PubMed]
- de Tena, J.G.; Abejon, L.; Horcajo, P. Vitamin D insufficiency. N. Engl. J. Med. 2011, 364, 1378. [Google Scholar] [CrossRef]
- Bacchetta, J.; Sea, J.L.; Chun, R.F.; Lisse, T.S.; Wesseling-Perry, K.; Gales, B.; Adams, J.S.; Salusky, I.B.; Hewison, M. Fibroblast growth factor 23 inhibits extrarenal synthesis of 1,25-dihydroxyvitamin D in human monocytes. J. Bone Miner. Res. 2013, 28, 46–55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henry, H.L. Regulation of vitamin D metabolism. Best Pract. Res. Clin. Endocrinol. Metab. 2011, 25, 531–541. [Google Scholar] [CrossRef]
- Haussler, M.R.; Haussler, C.A.; Bartik, L.; Whitfield, G.K.; Hsieh, J.C.; Slater, S.; Jurutka, P.W. Vitamin D receptor: Molecular signaling and actions of nutritional ligands in disease prevention. Nutr. Rev. 2008, 66, S98–S112. [Google Scholar] [CrossRef]
- Nagpal, S.; Na, S.; Rathnachalam, R. Noncalcemic actions of vitamin D receptor ligands. Endocr. Rev. 2005, 26, 662–687. [Google Scholar] [CrossRef]
- Mahon, B.D.; Wittke, A.; Weaver, V.; Cantorna, M.T. The targets of vitamin D depend on the differentiation and activation status of CD4 positive T cells. J. Cell. Biochem. 2003, 89, 922–932. [Google Scholar] [CrossRef]
- Adorini, L.; Penna, G.; Giarratana, N.; Roncari, A.; Amuchastegui, S.; Daniel, K.C.; Uskokovic, M. Dendritic cells as key targets for immunomodulation by Vitamin D receptor ligands. J. Steroid Biochem. Mol. Biol. 2004, 89–90, 437–441. [Google Scholar] [CrossRef]
- Li, Y.C. Vitamin D regulation of the renin-angiotensin system. J. Cell. Biochem. 2003, 88, 327–331. [Google Scholar] [CrossRef]
- Chiu, K.C.; Chu, A.; Go, V.L.; Saad, M.F. Hypovitaminosis D is associated with insulin resistance and beta cell dysfunction. Am. J. Clin. Nutr. 2004, 79, 820–825. [Google Scholar] [CrossRef] [Green Version]
- Zittermann, A. Vitamin D and disease prevention with special reference to cardiovascular disease. Prog. Biophys. Mol. Biol. 2006, 92, 39–48. [Google Scholar] [CrossRef] [PubMed]
- Panda, D.K.; Miao, D.; Tremblay, M.L.; Sirois, J.; Farookhi, R.; Hendy, G.N.; Goltzman, D. Targeted ablation of the 25-hydroxyvitamin D 1alpha-hydroxylase enzyme: Evidence for skeletal, reproductive, and immune dysfunction. Proc. Natl. Acad. Sci. USA 2001, 98, 7498–7503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bellomo, R.; Ronco, C.; Kellum, J.A.; Mehta, R.L.; Palevsky, P. Acute renal failure—Definition, outcome measures, animal models, fluid therapy and information technology needs: The second international consensus conference of the acute dialysis quality initiative (ADQI) group. Crit. Care 2004, 8, R204–R212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chertow, G.M.; Burdick, E.; Honour, M.; Bonventre, J.V.; Bates, D.W. Acute kidney injury, mortality, length of stay, and costs in hospitalized patients. J. Am. Soc. Nephrol. 2005, 16, 3365–3370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mehta, R.L.; Kellum, J.A.; Shah, S.V.; Molitoris, B.A.; Ronco, C.; Warnock, D.G.; Levin, A. Acute kidney injury network: Report of an initiative to improve outcomes in acute kidney injury. Crit. Care 2007, 11, R31. [Google Scholar] [CrossRef] [Green Version]
- Graidis, S.; Papavramidis, T.S.; Papaioannou, M. Vitamin D and acute kidney injury: A two-way causality relation and a predictive, prognostic, and therapeutic role of vitamin D. Front. Nutr. 2020, 7, 630951. [Google Scholar] [CrossRef]
- Lucidarme, O.; Messai, E.; Mazzoni, T.; Arcade, M.; du Cheyron, D. Incidence and risk factors of vitamin D deficiency in critically ill patients: Results from a prospective observational study. Intensive Care Med. 2010, 36, 1609–1611. [Google Scholar] [CrossRef]
- Aygencel, G.; Turkoglu, M.; Tuncel, A.F.; Candir, B.A.; Bildaci, Y.D.; Pasaoglu, H. Is vitamin d insufficiency associated with mortality of critically ill patients? Crit. Care Res. Pract. 2013, 2013, 856747. [Google Scholar] [CrossRef] [Green Version]
- Braun, A.; Chang, D.; Mahadevappa, K.; Gibbons, F.K.; Liu, Y.; Giovannucci, E.; Christopher, K.B. Association of low serum 25-hydroxyvitamin D levels and mortality in the critically ill. Crit. Care Med. 2011, 39, 671–677. [Google Scholar] [CrossRef] [Green Version]
- Braun, A.B.; Gibbons, F.K.; Litonjua, A.A.; Giovannucci, E.; Christopher, K.B. Low serum 25-hydroxyvitamin D at critical care initiation is associated with increased mortality. Crit. Care Med. 2012, 40, 63–72. [Google Scholar] [CrossRef]
- Watkins, R.R.; Yamshchikov, A.V.; Lemonovich, T.L.; Salata, R.A. The role of vitamin D deficiency in sepsis and potential therapeutic implications. J. Infect. 2011, 63, 321–326. [Google Scholar] [CrossRef] [PubMed]
- Braun, A.B.; Christopher, K.B. Vitamin D in acute kidney injury. Inflamm. Allergy Drug Targets 2013, 12, 262–272. [Google Scholar] [CrossRef] [PubMed]
- Martineau, A.R.; Jolliffe, D.A.; Hooper, R.L.; Greenberg, L.; Aloia, J.F.; Bergman, P.; Dubnov-Raz, G.; Esposito, S.; Ganmaa, D.; Ginde, A.A.; et al. Vitamin D supplementation to prevent acute respiratory tract infections: Systematic review and meta-analysis of individual participant data. BMJ 2017, 356, i6583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oristrell, J.; Oliva, J.C.; Casado, E.; Subiriana, I.; Domínguez, D.; Toloba, A.; Balado, A.; Grau, M. Vitamin D supplementation and COVID-19 risk: A population-based, cohort study. J. Endocrinol. Invest. 2022, 45, 167–179. [Google Scholar] [CrossRef] [PubMed]
- Seal, K.H.; Bertenthal, D.; Carey, E.; Grunfeld, C.; Bikle, D.D.; Lu, C.M. Association of Vitamin D Status and COVID-19-Related Hospitalization and Mortality. J. Gen. Intern. Med. 2022, 37, 853–861. [Google Scholar] [CrossRef]
- Dror, A.A.; Morozov, N.; Daoud, A.; Namir, Y.; Yakir, O.; Shachar, Y.; Lifshitz, M.; Segal, E.; Fisher, L.; Mizrachi, M.; et al. Pre-infection 25-hydroxyvitamin D3 levels and association with severity of COVID-19 illness. PLoS ONE 2022, 17, e0263069. [Google Scholar] [CrossRef]
- Villasis-Keever, M.A.; López-Alarcón, M.G.; Miranda-Novales, G.; Zurita-Cruz, J.N.; Barrada-Vázquez, A.S.; González-Ibarra, J.; Martínez-Reyes, M.; Grajales-Muñiz, C.; Santacruz-Tinoco, C.E.; Martínez-Miguel, B.; et al. Efficacy and Safety of Vitamin D Supplementation to Prevent COVID-19 in Frontline Healthcare Workers. A Randomized Clinical Trial. Arch. Med. Res. 2022, 18, 423–430. [Google Scholar] [CrossRef]
- Mercola, J.; Grant, W.B.; Wagner, C.L. Evidence regarding vitamin D and risk of COVID-19 and its severity. Nutrients 2020, 12, 3361. [Google Scholar] [CrossRef]
- Ari, E.; Kedrah, A.E.; Alahdab, Y.; Bulut, G.; Eren, Z.; Baytekin, O.; Odabasi, D. Antioxidant and renoprotective effects of paricalcitol on experimental contrast-induced nephropathy model. Br. J. Radiol. 2012, 85, 1038–1043. [Google Scholar] [CrossRef] [Green Version]
- Park, J.W.; Bae, E.H.; Kim, I.J.; Ma, S.K.; Choi, C.; Lee, J.; Kim, S.W. Renoprotective effects of paricalcitol on gentamicin-induced kidney injury in rats. Am. J. Physiol. Renal Physiol. 2010, 298, F301–F313. [Google Scholar] [CrossRef] [Green Version]
- Hur, E.; Garip, A.; Camyar, A.; Ilgun, S.; Ozisik, M.; Tuna, S.; Olukman, M.; Ozdemir, Z.N.; Sozmen, E.Y.; Sen, S.; et al. The effects of vitamin d on gentamicin-induced acute kidney injury in experimental rat model. Int. J. Endocrinol. 2013, 2013, 313528. [Google Scholar] [CrossRef] [PubMed]
- Su, C.M.; Wang, L.; Yoo, D. Activation of NF-κB and induction of proinflammatory cytokine expressions mediated by ORF7a protein of SARS-CoV-2. Sci. Rep. 2021, 11, 13464. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Chen, Y.H.; Tan, Z.X.; Xie, D.D.; Zhang, C.; Zhang, Z.H.; Wang, H.; Zhao, H.; Yu, D.X.; Xu, D.X. Vitamin D3 pretreatment regulates renal inflammatory responses during lipopolysaccharide-induced acute kidney injury. Sci. Rep. 2015, 5, 18687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, S.; Chen, Y.H.; Tan, Z.X.; Xie, D.D.; Zhang, C.; Xia, M.Z.; Wang, H.; Zhao, H.; Xu, D.X.; Yu, D.X. Vitamin D3 pretreatment alleviates renal oxidative stress in lipopolysaccharide-induced acute kidney injury. J. Steroid Biochem. Mol. Biol. 2015, 152, 133–141. [Google Scholar] [CrossRef] [PubMed]
- Forman, J.P.; Williams, J.S.; Fisher, N.D. Plasma 25-hydroxyvitamin D and regulation of the renin-angiotensin system in humans. Hypertension 2010, 55, 1283–1288. [Google Scholar] [CrossRef] [Green Version]
- Kong, J.; Qiao, G.; Zhang, Z.; Liu, S.Q.; Li, Y.C. Targeted vitamin D receptor expression in juxtaglomerular cells suppresses renin expression independent of parathyroid hormone and calcium. Kidney Int. 2008, 74, 1577–1581. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Zhang, Y.; Ning, G.; Deb, D.K.; Kong, J.; Li, Y.C. Combination therapy with AT1 blocker and vitamin D analog markedly ameliorates diabetic nephropathy: Blockade of compensatory renin increase. Proc. Natl. Acad. Sci. USA 2008, 105, 15896–15901. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.C.; Kong, J.; Wei, M.; Chen, Z.F.; Liu, S.Q.; Cao, L.P. 1,25-Dihydroxyvitamin D3 is a negative endocrine regulator of the renin-angiotensin system. J. Clin. Investig. 2002, 110, 229–238. [Google Scholar] [CrossRef]
- Yuan, W.; Pan, W.; Kong, J.; Zheng, W.; Szeto, F.L.; Wong, K.E.; Cohen, R.; Klopot, A.; Zhang, Z.; Li, Y.C. 1,25-dihydroxyvitamin D3 suppresses renin gene transcription by blocking the activity of the cyclic AMP response element in the renin gene promoter. J. Biol. Chem. 2007, 282, 29821–29830. [Google Scholar] [CrossRef] [Green Version]
- Mizobuchi, M.; Morrissey, J.; Finch, J.L.; Martin, D.R.; Liapis, H.; Akizawa, T.; Slatopolsky, E. Combination therapy with an angiotensin-converting enzyme inhibitor and a vitamin D analog suppresses the progression of renal insufficiency in uremic rats. J. Am. Soc. Nephrol. 2007, 18, 1796–1806. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Kong, J.; Deb, D.K.; Chang, A.; Li, Y.C. Vitamin D receptor attenuates renal fibrosis by suppressing the renin-angiotensin system. J. Am. Soc. Nephrol. 2010, 21, 966–973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Zeeuw, D.; Agarwal, R.; Amdahl, M.; Audhya, P.; Coyne, D.; Garimella, T.; Parving, H.H.; Pritchett, Y.; Remuzzi, G.; Ritz, E.; et al. Selective vitamin D receptor activation with paricalcitol for reduction of albuminuria in patients with type 2 diabetes (VITAL study): A randomised controlled trial. Lancet 2010, 376, 1543–1551. [Google Scholar] [CrossRef]
- Lucisano, S.; Buemi, M.; Passantino, A.; Aloisi, C.; Cernaro, V.; Santoro, D. New insights on the role of vitamin D in the progression of renal damage. Kidney Blood Press. Res. 2013, 37, 667–678. [Google Scholar] [CrossRef] [PubMed]
- Park, J.W.; Cho, J.W.; Joo, S.Y.; Kim, C.S.; Choi, J.S.; Bae, E.H.; Ma, S.K.; Kim, S.H.; Lee, J.; Kim, S.W. Paricalcitol prevents cisplatin-induced renal injury by suppressing apoptosis and proliferation. Eur. J. Pharmacol. 2012, 683, 301–309. [Google Scholar] [CrossRef]
- Park, J.W.; Bae, E.H.; Kim, I.J.; Ma, S.K.; Choi, C.; Lee, J.; Kim, S.W. Paricalcitol attenuates cyclosporine-induced kidney injury in rats. Kidney Int. 2010, 77, 1076–1085. [Google Scholar] [CrossRef] [Green Version]
- Kapil, A.; Singh, J.P.; Kaur, T.; Singh, B.; Singh, A.P. Involvement of peroxisome proliferator-activated receptor gamma in vitamin D-mediated protection against acute kidney injury in rats. J. Surg. Res. 2013, 185, 774–783. [Google Scholar] [CrossRef]
- Lee, J.W.; Kim, S.C.; Ko, Y.S.; Lee, H.Y.; Cho, E.; Kim, M.G.; Jo, S.K.; Cho, W.Y.; Kim, H.K. Renoprotective effect of paricalcitol via a modulation of the TLR4-NF-kappaB pathway in ischemia/reperfusion-induced acute kidney injury. Biochem. Biophys. Res. Commun. 2014, 444, 121–127. [Google Scholar] [CrossRef]
- Tan, X.; Wen, X.; Liu, Y. Paricalcitol inhibits renal inflammation by promoting vitamin D receptor-mediated sequestration of NF-kappaB signaling. J. Am. Soc. Nephrol. 2008, 19, 1741–1752. [Google Scholar] [CrossRef] [Green Version]
- Tan, X.; Li, Y.; Liu, Y. Paricalcitol attenuates renal interstitial fibrosis in obstructive nephropathy. J. Am. Soc. Nephrol. 2006, 17, 3382–3393. [Google Scholar] [CrossRef] [Green Version]
- de Borst, M.H.; Vervloet, M.G.; ter Wee, P.M.; Navis, G. Cross talk between the renin-angiotensin-aldosterone system and vitamin D-FGF-23-klotho in chronic kidney disease. J. Am. Soc. Nephrol. 2011, 22, 1603–1609. [Google Scholar] [CrossRef] [Green Version]
- Holick, M.F. High prevalence of vitamin D inadequacy and implications for health. Mayo Clin. Proc. 2006, 81, 353–373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Howe, W.R.; Dellavalle, R. Vitamin D deficiency. N. Engl. J. Med. 2007, 357, 266–281. [Google Scholar] [CrossRef]
- Zadshir, A.; Tareen, N.; Pan, D.; Norris, K.; Martins, D. The prevalence of hypovitaminosis D among US adults: Data from the NHANES III. Ethn. Dis. 2005, 15, S5-97–S5-101. [Google Scholar] [PubMed]
- Icardi, A.; Paoletti, E.; De Nicola, L.; Mazzaferro, S.; Russo, R.; Cozzolino, M. Renal anaemia and EPO hyporesponsiveness associated with vitamin D deficiency: The potential role of inflammation. Nephrol. Dial. Transplant. 2013, 28, 1672–1679. [Google Scholar] [CrossRef] [Green Version]
- Asakura, H.; Aoshima, K.; Suga, Y.; Yamazaki, M.; Morishita, E.; Saito, M.; Miyamoto, K.; Nakao, S. Beneficial effect of the active form of vitamin D3 against LPS-induced DIC but not against tissue-factor-induced DIC in rat models. Thromb. Haemost. 2001, 85, 287–290. [Google Scholar] [CrossRef]
- Moller, S.; Laigaard, F.; Olgaard, K.; Hemmingsen, C. Effect of 1,25-dihydroxy-vitamin D3 in experimental sepsis. Int. J. Med. Sci. 2007, 4, 190–195. [Google Scholar] [CrossRef] [Green Version]
- Jeng, L.; Yamshchikov, A.V.; Judd, S.E.; Blumberg, H.M.; Martin, G.S.; Ziegler, T.R.; Tangpricha, V. Alterations in vitamin D status and anti-microbial peptide levels in patients in the intensive care unit with sepsis. J. Transl. Med. 2009, 7, 28. [Google Scholar] [CrossRef] [Green Version]
- Arnson, Y.; Gringauz, I.; Itzhaky, D.; Amital, H. Vitamin D deficiency is associated with poor outcomes and increased mortality in severely ill patients. QJM 2012, 105, 633–639. [Google Scholar] [CrossRef] [Green Version]
- Flynn, L.; Zimmerman, L.H.; McNorton, K.; Dolman, M.; Tyburski, J.; Baylor, A.; Wilson, R.; Dolman, H. Effects of vitamin D deficiency in critically ill surgical patients. Am. J. Surg. 2012, 203, 379–382. [Google Scholar] [CrossRef]
- Shah, K.; Varna, P.V.; Sharma, U.; Mavalankar, D. Does vitamin D supplementation reduce COVID-19 severity?—A systematic review. QJM 2022, hcac040. [Google Scholar] [CrossRef]
- Braun, A.B.; Litonjua, A.A.; Moromizato, T.; Gibbons, F.K.; Giovannucci, E.; Christopher, K.B. Association of low serum 25-hydroxyvitamin D levels and acute kidney injury in the critically ill. Crit. Care Med. 2012, 40, 3170–3179. [Google Scholar] [CrossRef]
- Lai, L.; Qian, J.; Yang, Y.; Xie, Q.; You, H.; Zhou, Y.; Ma, S.; Hao, C.; Gu, Y.; Ding, F. Is the serum vitamin D level at the time of hospital-acquired acute kidney injury diagnosis associated with prognosis? PLoS ONE 2013, 8, e64964. [Google Scholar] [CrossRef]
- Leaf, D.E.; Waikar, S.S.; Wolf, M.; Cremers, S.; Bhan, I.; Stern, L. Dysregulated mineral metabolism in patients with acute kidney injury and risk of adverse outcomes. Clin. Endocrinol. 2013, 79, 491–498. [Google Scholar] [CrossRef] [Green Version]
- Adorini, L.; Penna, G. Dendritic cell tolerogenicity: A key mechanism in immunomodulation by vitamin D receptor agonists. Hum. Immunol. 2009, 70, 345–352. [Google Scholar] [CrossRef]
- Lang, C.L.; Wang, M.H.; Chiang, C.K.; Lu, K.C. Vitamin D and the immune system from the nephrologist’s viewpoint. ISRN Endocrinol. 2014, 2014, 105456. [Google Scholar] [CrossRef] [Green Version]
- Koeffler, H.P.; Amatruda, T.; Ikekawa, N.; Kobayashi, Y.; DeLuca, H.F. Induction of macrophage differentiation of human normal and leukemic myeloid stem cells by 1,25-dihydroxyvitamin D3 and its fluorinated analogues. Cancer Res. 1984, 44, 5624–5628. [Google Scholar]
- Stoffels, K.; Overbergh, L.; Bouillon, R.; Mathieu, C. Immune regulation of 1alpha-hydroxylase in murine peritoneal macrophages: Unravelling the IFNgamma pathway. J. Steroid Biochem. Mol. Biol. 2007, 103, 567–571. [Google Scholar] [CrossRef]
- Krutzik, S.R.; Hewison, M.; Liu, P.T.; Robles, J.A.; Stenger, S.; Adams, J.S.; Modlin, R.L. IL-15 links TLR2/1-induced macrophage differentiation to the vitamin D-dependent antimicrobial pathway. J. Immunol. 2008, 181, 7115–7120. [Google Scholar] [CrossRef]
- Bacchetta, J.; Salusky, I.B.; Hewison, M. Beyond mineral metabolism, is there an interplay between FGF23 and vitamin D in innate immunity? Pediatr. Nephrol. 2013, 28, 577–582. [Google Scholar] [CrossRef]
- Ramanathan, B.; Davis, E.G.; Ross, C.R.; Blecha, F. Cathelicidins: Microbicidal activity, mechanisms of action, and roles in innate immunity. Microbes Infect. 2002, 4, 361–372. [Google Scholar] [CrossRef]
- Hewison, M. Antibacterial effects of vitamin D. Nat. Rev. Endocrinol. 2011, 7, 337–345. [Google Scholar] [CrossRef]
- Yang, H.Y.; Lu, K.C.; Lee, H.S.; Huang, S.M.; Lin, Y.F.; Wu, C.C.; Salter, D.M.; Su, S.L. Role of the functional toll-like receptor-9 promoter polymorphism (-1237T/C) in increased risk of end-stage renal disease: A case-control study. PLoS ONE 2013, 8, e58444. [Google Scholar] [CrossRef] [Green Version]
- Sadeghi, K.; Wessner, B.; Laggner, U.; Ploder, M.; Tamandl, D.; Friedl, J.; Zugel, U.; Steinmeyer, A.; Pollak, A.; Roth, E.; et al. Vitamin D3 down-regulates monocyte TLR expression and triggers hyporesponsiveness to pathogen-associated molecular patterns. Eur. J. Immunol. 2006, 36, 361–370. [Google Scholar] [CrossRef]
- Ferreira, G.B.; van Etten, E.; Verstuyf, A.; Waer, M.; Overbergh, L.; Gysemans, C.; Mathieu, C. 1,25-Dihydroxyvitamin D3 alters murine dendritic cell behaviour in vitro and in vivo. Diabetes Metab. Res. Rev. 2011, 27, 933–941. [Google Scholar] [CrossRef]
- Zipitis, C.S.; Akobeng, A.K. Vitamin D supplementation in early childhood and risk of type 1 diabetes: A systematic review and meta-analysis. Arch. Dis. Child. 2008, 93, 512–517. [Google Scholar] [CrossRef] [Green Version]
- Hewison, M. An update on vitamin D and human immunity. Clin. Endocrinol. 2012, 76, 315–325. [Google Scholar] [CrossRef]
- Van Belle, T.L.; Gysemans, C.; Mathieu, C. Vitamin D in autoimmune, infectious and allergic diseases: A vital player? Best Pract. Res. Clin. Endocrinol. Metab. 2011, 25, 617–632. [Google Scholar] [CrossRef]
- Palmer, M.T.; Lee, Y.K.; Maynard, C.L.; Oliver, J.R.; Bikle, D.D.; Jetten, A.M.; Weaver, C.T. Lineage-specific effects of 1,25-dihydroxyvitamin D3 on the development of effector CD4 T cells. J. Biol. Chem. 2011, 286, 997–1004. [Google Scholar] [CrossRef] [Green Version]
- Boonstra, A.; Barrat, F.J.; Crain, C.; Heath, V.L.; Savelkoul, H.F.; O’Garra, A. 1alpha,25-Dihydroxyvitamin d3 has a direct effect on naive CD4(+) T cells to enhance the development of Th2 cells. J. Immunol. 2001, 167, 4974–4980. [Google Scholar] [CrossRef] [Green Version]
- Ivanov, I.I.; McKenzie, B.S.; Zhou, L.; Tadokoro, C.E.; Lepelley, A.; Lafaille, J.J.; Cua, D.J.; Littman, D.R. The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell 2006, 126, 1121–1133. [Google Scholar] [CrossRef] [Green Version]
- Jeffery, L.E.; Burke, F.; Mura, M.; Zheng, Y.; Qureshi, O.S.; Hewison, M.; Walker, L.S.; Lammas, D.A.; Raza, K.; Sansom, D.M. 1,25-Dihydroxyvitamin D3 and IL-2 combine to inhibit T cell production of inflammatory cytokines and promote development of regulatory T cells expressing CTLA-4 and FoxP3. J. Immunol. 2009, 183, 5458–5467. [Google Scholar] [CrossRef] [Green Version]
- Sakaguchi, S.; Ono, M.; Setoguchi, R.; Yagi, H.; Hori, S.; Fehervari, Z.; Shimizu, J.; Takahashi, T.; Nomura, T. Foxp3+ CD25+ CD4+ natural regulatory T cells in dominant self-tolerance and autoimmune disease. Immunol. Rev. 2006, 212, 8–27. [Google Scholar] [CrossRef]
- Rudensky, A.Y. Regulatory T cells and Foxp3. Immunol. Rev. 2011, 241, 260–268. [Google Scholar] [CrossRef] [Green Version]
- Gorman, S.; Kuritzky, L.A.; Judge, M.A.; Dixon, K.M.; McGlade, J.P.; Mason, R.S.; Finlay-Jones, J.J.; Hart, P.H. Topically applied 1,25-dihydroxyvitamin D3 enhances the suppressive activity of CD4+CD25+ cells in the draining lymph nodes. J. Immunol. 2007, 179, 6273–6283. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.; Sims, G.P.; Chen, X.X.; Gu, Y.Y.; Chen, S.; Lipsky, P.E. Modulatory effects of 1,25-dihydroxyvitamin D3 on human B cell differentiation. J. Immunol. 2007, 179, 1634–1647. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Quarles, L.D. How fibroblast growth factor 23 works. J. Am. Soc. Nephrol. 2007, 18, 1637–1647. [Google Scholar] [CrossRef] [Green Version]
- Yamazaki, Y.; Tamada, T.; Kasai, N.; Urakawa, I.; Aono, Y.; Hasegawa, H.; Fujita, T.; Kuroki, R.; Yamashita, T.; Fukumoto, S.; et al. Anti-FGF23 neutralizing antibodies show the physiological role and structural features of FGF23. J. Bone Miner. Res. 2008, 23, 1509–1518. [Google Scholar] [CrossRef]
- Shimada, T.; Hasegawa, H.; Yamazaki, Y.; Muto, T.; Hino, R.; Takeuchi, Y.; Fujita, T.; Nakahara, K.; Fukumoto, S.; Yamashita, T. FGF-23 is a potent regulator of vitamin D metabolism and phosphate homeostasis. J. Bone Miner. Res. 2004, 19, 429–435. [Google Scholar] [CrossRef] [Green Version]
- Razzaque, M.S. Does FGF23 toxicity influence the outcome of chronic kidney disease? Nephrol. Dial. Transplant. 2009, 24, 4–7. [Google Scholar] [CrossRef]
- Faul, C.; Amaral, A.P.; Oskouei, B.; Hu, M.C.; Sloan, A.; Isakova, T.; Gutierrez, O.M.; Aguillon-Prada, R.; Lincoln, J.; Hare, J.M.; et al. FGF23 induces left ventricular hypertrophy. J. Clin. Investig. 2011, 121, 4393–4408. [Google Scholar] [CrossRef] [Green Version]
- Isakova, T.; Xie, H.; Yang, W.; Xie, D.; Anderson, A.H.; Scialla, J.; Wahl, P.; Gutierrez, O.M.; Steigerwalt, S.; He, J.; et al. Fibroblast growth factor 23 and risks of mortality and end-stage renal disease in patients with chronic kidney disease. JAMA 2011, 305, 2432–2439. [Google Scholar] [CrossRef] [Green Version]
- Bhan, I.; Shah, A.; Holmes, J.; Isakova, T.; Gutierrez, O.; Burnett, S.M.; Juppner, H.; Wolf, M. Post-transplant hypophosphatemia: Tertiary ‘Hyper-Phosphatoninism’? Kidney Int. 2006, 70, 1486–1494. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; Hsu, R.; Hsu, C.Y.; Kordesch, K.; Nicasio, E.; Cortez, A.; McAlpine, I.; Brady, S.; Zhuo, H.; Kangelaris, K.N.; et al. FGF-23 and PTH levels in patients with acute kidney injury: A cross-sectional case series study. Ann. Intensive Care 2011, 1, 21. [Google Scholar] [CrossRef] [Green Version]
- Leaf, D.E.; Wolf, M.; Waikar, S.S.; Chase, H.; Christov, M.; Cremers, S.; Stern, L. FGF-23 levels in patients with AKI and risk of adverse outcomes. Clin. J. Am. Soc. Nephrol. 2012, 7, 1217–1223. [Google Scholar] [CrossRef] [Green Version]
- Leaf, D.E.; Jacob, K.A.; Srivastava, A.; Chen, M.E.; Christov, M.; Juppner, H.; Sabbisetti, V.S.; Martin, A.; Wolf, M.; Waikar, S.S. Fibroblast growth factor 23 levels associate with AKI and death in critical illness. J. Am. Soc. Nephrol. 2017, 28, 1877–1885. [Google Scholar] [CrossRef]
- Christov, M.; Waikar, S.S.; Pereira, R.C.; Havasi, A.; Leaf, D.E.; Goltzman, D.; Pajevic, P.D.; Wolf, M.; Juppner, H. Plasma FGF23 levels increase rapidly after acute kidney injury. Kidney Int. 2013, 84, 776–785. [Google Scholar] [CrossRef] [Green Version]
- Kelly, K.J.; Williams, W.W., Jr.; Colvin, R.B.; Meehan, S.M.; Springer, T.A.; Gutierrez-Ramos, J.C.; Bonventre, J.V. Intercellular adhesion molecule-1-deficient mice are protected against ischemic renal injury. J. Clin. Investig. 1996, 97, 1056–1063. [Google Scholar] [CrossRef]
- Sugden, J.A.; Davies, J.I.; Witham, M.D.; Morris, A.D.; Struthers, A.D. Vitamin D improves endothelial function in patients with type 2 diabetes mellitus and low vitamin D levels. Diabet. Med. 2008, 25, 320–325. [Google Scholar] [CrossRef]
- Al Mheid, I.; Patel, R.; Murrow, J.; Morris, A.; Rahman, A.; Fike, L.; Kavtaradze, N.; Uphoff, I.; Hooper, C.; Tangpricha, V.; et al. Vitamin D status is associated with arterial stiffness and vascular dysfunction in healthy humans. J. Am. Coll. Cardiol. 2011, 58, 186–192. [Google Scholar] [CrossRef] [Green Version]
- Talmor, Y.; Golan, E.; Benchetrit, S.; Bernheim, J.; Klein, O.; Green, J.; Rashid, G. Calcitriol blunts the deleterious impact of advanced glycation end products on endothelial cells. Am. J. Physiol. Renal Physiol. 2008, 294, F1059–F1064. [Google Scholar] [CrossRef] [Green Version]
- Abuelo, J.G. Normotensive ischemic acute renal failure. N. Engl. J. Med. 2007, 357, 797–805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aksu, U.; Demirci, C.; Ince, C. The pathogenesis of acute kidney injury and the toxic triangle of oxygen, reactive oxygen species and nitric oxide. Contrib. Nephrol. 2011, 174, 119–128. [Google Scholar] [CrossRef] [PubMed]
- Chueh, T.-I.; Zheng, C.-M.; Hou, Y.-C.; Lu, K.-C. Novel evidence of acute kidney injury in COVID-19. J. Clin. Med. 2020, 9, 3547. [Google Scholar] [CrossRef]
- Nadim, M.K.; Forni, L.G.; Mehta, R.L.; Connor, M.J., Jr.; Liu, K.D.; Ostermann, M.; Rimmelé, T.; Zarbock, A.; Bell, S.; Bihorac, A.; et al. COVID-19-associated acute kidney injury: Consensus report of the 25th Acute Disease Quality Initiative (ADQI) workgroup. Nat. Rev. Nephrol. 2020, 16, 747–764. [Google Scholar] [CrossRef]
- Kellum, J.A.; van Till, J.W.O.; Mulligan, G. Targeting acute kidney injury in COVID-19. Nephrol. Dial. Transplant. 2020, 35, 1652–1662. [Google Scholar] [CrossRef]
- Kopustinskiene, D.M.; Bernatoniene, J. Molecular mechanisms of melatonin-mediated cell protection and signaling in health and disease. Pharmaceutics 2021, 13, 129. [Google Scholar] [CrossRef]
- Peng, M.-Y.; Liu, W.-C.; Zheng, J.-Q.; Lu, C.-L.; Hou, Y.-C.; Zheng, C.-M.; Song, J.-Y.; Lu, K.-C.; Chao, Y.-C. Immunological aspects of SARS-CoV-2 infection and the putative beneficial role of vitamin-D. Int. J. Mol. Sci. 2021, 22, 5251. [Google Scholar] [CrossRef]
- Al Kiyumi, M.H.; Kalra, S.; Davies, J.S.; Kalhan, A. The impact of vitamin D deficiency on the severity of symptoms and mortality rate among adult patients with covid-19: A systematic review and meta-analysis. Indian J. Endocrinol. Metab. 2021, 25, 261–282. [Google Scholar] [CrossRef]
- Ranjbar, M.; Niya, M.H.K.; Roham, M.; Rezaie, N.; Yadollahzadeh, M.; Farrokhpour, M.; Azimi, M.; Motamed, N.; Perumal, D.; Tameshkel, F.S.; et al. Serum level of Vitamin D is associated with COVID-19 mortality rate in hospitalized patients. J. Res. Med. Sci. 2021, 26, 112. [Google Scholar] [CrossRef]
- Sempos, C.T.; Durazo-Arvizu, R.A.; Dawson-Hughes, B.; Yetley, E.A.; Looker, A.C.; Schleicher, R.L.; Cao, G.; Burt, V.; Kramer, H.; Bailey, R.L.; et al. Is there a reverse J-shaped association between 25-hydroxyvitamin D and all-cause mortality? Results from the U.S. nationally representative NHANES. J. Clin. Endocrinol. Metab. 2013, 98, 3001–3009. [Google Scholar] [CrossRef] [Green Version]
- Melamed, M.L.; Michos, E.D.; Post, W.; Astor, B. 25-hydroxyvitamin D levels and the risk of mortality in the general population. Arch. Intern. Med. 2008, 168, 1629–1637. [Google Scholar] [CrossRef] [PubMed]
- Kendrick, J.; Cheung, A.K.; Kaufman, J.S.; Greene, T.; Roberts, W.L.; Smits, G.; Chonchol, M. Associations of plasma 25-hydroxyvitamin D and 1,25-dihydroxyvitamin D concentrations with death and progression to maintenance dialysis in patients with advanced kidney disease. Am. J. Kidney Dis. 2012, 60, 567–575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chonchol, M.; Scragg, R. 25-Hydroxyvitamin D, insulin resistance, and kidney function in the Third National Health and Nutrition Examination Survey. Kidney Int. 2007, 71, 134–139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolland, M.J.; Avenell, A.; Baron, J.A.; Grey, A.; MacLennan, G.S.; Gamble, G.D.; Reid, I.R. Effect of calcium supplements on risk of myocardial infarction and cardiovascular events: Meta-analysis. BMJ 2010, 341, c3691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Durup, D.; Jørgensen, H.L.; Christensen, J.; Schwarz, P.; Heegaard, A.M.; Lind, B. A reverse J-shaped association of all-cause mortality with serum 25-hydroxyvitamin D in general practice: The CopD study. J. Clin. Endocrinol. Metab. 2012, 97, 2644–2652. [Google Scholar] [CrossRef] [Green Version]
- Durup, D.; Jørgensen, H.L.; Christensen, J.; Tjønneland, A.; Olsen, A.; Halkjær, J.; Lind, B.; Heegaard, A.-M.; Schwarz, P. A Reverse J-Shaped Association Between Serum 25-Hydroxyvitamin D and Cardiovascular Disease Mortality: The CopD Study. J. Clin. Endocrinol. Metab. 2015, 100, 2339–2346. [Google Scholar] [CrossRef]
- Park, J.M.; Lee, B.; Kim, Y.S.; Hong, K.-W.; Park, Y.C.; Shin, D.H.; Kim, Y.; Han, K.; Kim, K.; Shin, J.; et al. Calcium Supplementation, Risk of Cardiovascular Diseases, and Mortality: A Real-World Study of the Korean National Health Insurance Service Data. Nutrients 2022, 14, 2538. [Google Scholar] [CrossRef]
- Jung, C.Y.; Yun, H.R.; Park, J.T.; Joo, Y.S.; Kim, H.W.; Yoo, T.-H.; Kang, S.-W.; Lee, J.; Chae, D.-W.; Chung, W.; et al. Association of coronary artery calcium with adverse cardiovascular outcomes and death in patients with chronic kidney disease: Results from the KNOW-CKD. Nephrol. Dial. Transplant. 2022, gfac194. [Google Scholar] [CrossRef]
- Maresz, K. Proper Calcium Use: Vitamin K2 as a Promoter of Bone and Cardiovascular Health. Integr. Med. 2015, 14, 34–39. [Google Scholar]
- Morris, H.A. Vitamin D: Can you have too much of a good thing in chronic kidney disease? Kidney Int. 2015, 88, 936–938. [Google Scholar] [CrossRef] [Green Version]
- Kusunoki, Y.; Matsui, I.; Hamano, T.; Shimomura, A.; Mori, D.; Yonemoto, S.; Takabatake, Y.; Tsubakihara, Y.; St-Arnaud, R.; Isaka, Y.; et al. Excess 25-hydroxyvitamin D3 exacerbates tubulointerstitial injury in mice by modulating macrophage phenotype. Kidney Int. 2015, 88, 1013–1029. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| AKI Animal Models | Intervention | Outcomes | Summary of Results |
|---|---|---|---|
| Contrast induced (Wistar albino rats) [33] | Paricalcitol i.p. for 5 days | Attenuated the increase in oxidative biomarkers; histological improvement | Antioxidant effect via the inhibition of lipid oxidation |
| Gentamicin induced (Sprague–Dawley rats) [34] | Paricalcitol s.c. for 14 days | Attenuated the increase in inflammatory cytokines and adhesion molecules; reversed the TGF-1-induced EMT process and extracellular matrix accumulation | Inhibition of renal inflammation and fibrosis through the interruption of the NF-κB/ERK signaling pathway, and preservation of tubular epithelial integrity via inhibition of the EMT process |
| Gentamicin induced (Wistar albino rats) [35] | 1α,25(OH)2D3 s.c. for 8 days | Lowered blood pressure and increased urine volume by increasing GSH levels; no histological improvement | Antioxidant effect; beneficial effects via the RAS system |
| Ischemia/reperfusion induced (C57BL/6 mice) [36] | Paricalcitol i.p. 24 h before ischemia | Attenuated functional deterioration and histological damage; decreased Toll-like receptor 4 and nuclear translocation of the p65 subunit of NF-κB | Suppression of TLR4/NF-κB-mediated inflammation |
| Ischemia/reperfusion induced (Wistar albino rats) [37] | Vitamin D (0.25, 0.5, and 1 mg/kg) for 7 days before ischemia/reperfusion | Attenuated the increase in oxidative biomarkers | Activation of PPAR-γ |
| Cisplatin induced (Sprague–Dawley rats) [38] | Paricalcitol s.c. for 4 days | Attenuated the increase in the expression of p-ERK1/2, P-p38, fibronectin, and CTGF and proapoptotic markers CDK2, cyclin E, and PCNA | Suppression of fibrotic, apoptotic, and proliferative factors via the inhibition of TGF-β1, MAPK signaling, p53-induced apoptosis, and augmentation of p27kip1 |
| Cyclosporin induced (Sprague–Dawley rats) [39] | Paricalcitol s.c. for 28 days | Prevented TGF-β1-induced EMT and extracellular matrix accumulation | Suppression of inflammatory, profibrotic, and apoptotic factors via the inhibition of the NF-κB, Smad, and MAPK signaling pathways |
| Obstructive nephropathy (CD-1 mice) [40] | Paricalcitol s.c. for 7 days | Inhibited RANTES mRNA and protein expression and abolished the ability of tubular cells to recruit lymphocytes and monocytes after TNF-β stimulation | Inhibition of renal inflammatory infiltration and RANTES expression by promoting the VDR-mediated sequestration of NF-κB signaling |
| Obstructive nephropathy (CD-1 mice) [41] | Paricalcitol s.c. for 7 days | Abolished TGF-β1-mediated E-cadherin suppression and α-smooth muscle actin and fibronectin induction in tubular epithelial cells by blocking the EMT directly; completely suppressed the renal induction of Snail | Preservation of tubular epithelial integrity via the suppression of the EMT |
| Lipopolysaccharide (LPS) induced nephropathy (CD-1 mice) [42] | Vitamin D3 (each 25 μg/kg) by gavage at 1, 24, and 48 h before LPS injection | Attenuated LPS-induced inflammatory cytokines and chemokines and adhesion molecules; reinforced the interaction between VDR and NF-κB p65 subunit in the kidney | Vitamin D3 pretreatment downregulated the renal inflammatory response, and the interaction between VDR and the NF-κB p65 subunit provided an explanation |
| Lipopolysaccharide (LPS) induced nephropathy (CD-1 mice) [43] | Vitamin D3 (each 25 μg/kg) by gavage at 1, 24, and 48 h before LPS injection | Alleviated LPS-induced renal GSH depletion, lipid peroxidation, serum and renal NO production, and protein nitration through regulating oxidant and antioxidant enzyme genes | Vitamin D3 pretreatment alleviated LPS-induced renal oxidative stress through regulating oxidant and antioxidant enzyme genes |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hsieh, M.-C.; Hsiao, P.-J.; Liao, M.-T.; Hou, Y.-C.; Chang, Y.-C.; Chiang, W.-F.; Wu, K.-L.; Chan, J.-S.; Lu, K.-C. The Role of Vitamin D in SARS-CoV-2 Infection and Acute Kidney Injury. Int. J. Mol. Sci. 2022, 23, 7368. https://doi.org/10.3390/ijms23137368
Hsieh M-C, Hsiao P-J, Liao M-T, Hou Y-C, Chang Y-C, Chiang W-F, Wu K-L, Chan J-S, Lu K-C. The Role of Vitamin D in SARS-CoV-2 Infection and Acute Kidney Injury. International Journal of Molecular Sciences. 2022; 23(13):7368. https://doi.org/10.3390/ijms23137368
Chicago/Turabian StyleHsieh, Ming-Chun, Po-Jen Hsiao, Min-Tser Liao, Yi-Chou Hou, Ya-Chieh Chang, Wen-Fang Chiang, Kun-Lin Wu, Jenq-Shyong Chan, and Kuo-Cheng Lu. 2022. "The Role of Vitamin D in SARS-CoV-2 Infection and Acute Kidney Injury" International Journal of Molecular Sciences 23, no. 13: 7368. https://doi.org/10.3390/ijms23137368
APA StyleHsieh, M.-C., Hsiao, P.-J., Liao, M.-T., Hou, Y.-C., Chang, Y.-C., Chiang, W.-F., Wu, K.-L., Chan, J.-S., & Lu, K.-C. (2022). The Role of Vitamin D in SARS-CoV-2 Infection and Acute Kidney Injury. International Journal of Molecular Sciences, 23(13), 7368. https://doi.org/10.3390/ijms23137368