
Full-Length Transcriptome Sequencing Reveals Alternative Splicing and lncRNA Regulation during Nodule Development in Glycine max

 and
and
Abstract
:1. Introduction
2. Results
2.1. Genome-Wide Characterization of Full-Length Transcripts in Soybean by Iso-Seq
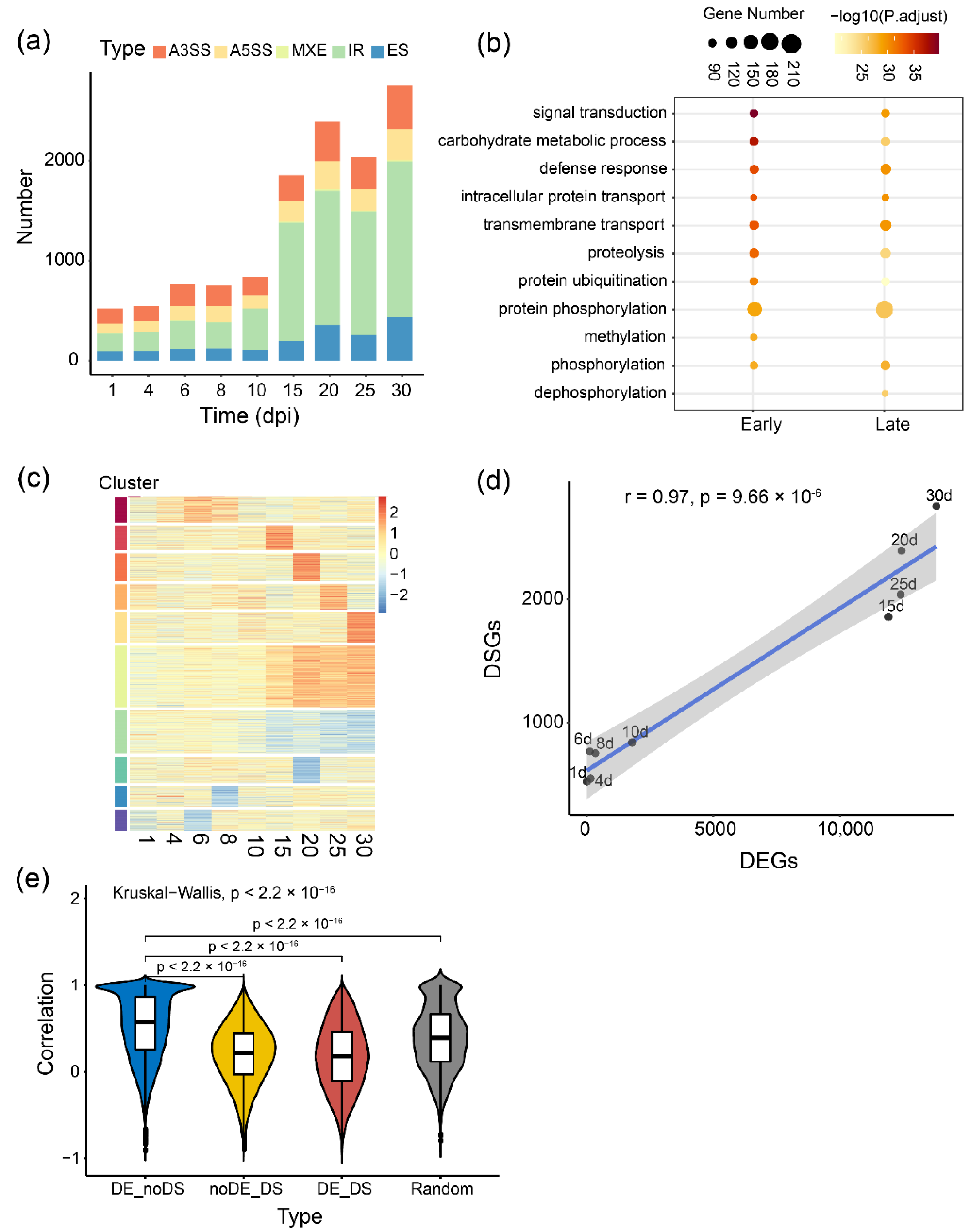
2.2. Comprehensive Temporal Analysis of Alternative Splicing Dynamics during Nodule and Root Development
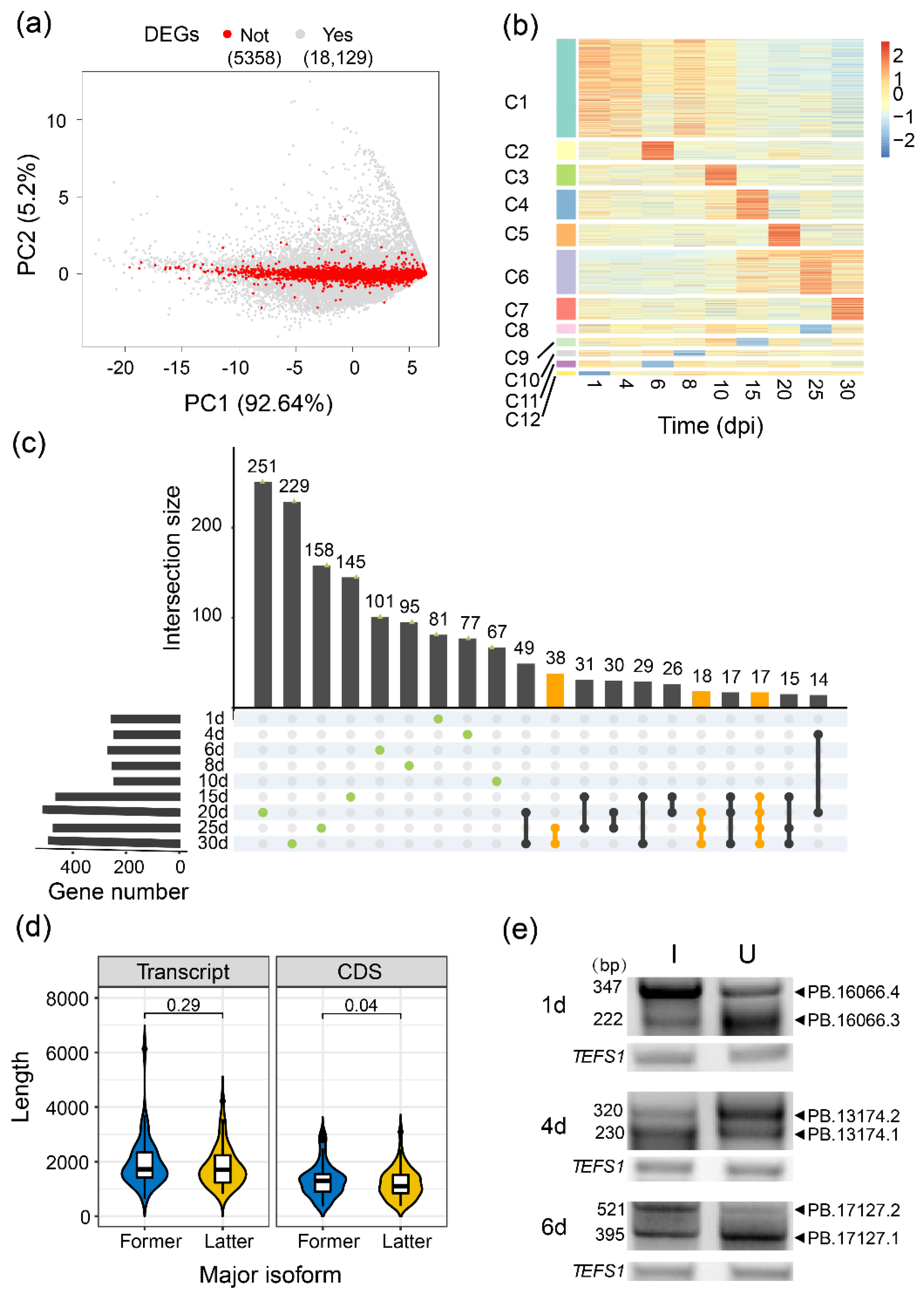
2.3. Differential Transcript Usage Evaluated by Isoform Expression Levels
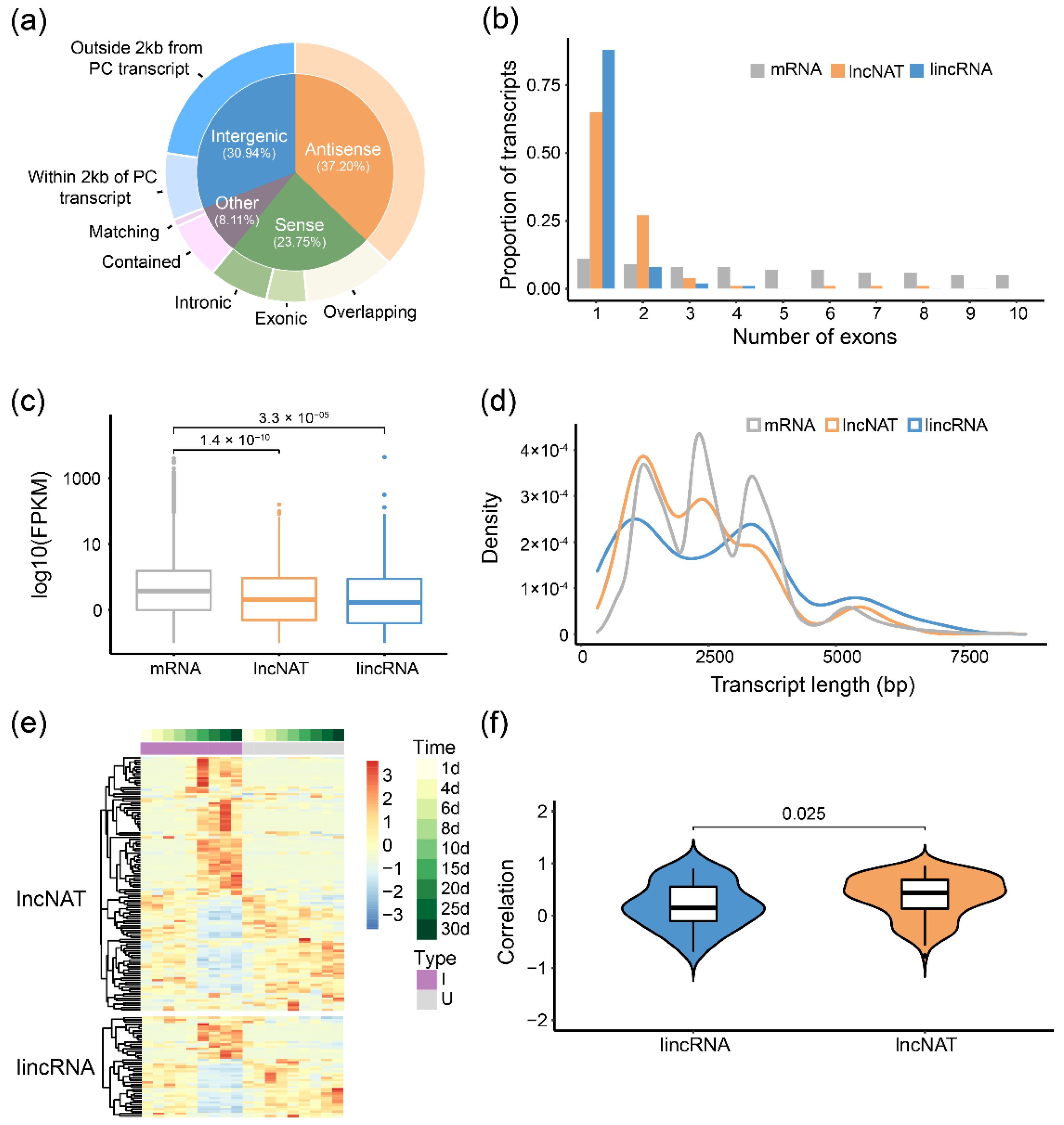
2.4. Long Non-Coding RNA Identified from Iso-Seq Transcripts
3. Discussion
4. Materials and Methods
4.1. Materials and Growth Conditions
4.2. RNA Preparation
4.3. Library Preparation and Sequencing
4.4. RT-PCR for Validation
4.5. Pacbio Data Analysis
4.6. Integrative Analysis of Iso-Seq with Illumina Short Reads
4.7. Annotation of Pacbio Isoforms
4.8. LncRNAs Identification
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hartman, G.L.; West, E.D.; Herman, T.K. Crops that feed the World 2. Soybean—Worldwide production, use, and constraints caused by pathogens and pests. Food Secur. 2011, 3, 5–17. [Google Scholar] [CrossRef]
- Lindstrom, K.; Mousavi, S.A. Effectiveness of nitrogen fixation in rhizobia. Microb. Biotechnol. 2020, 13, 1314–1335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Libault, M.; Farmer, A.; Brechenmacher, L.; Drnevich, J.; Langley, R.J.; Bilgin, D.D.; Radwan, O.; Neece, D.J.; Clough, S.J.; May, G.D.; et al. Complete Transcriptome of the Soybean Root Hair Cell, a Single-Cell Model, and Its Alteration in Response to Bradyrhizobium japonicum Infection. Plant Physiol. 2010, 152, 541–552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, S.L.; Li, R.; Chen, H.F.; Zhang, C.J.; Chen, L.M.; Hao, Q.N.; Chen, S.L.; Shan, Z.H.; Yang, Z.L.; Zhang, X.J.; et al. RNA-Seq analysis of nodule development at five different developmental stages of soybean (Glycine max) inoculated with Bradyrhizobium japonicum strain 113-2. Sci. Rep. 2017, 7, 42248. [Google Scholar] [CrossRef] [PubMed]
- Niyikiza, D.; Piya, S.; Routray, P.; Miao, L.; Kim, W.S.; Burch-Smith, T.; Gill, T.; Sams, C.; Arelli, P.R.; Pantalone, V.; et al. Interactions of gene expression, alternative splicing, and DNA methylation in determining nodule identity. Plant J. 2020, 103, 1744–1766. [Google Scholar] [CrossRef]
- Feng, J.; Lee, T.; Schiessl, K.; Oldroyd, G.E.D. Processing of NODULE INCEPTION controls the transition to nitrogen fixation in root nodules. Science 2021, 374, 629–632. [Google Scholar] [CrossRef]
- Fu, M.; Sun, J.; Li, X.; Guan, Y.; Xie, F. Asymmetric redundancy of soybean Nodule Inception (NIN) genes in root nodule symbiosis. Plant Physiol. 2021, 188, 477–489. [Google Scholar] [CrossRef]
- Pan, Q.; Shai, O.; Lee, L.J.; Frey, B.J.; Blencowe, B.J. Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat. Genet. 2008, 40, 1413–1415. [Google Scholar] [CrossRef]
- Reddy, A.S.; Marquez, Y.; Kalyna, M.; Barta, A. Complexity of the alternative splicing landscape in plants. Plant Cell 2013, 25, 3657–3683. [Google Scholar] [CrossRef] [Green Version]
- Gao, P.; Quilichini, T.D.; Zhai, C.; Qin, L.; Nilsen, K.T.; Li, Q.; Sharpe, A.G.; Kochian, L.V.; Zou, J.; Reddy, A.S.N.; et al. Alternative splicing dynamics and evolutionary divergence during embryogenesis in wheat species. Plant Biotechnol. J. 2021, 19, 1624–1643. [Google Scholar] [CrossRef]
- Chen, M.X.; Zhu, F.Y.; Wang, F.Z.; Ye, N.H.; Gao, B.; Chen, X.; Zhao, S.S.; Fan, T.; Cao, Y.Y.; Liu, T.Y.; et al. Alternative splicing and translation play important roles in hypoxic germination in rice. J. Exp. Bot. 2019, 70, 817–833. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Qin, J.; Tian, X.; Xu, S.; Wang, Y.; Li, H.; Wang, X.; Peng, H.; Yao, Y.; Hu, Z.; et al. Global profiling of alternative splicing landscape responsive to drought, heat and their combination in wheat (Triticum aestivum L.). Plant Biotechnol. J. 2018, 16, 714–726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Guo, Q.; Liu, P.; Huang, J.; Zhang, S.; Yang, G.; Wu, C.; Zheng, C.; Yan, K. Dual roles of the Serine/Arginine-rich splicing factor SR45a in promoting and interacting with nuclear cap-binding complex to modulate the salt stress response in Arabidopsis. New Phytol. 2021, 230, 641–655. [Google Scholar] [CrossRef] [PubMed]
- Dressano, K.; Weckwerth, P.R.; Poretsky, E.; Takahashi, Y.; Villarreal, C.; Shen, Z.; Schroeder, J.I.; Briggs, S.P.; Huffaker, A. Dynamic regulation of Pep-induced immunity through post-translational control of defence transcript splicing. Nat. Plants 2020, 6, 1008–1019. [Google Scholar] [CrossRef]
- Huisman, R.; Hontelez, J.; Mysore, K.S.; Wen, J.; Bisseling, T.; Limpens, E. A symbiosis-dedicated SYNTAXIN OF PLANTS 13II isoform controls the formation of a stable host-microbe interface in symbiosis. New Phytol. 2016, 211, 1338–1351. [Google Scholar] [CrossRef] [PubMed]
- Pan, H.; Oztas, O.; Zhang, X.; Wu, X.; Stonoha, C.; Wang, E.; Wang, B.; Wang, D. A symbiotic SNARE protein generated by alternative termination of transcription. Nat. Plants 2016, 2, 15197. [Google Scholar] [CrossRef]
- Bourcy, M.; Brocard, L.; Pislariu, C.I.; Cosson, V.; Mergaert, P.; Tadege, M.; Mysore, K.S.; Udvardi, M.K.; Gourion, B.; Ratet, P. Medicago truncatula DNF2 is a PI-PLC-XD-containing protein required for bacteroid persistence and prevention of nodule early senescence and defense-like reactions. New Phytol. 2013, 197, 1250–1261. [Google Scholar] [CrossRef]
- Combier, J.P.; de Billy, F.; Gamas, P.; Niebel, A.; Rivas, S. Trans-regulation of the expression of the transcription factor MtHAP2-1 by a uORF controls root nodule development. Genes Dev. 2008, 22, 1549–1559. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Zhu, H.; Jin, L.; Chen, T.; Wang, L.; Kang, H.; Hong, Z.; Zhang, Z. Splice variants of the SIP1 transcripts play a role in nodule organogenesis in Lotus japonicus. Plant Mol. Biol. 2013, 82, 97–111. [Google Scholar] [CrossRef]
- Liu, Y.; Gonzalez-Porta, M.; Santos, S.; Brazma, A.; Marioni, J.C.; Aebersold, R.; Venkitaraman, A.R.; Wickramasinghe, V.O. Impact of Alternative Splicing on the Human Proteome. Cell Rep. 2017, 20, 1229–1241. [Google Scholar] [CrossRef] [Green Version]
- Kim, E.; Magen, A.; Ast, G. Different levels of alternative splicing among eukaryotes. Nucleic Acids Res. 2007, 35, 125–131. [Google Scholar] [CrossRef] [Green Version]
- Martin, G.; Marquez, Y.; Mantica, F.; Duque, P.; Irimia, M. Alternative splicing landscapes in Arabidopsis thaliana across tissues and stress conditions highlight major functional differences with animals. Genome Biol. 2021, 22, 35. [Google Scholar] [CrossRef] [PubMed]
- Gonzàlez-Porta, M.; Frankish, A.; Rung, J.; Harrow, J.; Brazma, A. Transcriptome analysis of human tissues and cell lines reveals one dominant transcript per gene. Genome Biol. 2013, 14, R70. [Google Scholar] [CrossRef] [Green Version]
- Vaneechoutte, D.; Estrada, A.R.; Lin, Y.C.; Loraine, A.E.; Vandepoele, K. Genome-wide characterization of differential transcript usage in Arabidopsis thaliana. Plant J. 2017, 92, 1218–1231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, X.; Li, J.; Lian, B.; Gu, H.; Li, Y.; Qi, Y. Global identification of Arabidopsis lncRNAs reveals the regulation of MAF4 by a natural antisense RNA. Nat. Commun. 2018, 9, 5056. [Google Scholar] [CrossRef] [Green Version]
- Johnsson, P.; Ziegenhain, C.; Hartmanis, L.; Hendriks, G.J.; Hagemann-Jensen, M.; Reinius, B.; Sandberg, R. Transcriptional kinetics and molecular functions of long noncoding RNAs. Nat. Genet. 2022, 54, 306–317. [Google Scholar] [CrossRef] [PubMed]
- Derrien, T.; Johnson, R.; Bussotti, G.; Tanzer, A.; Djebali, S.; Tilgner, H.; Guernec, G.; Martin, D.; Merkel, A.; Knowles, D.G.; et al. The GENCODE v7 catalog of human long noncoding RNAs: Analysis of their gene structure, evolution, and expression. Genome Res. 2012, 22, 1775–1789. [Google Scholar] [CrossRef] [Green Version]
- Chekanova, J.A. Long non-coding RNAs and their functions in plants. Curr. Opin. Plant Biol. 2015, 27, 207–216. [Google Scholar] [CrossRef] [Green Version]
- Kopp, F.; Mendell, J.T. Functional Classification and Experimental Dissection of Long Noncoding RNAs. Cell 2018, 172, 393–407. [Google Scholar] [CrossRef] [Green Version]
- Zou, C.; Wang, Q.; Lu, C.; Yang, W.; Zhang, Y.; Cheng, H.; Feng, X.; Prosper, M.A.; Song, G. Transcriptome analysis reveals long noncoding RNAs involved in fiber development in cotton (Gossypium arboreum). Sci. China Life Sci. 2016, 59, 164–171. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Q.; Wan, Q.; Jiang, Y.; Liu, J.; Qiang, L.; Sun, L. A Landscape of Murine Long Non-Coding RNAs Reveals the Leading Transcriptome Alterations in Adipose Tissue during Aging. Cell Rep. 2020, 31, 107694. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Lin, W.; Ku, Y.S.; Wong, F.L.; Li, M.W.; Lam, H.M.; Ngai, S.M.; Chan, T.F. Analysis of Soybean Long Non-Coding RNAs Reveals a Subset of Small Peptide-Coding Transcripts. Plant Physiol. 2020, 182, 1359–1374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Traubenik, S.; Reynoso, M.A.; Hobecker, K.; Lancia, M.; Hummel, M.; Rosen, B.; Town, C.; Bailey-Serres, J.; Blanco, F.; Zanetti, M.E. Reprogramming of Root Cells during Nitrogen-Fixing Symbiosis Involves Dynamic Polysome Association of Coding and Noncoding RNAs. Plant Cell 2020, 32, 352–373. [Google Scholar] [CrossRef] [PubMed]
- Ganguly, P.; Roy, D.; Das, T.; Kundu, A.; Cartieaux, F.; Ghosh, Z.; DasGupta, M. The Natural Antisense Transcript DONE40 Derived from the lncRNA ENOD40 Locus Interacts with SET Domain Protein ASHR3 During Inception of Symbiosis in Arachis hypogaea. Mol. Plant-Microbe Interact. MPMI 2021, 34, 1057–1070. [Google Scholar] [CrossRef]
- Roy, S.W.; Gilbert, W. The evolution of spliceosomal introns: Patterns, puzzles and progress. Nat. Rev. Genet. 2006, 7, 211–221. [Google Scholar]
- Parikshak, N.N.; Swarup, V.; Belgard, T.G.; Irimia, M.; Ramaswami, G.; Gandal, M.J.; Hartl, C.; Leppa, V.; de la Torre Ubieta, L.; Huang, J.; et al. Genome-wide changes in lncRNA, splicing, and regional gene expression patterns in autism. Nature 2016, 540, 423–427. [Google Scholar] [CrossRef]
- Shen, Y.; Zhou, Z.; Wang, Z.; Li, W.; Fang, C.; Wu, M.; Ma, Y.; Liu, T.; Kong, L.A.; Peng, D.L.; et al. Global dissection of alternative splicing in paleopolyploid soybean. Plant Cell 2014, 26, 996–1008. [Google Scholar] [CrossRef] [Green Version]
- Hughes, T.A. Regulation of gene expression by alternative untranslated regions. Trends Genet. 2006, 22, 119–122. [Google Scholar] [CrossRef]
- Kajdasz, A.; Niewiadomska, D.; Sekrecki, M.; Sobczak, K. Distribution of alternative untranslated regions within the mRNA of the CELF1 splicing factor affects its expression. Sci. Rep. 2022, 12, 190. [Google Scholar] [CrossRef]
- Yi, X.; Liu, J.; Chen, S.; Wu, H.; Liu, M.; Xu, Q.; Lei, L.; Lee, S.; Zhang, B.; Kudrna, D.; et al. Genome assembly of the JD17 soybean provides a new reference genome for Comparative genomics. G3 Genes Genomes Genet. 2022, 12, jkac017. [Google Scholar] [CrossRef]
- Chu, J.S.; Peng, B.; Tang, K.; Yi, X.; Zhou, H.; Wang, H.; Li, G.; Leng, J.; Chen, N.; Feng, X. Eight soybean reference genome resources from varying latitudes and agronomic traits. Sci. Data 2021, 8, 164. [Google Scholar] [CrossRef] [PubMed]
- Murray, J.D.; Karas, B.J.; Sato, S.; Tabata, S.; Amyot, L.; Szczyglowski, K. A cytokinin perception mutant colonized by Rhizobium in the absence of nodule organogenesis. Science 2007, 315, 101–104. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Rizzo, S.; Crespi, M.; Frugier, F. The Medicago truncatula CRE1 cytokinin receptor regulates lateral root development and early symbiotic interaction with Sinorhizobium meliloti. Plant Cell 2006, 18, 2680–2693. [Google Scholar] [CrossRef] [Green Version]
- Tirichine, L.; Sandal, N.; Madsen, L.H.; Radutoiu, S.; Albrektsen, A.S.; Sato, S.; Asamizu, E.; Tabata, S.; Stougaard, J. A gain-of-function mutation in a cytokinin receptor triggers spontaneous root nodule organogenesis. Science 2007, 315, 104–107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vernie, T.; Kim, J.; Frances, L.; Ding, Y.; Sun, J.; Guan, D.; Niebel, A.; Gifford, M.L.; de Carvalho-Niebel, F.; Oldroyd, G.E. The NIN Transcription Factor Coordinates Diverse Nodulation Programs in Different Tissues of the Medicago truncatula Root. Plant Cell 2015, 27, 3410–3424. [Google Scholar] [CrossRef] [Green Version]
- Tsikou, D.; Yan, Z.; Holt, D.B.; Abel, N.B.; Reid, D.E.; Madsen, L.H.; Bhasin, H.; Sexauer, M.; Stougaard, J.; Markmann, K. Systemic control of legume susceptibility to rhizobial infection by a mobile microRNA. Science 2018, 362, 233–236. [Google Scholar] [CrossRef]
- Miri, M.; Janakirama, P.; Huebert, T.; Ross, L.; McDowell, T.; Orosz, K.; Markmann, K.; Szczyglowski, K. Inside out: Root cortex-localized LHK1 cytokinin receptor limits epidermal infection of Lotus japonicus roots by Mesorhizobium loti. New Phytol. 2019, 222, 1523–1537. [Google Scholar] [CrossRef]
- Cortes, A.J.; Lopez-Hernandez, F.; Osorio-Rodriguez, D. Predicting Thermal Adaptation by Looking Into Populations’ Genomic Past. Front. Genet. 2020, 11, 564515. [Google Scholar] [CrossRef]
- Shen, Y.; Zhou, G.; Liang, C.; Tian, Z. Omics-based interdisciplinarity is accelerating plant breeding. Curr. Opin. Plant Biol. 2022, 66, 102167. [Google Scholar] [CrossRef]
- Di, C.; Yuan, J.; Wu, Y.; Li, J.; Lin, H.; Hu, L.; Zhang, T.; Qi, Y.; Gerstein, M.B.; Guo, Y.; et al. Characterization of stress-responsive lncRNAs in Arabidopsis thaliana by integrating expression, epigenetic and structural features. Plant J. 2014, 80, 848–861. [Google Scholar] [CrossRef]
- Wang, B.; Tseng, E.; Regulski, M.; Clark, T.A.; Hon, T.; Jiao, Y.; Lu, Z.; Olson, A.; Stein, J.C.; Ware, D. Unveiling the complexity of the maize transcriptome by single-molecule long-read sequencing. Nat. Commun. 2016, 7, 11708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melé, M.; Mattioli, K.; Mallard, W.; Shechner, D.M.; Gerhardinger, C.; Rinn, J.L. Chromatin environment, transcriptional regulation, and splicing distinguish lincRNAs and mRNAs. Genome Res. 2017, 27, 27–37. [Google Scholar] [CrossRef] [Green Version]
- Bardou, F.; Ariel, F.; Simpson, C.G.; Romero-Barrios, N.; Laporte, P.; Balzergue, S.; Brown, J.W.; Crespi, M. Long noncoding RNA modulates alternative splicing regulators in Arabidopsis. Dev. Cell 2014, 30, 166–176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krystal, G.W.; Armstrong, B.C.; Battey, J.F. N-myc mRNA forms an RNA-RNA duplex with endogenous antisense transcripts. Mol. Cell Biol. 1990, 10, 4180–4191. [Google Scholar] [PubMed] [Green Version]
- Drechsel, G.; Kahles, A.; Kesarwani, A.K.; Stauffer, E.; Behr, J.; Drewe, P.; Ratsch, G.; Wachter, A. Nonsense-mediated decay of alternative precursor mRNA splicing variants is a major determinant of the Arabidopsis steady state transcriptome. Plant Cell 2013, 25, 3726–3742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lykke-Andersen, S.; Jensen, T.H. Nonsense-mediated mRNA decay: An intricate machinery that shapes transcriptomes. Nat. Rev. Mol. Cell Biol. 2015, 16, 665–677. [Google Scholar] [CrossRef] [Green Version]
- Kalyna, M.; Simpson, C.G.; Syed, N.H.; Lewandowska, D.; Marquez, Y.; Kusenda, B.; Marshall, J.; Fuller, J.; Cardle, L.; McNicol, J.; et al. Alternative splicing and nonsense-mediated decay modulate expression of important regulatory genes in Arabidopsis. Nucleic Acids Res. 2012, 40, 2454–2469. [Google Scholar] [CrossRef] [Green Version]
- Vattem, K.M.; Wek, R.C. Reinitiation involving upstream ORFs regulates ATF4 mRNA translation in mammalian cells. Proc. Natl. Acad. Sci. USA 2004, 101, 11269–11274. [Google Scholar] [CrossRef] [Green Version]
- Kereszt, A.; Li, D.; Indrasumunar, A.; Nguyen, C.D.; Nontachaiyapoom, S.; Kinkema, M.; Gresshoff, P.M. Agrobacterium rhizogenes-mediated transformation of soybean to study root biology. Nat. Protoc. 2007, 2, 948–952. [Google Scholar] [CrossRef]
- Fahraeus, G. The infection of clover root hairs by nodule bacteria studied by a simple glass slide technique. J. Gen. Microbiol. 1957, 16, 374–381. [Google Scholar] [CrossRef] [Green Version]
- Cole, M.A.; Elkan, G.H. Transmissible resistance to penicillin G, neomycin, and chloramphenicol in Rhizobium japonicum. Antmicrob. Agents Chemother. 1973, 4, 248–253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, T.D.; Watanabe, C.K. GMAP: A genomic mapping and alignment program for mRNA and EST sequences. Bioinformatics 2005, 21, 1859–1875. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foissac, S.; Sammeth, M. ASTALAVISTA: Dynamic and flexible analysis of alternative splicing events in custom gene datasets. Nucleic Acids Res. 2007, 35, W297–W299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzberg, S.L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 2019, 37, 907–915. [Google Scholar] [CrossRef]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef] [Green Version]
- Trapnell, C.; Roberts, A.; Goff, L.; Pertea, G.; Kim, D.; Kelley, D.R.; Pimentel, H.; Salzberg, S.L.; Rinn, J.L.; Pachter, L. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 2012, 7, 562–578. [Google Scholar] [CrossRef] [Green Version]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Shen, S.; Park, J.W.; Lu, Z.X.; Lin, L.; Henry, M.D.; Wu, Y.N.; Zhou, Q.; Xing, Y. rMATS: Robust and flexible detection of differential alternative splicing from replicate RNA-Seq data. Proc. Natl. Acad. Sci. USA 2014, 111, E5593–E5601. [Google Scholar] [CrossRef] [Green Version]
- Haas, B.J.; Papanicolaou, A.; Yassour, M.; Grabherr, M.; Blood, P.D.; Bowden, J.; Couger, M.B.; Eccles, D.; Li, B.; Lieber, M.; et al. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat. Protoc. 2013, 8, 1494–1512. [Google Scholar] [CrossRef]
- Finn, R.D.; Attwood, T.K.; Babbitt, P.C.; Bateman, A.; Bork, P.; Bridge, A.J.; Chang, H.-Y.; Dosztányi, Z.; El-Gebali, S.; Fraser, M.; et al. InterPro in 2017—beyond protein family and domain annotations. Nucleic Acids Res. 2017, 45, D190–D199. [Google Scholar] [CrossRef] [PubMed]
- Conesa, A.; Gotz, S.; Garcia-Gomez, J.M.; Terol, J.; Talon, M.; Robles, M. Blast2GO: A universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 2005, 21, 3674–3676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, G.; Wang, L.G.; Han, Y.; He, Q.Y. clusterProfiler: An R package for comparing biological themes among gene clusters. OMICS 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Letunic, I.; Khedkar, S.; Bork, P. SMART: Recent updates, new developments and status in 2020. Nucleic Acids Res. 2020, 49, D458–D460. [Google Scholar] [CrossRef] [PubMed]
- Li, A.; Zhang, J.; Zhou, Z. PLEK: A tool for predicting long non-coding RNAs and messenger RNAs based on an improved k-mer scheme. BMC Bioinform. 2014, 15, 311. [Google Scholar] [CrossRef] [Green Version]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Library Size (kb) | SMRT Cells | Polymerase Reads | FLNC 1 | High Quality Isoforms |
|---|---|---|---|---|
| 1–2 | 12 | 905,500 | 419,736 | 117,970 |
| 2–4 | 12 | 1,231,202 | 612,254 | 144,968 |
| 3–5 | 12 | 1,238,704 | 542,507 | 105,423 |
| 3.5–6 | 8 | 697,016 | 161,752 | 55,532 |
| 4–10 | 8 | 818,271 | 246,844 | 28,810 |
| Total | 52 | 4,890,693 | 1,983,093 | 452,703 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, J.; Chen, S.; Liu, M.; Chen, Y.; Fan, W.; Lee, S.; Xiao, H.; Kudrna, D.; Li, Z.; Chen, X.; et al. Full-Length Transcriptome Sequencing Reveals Alternative Splicing and lncRNA Regulation during Nodule Development in Glycine max. Int. J. Mol. Sci. 2022, 23, 7371. https://doi.org/10.3390/ijms23137371
Liu J, Chen S, Liu M, Chen Y, Fan W, Lee S, Xiao H, Kudrna D, Li Z, Chen X, et al. Full-Length Transcriptome Sequencing Reveals Alternative Splicing and lncRNA Regulation during Nodule Development in Glycine max. International Journal of Molecular Sciences. 2022; 23(13):7371. https://doi.org/10.3390/ijms23137371
Chicago/Turabian StyleLiu, Jing, Shengcai Chen, Min Liu, Yimian Chen, Wei Fan, Seunghee Lee, Han Xiao, Dave Kudrna, Zixin Li, Xu Chen, and et al. 2022. "Full-Length Transcriptome Sequencing Reveals Alternative Splicing and lncRNA Regulation during Nodule Development in Glycine max" International Journal of Molecular Sciences 23, no. 13: 7371. https://doi.org/10.3390/ijms23137371
APA StyleLiu, J., Chen, S., Liu, M., Chen, Y., Fan, W., Lee, S., Xiao, H., Kudrna, D., Li, Z., Chen, X., Peng, Y., Tian, K., Zhang, B., Wing, R. A., Zhang, J., & Wang, X. (2022). Full-Length Transcriptome Sequencing Reveals Alternative Splicing and lncRNA Regulation during Nodule Development in Glycine max. International Journal of Molecular Sciences, 23(13), 7371. https://doi.org/10.3390/ijms23137371