
The Specific Role of Dermatan Sulfate as an Instructive Glycosaminoglycan in Tissue Development



Abstract
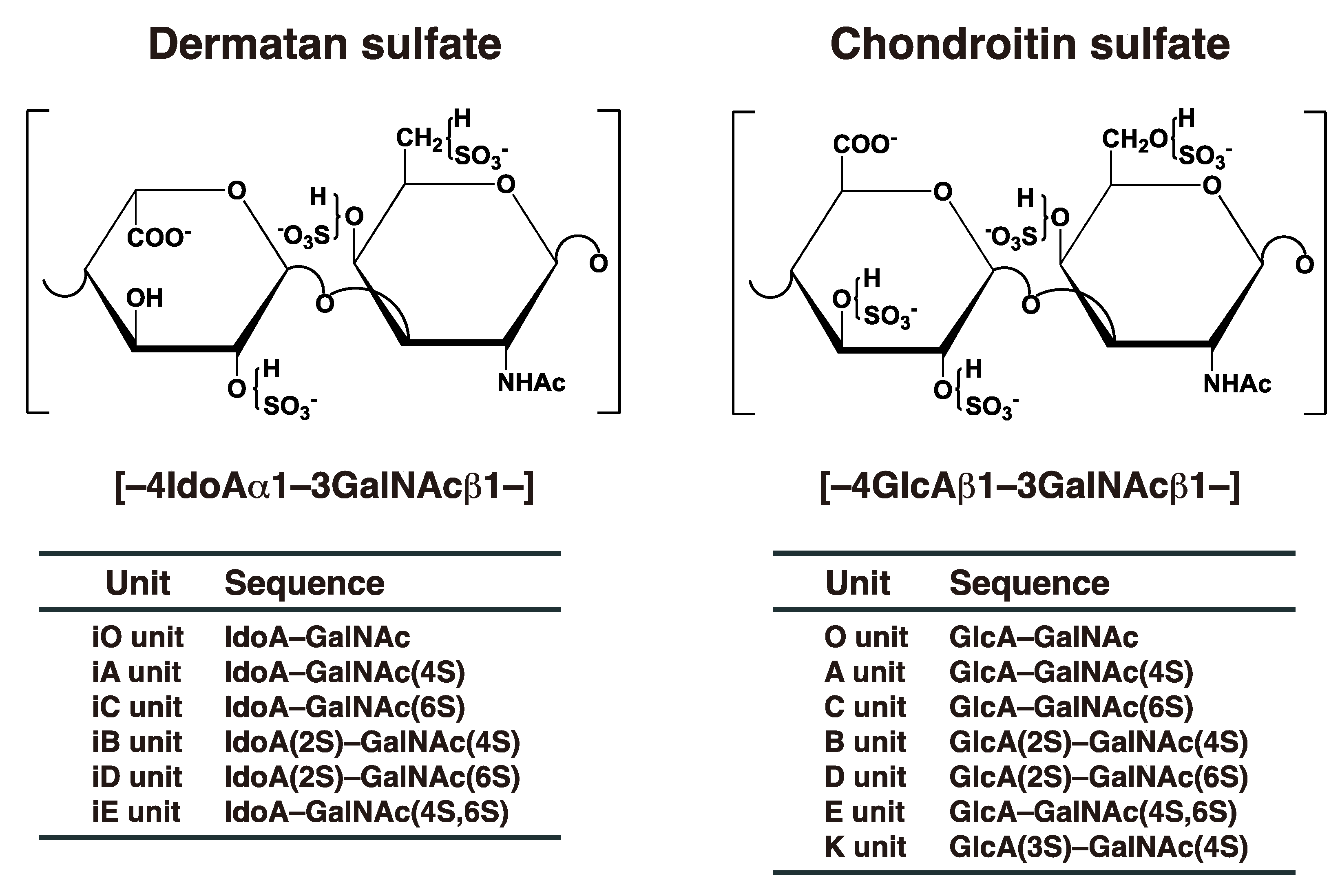
:1. Introduction
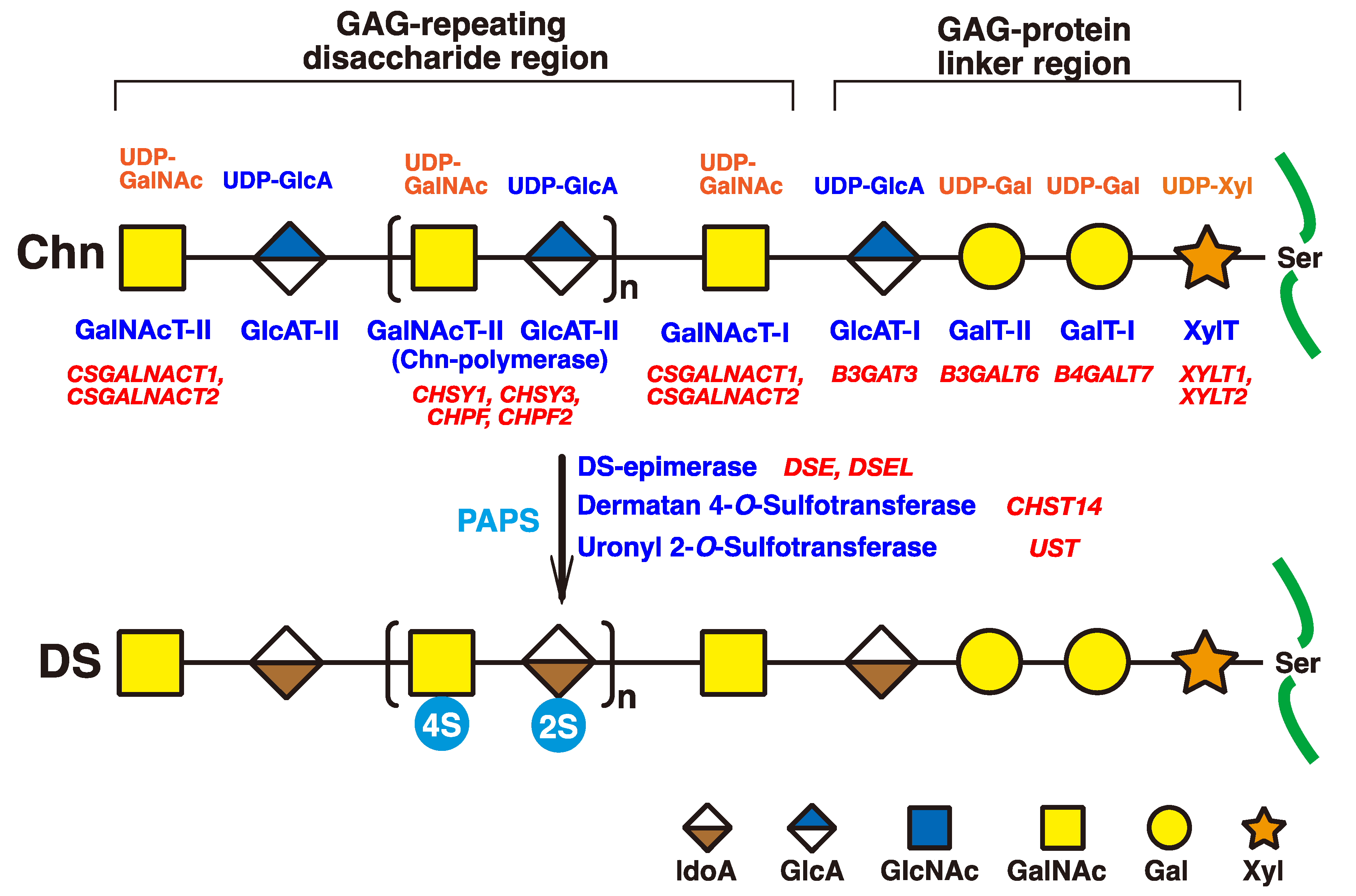
2. Biosynthesis of DS Chains
3. Classical and Additional Functions of DS
3.1. Classical Functions of DS

{kind=link}
{kind=link}
| DS Origin | Molecular Weight | IdoA Content | Binding Protein(s) | Biological Activities | Reference |
|---|---|---|---|---|---|
| Porcine skin | 11–25 kDa | ~75% | Heparin cofactor II, FGF2, FGF7, collagen | Anti-coagulation, cell growth, assembly of extracellular matrix | [2,47,51,66] |
| Ascidian (A. nigra) | –– | ~100% | Heparin cofactor II | Anti-coagulation, neurite outgrowth-promoting activity | [57,67] |
| Ascidian (S. plicata) | –– | ~70% | Heparin cofactor II | Anti-coagulation, neurite outgrowth-promoting activity | [57,68] |
| Embryonic sea urcin | –– | ~100% | –– | Neurite outgrowth-promoting activity | [57,69] |
| Hagfish notochord | 18 kDa | 60~75% | FGF2, FGF10, FGF16, FGF18, Midkine, Pleiotrophin, Heparin-binding EGF-like growth factor (HB-EGF), Vascular endothelial growth factor (VEGF), BDNF, GDNF | Neurite outgrowth-promoting activity | [58] |
| Shark skin | 70 kDa | 42% | FGF2, FGF10, FGF16, FGF18, Midkine, Pleiotrophin, HB-EGF, VEGF, BDNF, GDNF, heparin cofactor II | Neurite outgrowth-promoting activity, anti-coagulation | [59,61,70] |
3.2. Recent Additional Functions of DS
4. Knockout and Mutant Mice of Biosynthetic Enzymes of DS
4.1. Dse
| Coding Genes | Phenotypes of Knockout or Mutant Mice | Human Genetic Disorders (MIM Numbers) | Ref. for Knockout Mice | Ref. for Human Disorders |
|---|---|---|---|---|
| Dse | Thicker collagen fibrils in the dermis and hypodermis, smaller body weight, kinked tail, defects in fetal abdominal wall, exencephaly, and spina bifida. | Ehlers–Danlos syndrome musculocontractural type 2 (615539, 605942) | [28,77] | [21,81,82,83,84] |
| Dsel | Normal extracellular matrix features in the brain. | Bipolar disorder; depressive disorder; diaphragmatic hernia; microphthalmia (611125) | [77,78] | [75,85,86] |
| Chst14 | Increased skin fragility, disorganized collagen fibers, thoracic kyphosis, reduced fertility, kinked tail, myopathy-related phenotypes such as variation in fiber size and spread of the muscle interstitium, smaller body mass, alterations in the vascular structure of the placenta, an abnormal structure of the basement membrane of capillaries in the placental villus, better recovery after femoral nerve injury. | Ehlers–Danlos syndrome musculocontractural type 1; Ehlers–Danlos syndrome, type VIB; adducted thumb-clubfoot syndrome (601776, 608429) | [29,30,31,32,79,80] | [22,23,24,81,87,88,89,90,91,92,93,94,95,96] |
4.2. Dsel
4.3. Dse/Dsel
4.4. Chst14
5. Human Disorders Affecting the Skeleton and Skin Caused by Disturbances in DS Biosynthetic Enzymes
5.1. DSE
5.2. DSEL
5.3. CHST14
6. Conclusions and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Karl, M.; Chaffee, E. The mucopolysaccharides of skin. J. Biol. Chem. 1941, 138, 491–499. [Google Scholar]
- Fransson, L.-A.; Cheng, F.; Yoshida, K.; Heinegård, D.; Malmström, A.; Schmidtchen, A. Patterns of epimerization and sulphation in dermatan sulphate chains. In Dermatan Sulphate Proteoglycans: Chemistry, Biology, Chemical Pathology; Scott, J.E., Ed.; Portland Press: London, UK, 1993; pp. 11–25. [Google Scholar]
- Thelin, M.A.; Bartolini, B.; Axelsson, J.; Gustafsson, R.; Tykesson, E.; Pera, E.; Oldberg, Å.; Maccarana, M.; Malmström, A. Biological functions of iduronic acid in chondroitin/dermatan sulfate. FEBS J. 2013, 280, 2431–2446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trowbridge, J.M.; Gallo, R.L. Dermatan sulfate: New functions from an old glycosaminoglycan. Glycobiology 2002, 12, 117R–125R. [Google Scholar] [CrossRef] [PubMed]
- Mizumoto, S.; Yamada, S.; Sugahara, K. Molecular interactions between chondroitin-dermatan sulfate and growth factors/receptors/matrix proteins. Curr. Opin. Struct. Biol. 2015, 34, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Sugahara, K.; Mikami, T. Chondroitin/dermatan sulfate in the central nervous system. Curr. Opin. Struct. Biol. 2007, 17, 536–545. [Google Scholar] [CrossRef]
- Hayes, A.J.; Melrose, J. Glycans and glycosaminoglycans in neurobiology: Key regulators of neuronal cell function and fate. Biochem. J. 2018, 475, 2511–2545. [Google Scholar] [CrossRef]
- Hayes, A.; Sugahara, K.; Farrugia, B.; Whitelock, J.M.; Caterson, B.; Melrose, J. Biodiversity of CS-proteoglycan sulphation motifs: Chemical messenger recognition modules with roles in information transfer, control of cellular behaviour and tissue morphogenesis. Biochem. J. 2018, 475, 587–620. [Google Scholar] [CrossRef] [Green Version]
- Hayes, A.J.; Melrose, J. Neural Tissue Homeostasis and Repair Is Regulated via CS and DS Proteoglycan Motifs. Front. Cell Dev. Biol. 2021, 9, 696640. [Google Scholar] [CrossRef]
- Iozzo, R.V. Matrix proteoglycans: From molecular design to cellular function. Annu. Rev. Biochem. 1998, 67, 609–652. [Google Scholar] [CrossRef] [Green Version]
- Kinoshita, A.; Sugahara, K. Microanalysis of glycosaminoglycan-derived oligosaccharides labeled with a fluorophore 2-aminobenzamide by high-performance liquid chromatography: Application to disaccharide composition analysis and exosequencing of oligosaccharides. Anal. Biochem. 1999, 269, 367–378. [Google Scholar] [CrossRef]
- Malfait, F.; Francomano, C.; Byers, P.; Belmont, J.; Berglund, B.; Black, J.; Bloom, L.; Bowen, J.M.; Brady, A.F.; Burrows, N.P.; et al. The 2017 international classification of the Ehlers-Danlos syndromes. Am. J. Med. Genet. C Semin. Med. Genet. 2017, 175, 8–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malfait, F.; Castori, M.; Francomano, C.A.; Giunta, C.; Kosho, T.; Byers, P.H. The Ehlers-Danlos syndromes. Nat. Rev. Dis. Primers 2020, 6, 64. [Google Scholar] [CrossRef] [PubMed]
- Quentin, E.; Gladen, A.; Rodén, L.; Kresse, H. A genetic defect in the biosynthesis of dermatan sulfate proteoglycan: Galactosyltransferase I deficiency in fibroblasts from a patient with a progeroid syndrome. Proc. Natl. Acad. Sci. USA 1990, 87, 1342–1346. [Google Scholar] [PubMed] [Green Version]
- Almeida, R.; Levery, S.B.; Mandel, U.; Kresse, H.; Schwientek, T.; Bennett, E.P.; Clausen, H. Cloning and expression of a proteoglycan UDP-galactose:beta-xylose beta1,4-galactosyltransferase I. A seventh member of the human beta4-galactosyltransferase gene family. J. Biol. Chem. 1999, 274, 26165–26171. [Google Scholar] [CrossRef] [Green Version]
- Okajima, T.; Fukumoto, S.; Furukawa, K.; Urano, T. Molecular basis for the progeroid variant of Ehlers-Danlos syndrome. Identification and characterization of two mutations in galactosyltransferase I gene. J. Biol. Chem. 1999, 274, 28841–28844. [Google Scholar] [CrossRef] [Green Version]
- Nakajima, M.; Mizumoto, S.; Miyake, N.; Kogawa, R.; Iida, A.; Ito, H.; Kitoh, H.; Hirayama, A.; Mitsubuchi, H.; Miyazaki, O.; et al. Mutations in B3GALT6, which encodes a glycosaminoglycan linker region enzyme, cause a spectrum of skeletal and connective tissue disorders. Am. J. Hum. Genet. 2013, 92, 927–934. [Google Scholar] [CrossRef] [Green Version]
- Malfait, F.; Kariminejad, A.; Van Damme, T.; Gauche, C.; Syx, D.; Merhi-Soussi, F.; Gulberti, S.; Symoens, S.; Vanhauwaert, S.; Willaert, A.; et al. Defective initiation of glycosaminoglycan synthesis due to B3GALT6 mutations causes a pleiotropic Ehlers-Danlos-syndrome-like connective tissue disorder. Am. J. Hum. Genet. 2013, 92, 935–945. [Google Scholar]
- Okajima, T.; Yoshida, K.; Kondo, T.; Furukawa, K. Human homolog of Caenorhabditis elegans sqv-3 gene is galactosyltransferase I involved in the biosynthesis of the glycosaminoglycan-protein linkage region of proteoglycans. J. Biol. Chem. 1999, 274, 22915–22918. [Google Scholar]
- Bai, X.; Zhou, D.; Brown, J.R.; Crawford, B.E.; Hennet, T.; Esko, J.D. Biosynthesis of the linkage region of glycosaminoglycans: Cloning and activity of galactosyltransferase II, the sixth member of the β1,3-galactosyltransferase family (beta3GalT6). J. Biol. Chem. 2001, 276, 48189–48195. [Google Scholar] [CrossRef] [Green Version]
- Müller, T.; Mizumoto, S.; Suresh, I.; Komatsu, Y.; Vodopiutz, J.; Dundar, M.; Straub, V.; Lingenhel, A.; Melmer, A.; Lechner, S.; et al. Loss of dermatan sulfate epimerase (DSE) function results in musculocontractural Ehlers-Danlos syndrome. Hum. Mol. Genet. 2013, 22, 3761–3772. [Google Scholar] [CrossRef] [Green Version]
- Dündar, M.; Müller, T.; Zhang, Q.; Pan, J.; Steinmann, B.; Vodopiutz, J.; Gruber, R.; Sonoda, T.; Krabichler, B.; Utermann, G.; et al. Loss of dermatan-4-sulfotransferase 1 function results in adducted thumb-clubfoot syndrome. Am. J. Hum. Genet. 2009, 85, 873–882. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyake, N.; Kosho, T.; Mizumoto, S.; Furuichi, T.; Hatamochi, A.; Nagashima, Y.; Arai, E.; Takahashi, K.; Kawamura, R.; Wakui, K.; et al. Loss-of-function mutations of CHST14 in a new type of Ehlers-Danlos syndrome. Hum. Mutat. 2010, 31, 966–974. [Google Scholar] [CrossRef] [PubMed]
- Malfait, F.; Syx, D.; Vlummens, P.; Symoens, S.; Nampoothiri, S.; Hermanns-Lê, T.; Van Laer, L.; De Paepe, A. Musculocontractural Ehlers-Danlos Syndrome (former EDS type VIB) and adducted thumb clubfoot syndrome (ATCS) represent a single clinical entity caused by mutations in the dermatan-4-sulfotransferase 1 encoding CHST14 gene. Hum. Mutat. 2010, 31, 1233–1239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maccarana, M.; Olander, B.; Malmström, J.; Tiedemann, K.; Aebersold, R.; Lindahl, U.; Li, J.P.; Malmström, A. Biosynthesis of dermatan sulfate: Chondroitin-glucuronate C5-epimerase is identical to SART2. J. Biol. Chem. 2006, 281, 11560–11568. [Google Scholar] [CrossRef] [Green Version]
- Evers, M.R.; Xia, G.; Kang, H.G.; Schachner, M.; Baenziger, J.U. Molecular cloning and characterization of a dermatan-specific N-acetylgalactosamine 4-O-sulfotransferase. J. Biol. Chem. 2001, 276, 36344–36353. [Google Scholar]
- Mikami, T.; Mizumoto, S.; Kago, N.; Kitagawa, H.; Sugahara, K. Specificities of three distinct human chondroitin/dermatan N-acetylgalactosamine 4-O-sulfotransferases demonstrated using partially desulfated dermatan sulfate as an acceptor: Implication of differential roles in dermatan sulfate biosynthesis. J. Biol. Chem. 2003, 278, 36115–36127. [Google Scholar] [CrossRef] [Green Version]
- Maccarana, M.; Kalamajski, S.; Kongsgaard, M.; Magnusson, S.P.; Oldberg, A.; Malmström, A. Dermatan sulfate epimerase 1-deficient mice have reduced content and changed distribution of iduronic acids in dermatan sulfate and an altered collagen structure in skin. Mol. Cell. Biol. 2009, 29, 5517–5528. [Google Scholar] [CrossRef] [Green Version]
- Akyüz, N.; Rost, S.; Mehanna, A.; Bian, S.; Loers, G.; Oezen, I.; Mishra, B.; Hoffmann, K.; Guseva, D.; Laczynska, E.; et al. Dermatan 4-O-sulfotransferase1 ablation accelerates peripheral nerve regeneration. Exp. Neurol. 2013, 247, 517–530. [Google Scholar] [CrossRef]
- Hirose, T.; Mizumoto, S.; Hashimoto, A.; Takahashi, Y.; Yoshizawa, T.; Nitahara-Kasahara, Y.; Takahashi, N.; Nakayama, J.; Takehana, K.; Okada, T.; et al. Systematic investigation of the skin in Chst14-/- mice: A model for skin fragility in musculocontractural Ehlers-Danlos syndrome caused by CHST14 variants (mcEDS-CHST14). Glycobiology 2021, 31, 137–150. [Google Scholar] [CrossRef]
- Nitahara-Kasahara, Y.; Mizumoto, S.; Inoue, Y.U.; Saka, S.; Posadas-Herrera, G.; Nakamura-Takahashi, A.; Takahashi, Y.; Hashimoto, A.; Konishi, K.; Miyata, S.; et al. Muscle pathophysiology in mouse models of musculocontractural Ehlers-Danlos syndrome due to CHST14 mutations (mcEDS-CHST14), generated through CRISPR/Cas9-mediated genomic editing. Dis. Model. Mech. 2021, 14, dmm048963. [Google Scholar] [CrossRef]
- Nitahara-Kasahara, Y.; Posadas-Herrera, G.; Mizumoto, S.; Nakamura-Takahashi, A.; Inoue, Y.U.; Inoue, T.; Nomura, Y.; Takeda, S.; Yamada, S.; Kosho, T.; et al. Myopathy associated with dermatan sulfate-deficient decorin and myostatin in musculocontractural Ehlers-Danlos syndrome: A mouse model investigation. Front. Cell Dev. Biol. 2021, 9, 695021. [Google Scholar] [CrossRef]
- Götting, C.; Kuhn, J.; Zahn, R.; Brinkmann, T.; Kleesiek, K. Molecular cloning and expression of human UDP-D-Xylose:proteoglycan core protein beta-D-xylosyltransferase and its first isoform XT-II. J. Mol. Biol. 2000, 304, 517–528. [Google Scholar] [CrossRef]
- Pönighaus, C.; Ambrosius, M.; Casanova, J.C.; Prante, C.; Kuhn, J.; Esko, J.D.; Kleesiek, K.; Götting, C. Human xylosyltransferase II is involved in the biosynthesis of the uniform tetrasaccharide linkage region in chondroitin sulfate and heparan sulfate proteoglycans. J. Biol. Chem. 2007, 282, 5201–5206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kitagawa, H.; Tone, Y.; Tamura, J.; Neumann, K.W.; Ogawa, T.; Oka, S.; Kawasaki, T.; Sugahara, K. Molecular cloning and expression of glucuronyltransferase I involved in the biosynthesis of the glycosaminoglycan-protein linkage region of proteoglycans. J. Biol. Chem. 1998, 273, 6615–6618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindahl, U.; Rodén, L. Carbohydrate-protein linkages in proteoglycans of animal, plant and bacterial origin. In Glycoproteins: Their Composition, Structure and Function; Gottschalk, A., Ed.; Elsevier: Amsterdam, The Netherlands, 1972; pp. 491–517. [Google Scholar]
- Kjellén, L.; Lindahl, U. Proteoglycans: Structures and interactions. Annu. Rev. Biochem. 1991, 60, 443–475. [Google Scholar] [CrossRef] [PubMed]
- Kitagawa, H.; Uyama, T.; Sugahara, K. Molecular cloning and expression of a human chondroitin synthase. J. Biol. Chem. 2001, 276, 38721–38726. [Google Scholar] [CrossRef] [Green Version]
- Kitagawa, H.; Izumikawa, T.; Uyama, T.; Sugahara, K. Molecular cloning of a chondroitin polymerizing factor that cooperates with chondroitin synthase for chondroitin polymerization. J. Biol. Chem. 2003, 278, 23666–23671. [Google Scholar] [CrossRef] [Green Version]
- Izumikawa, T.; Uyama, T.; Okuura, Y.; Sugahara, K.; Kitagawa, H. Involvement of chondroitin sulfate synthase-3 (chondroitin synthase-2) in chondroitin polymerization through its interaction with chondroitin synthase-1 or chondroitin polymerizing factor. Biochem. J. 2007, 403, 545–552. [Google Scholar] [CrossRef]
- Izumikawa, T.; Koike, T.; Shiozawa, S.; Sugahara, K.; Tamura, J.; Kitagawa, H. Identification of chondroitin sulfate glucuronyltransferase as chondroitin synthase-3 involved in chondroitin polymerization: Chondroitin polymerization is achieved by multiple enzyme complexes consisting of chondroitin synthase family members. J. Biol. Chem. 2008, 283, 11396–11406. [Google Scholar] [CrossRef] [Green Version]
- Uyama, T.; Kitagawa, H.; Tamura, J.; Sugahara, K. Molecular cloning and expression of human chondroitin N-acetylgalactosaminyltransferase: The key enzyme for chain initiation and elongation of chondroitin/dermatan sulfate on the protein linkage region tetrasaccharide shared by heparin/heparan sulfate. J. Biol. Chem. 2002, 277, 8841–8846. [Google Scholar] [CrossRef] [Green Version]
- Uyama, T.; Kitagawa, H.; Tanaka, J.; Tamura, J.; Ogawa, T.; Sugahara, K. Molecular cloning and expression of a second chondroitin N-acetylgalactosaminyltransferase involved in the initiation and elongation of chondroitin/dermatan sulfate. J. Biol. Chem. 2003, 278, 3072–3078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pacheco, B.; Malmström, A.; Maccarana, M. Two dermatan sulfate epimerases form iduronic acid domains in dermatan sulfate. J. Biol. Chem. 2009, 284, 9788–9795. [Google Scholar] [PubMed] [Green Version]
- Kobayashi, M.; Sugumaran, G.; Liu, J.; Shworak, N.W.; Silbert, J.E.; Rosenberg, R.D. Molecular cloning and characterization of a human uronyl 2-sulfotransferase that sulfates iduronyl and glucuronyl residues in dermatan/chondroitin sulfate. J. Biol. Chem. 1999, 274, 10474–10480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scott, J.E. (Ed.) Dermatan Sulphate Proteoglycans: Chemistry, Biology, Chemical Pathology; Portland Press: London, UK, 1993. [Google Scholar]
- Toole, B.P.; Lowther, D.A. Dermatan sulfate-protein: Isolation from and interaction with collagen. Arch. Biochem. Biophys. 1968, 128, 567–578. [Google Scholar] [PubMed]
- Scott, J.E.; Orford, C.R. Dermatan sulphate-rich proteoglycan associates with rat tail-tendon collagen at the d band in the gap region. Biochem. J. 1981, 197, 213–216. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, T.; Kametani, K.; Koyama, Y.I.; Suzuki, D.; Imamura, Y.; Takehana, K.; Hiramatsu, K. Ring-Mesh Model of Proteoglycan Glycosaminoglycan Chains in Tendon based on Three-dimensional Reconstruction by Focused Ion Beam Scanning Electron Microscopy. J. Biol. Chem. 2016, 291, 23704–23708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirose, T.; Takahashi, N.; Tangkawattana, P.; Minaguchi, J.; Mizumoto, S.; Yamada, S.; Miyake, N.; Hayashi, S.; Hatamochi, A.; Nakayama, J.; et al. Structural alteration of glycosaminoglycan side chains and spatial disorganization of collagen networks in the skin of patients with mcEDS-CHST14. Biochim. Biophys. Acta Gen. Subj. 2019, 1863, 623–631. [Google Scholar] [CrossRef] [PubMed]
- Tollefsen, D.M.; Pestka, C.A.; Monafo, W.J. Activation of heparin cofactor II by dermatan sulfate. J. Biol. Chem. 1983, 258, 6713–6716. [Google Scholar] [CrossRef]
- Maimone, M.M.; Tollefsen, D.M. Structure of a dermatan sulfate hexasaccharide that binds to heparin cofactor II with high affinity. J. Biol. Chem. 1990, 265, 18263–18271. [Google Scholar] [CrossRef]
- Penc, S.F.; Pomahac, B.; Winkler, T.; Dorschner, R.A.; Eriksson, E.; Herndon, M.; Gallo, R.L. Dermatan sulfate released after injury is a potent promoter of fibroblast growth factor-2 function. J. Biol. Chem. 1998, 273, 28116–28121. [Google Scholar] [CrossRef] [Green Version]
- Trowbridge, J.M.; Rudisill, J.A.; Ron, D.; Gallo, R.L. Dermatan sulfate binds and potentiates activity of keratinocyte growth factor (FGF-7). J. Biol. Chem. 2002, 277, 42815–42820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lyon, M.; Deakin, J.A.; Rahmoune, H.; Fernig, D.G.; Nakamura, T.; Gallagher, J.T. Hepatocyte growth factor/scatter factor binds with high affinity to dermatan sulfate. J. Biol. Chem. 1998, 273, 271–278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor, K.R.; Rudisill, J.A.; Gallo, R.L. Structural and sequence motifs in dermatan sulfate for promoting fibroblast growth factor-2 (FGF-2) and FGF-7 activity. J. Biol. Chem. 2005, 280, 5300–5306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hikino, M.; Mikami, T.; Faissner, A.; Vilela-Silva, A.C.; Pavão, M.S.; Sugahara, K. Oversulfated dermatan sulfate exhibits neurite outgrowth-promoting activity toward embryonic mouse hippocampal neurons: Implications of dermatan sulfate in neuritogenesis in the brain. J. Biol. Chem. 2003, 278, 43744–43754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nandini, C.D.; Mikami, T.; Ohta, M.; Itoh, N.; Akiyama-Nambu, F.; Sugahara, K. Structural and functional characterization of oversulfated chondroitin sulfate/dermatan sulfate hybrid chains from the notochord of hagfish. Neuritogenic and binding activities for growth factors and neurotrophic factors. J. Biol. Chem. 2004, 279, 50799–50809. [Google Scholar] [CrossRef] [Green Version]
- Nandini, C.D.; Itoh, N.; Sugahara, K. Novel 70-kDa chondroitin sulfate/dermatan sulfate hybrid chains with a unique heterogeneous sulfation pattern from shark skin, which exhibit neuritogenic activity and binding activities for growth factors and neurotrophic factors. J. Biol. Chem. 2005, 280, 4058–4069. [Google Scholar] [CrossRef] [Green Version]
- Bao, X.; Mikami, T.; Yamada, S.; Faissner, A.; Muramatsu, T.; Sugahara, K. Heparin-binding growth factor, pleiotrophin, mediates neuritogenic activity of embryonic pig brain-derived chondroitin sulfate/dermatan sulfate hybrid chains. J. Biol. Chem. 2005, 280, 9180–9191. [Google Scholar] [CrossRef] [Green Version]
- Li, F.; Nandini, C.D.; Hattori, T.; Bao, X.; Murayama, D.; Nakamura, T.; Fukushima, N.; Sugahara, K. Structure of pleiotrophin- and hepatocyte growth factor-binding sulfated hexasaccharide determined by biochemical and computational approaches. J. Biol. Chem. 2010, 285, 27673–27685. [Google Scholar] [CrossRef] [Green Version]
- Machino, M.; Gong, Y.; Ozaki, T.; Suzuki, Y.; Watanabe, E.; Imagama, S.; Kadomatsu, K.; Sakamoto, K. Dermatan sulphate is an activating ligand of anaplastic lymphoma kinase. J. Biochem. 2021, 170, 631–637. [Google Scholar] [CrossRef]
- Fukatsu, T.; Sobue, M.; Nagasaka, T.; Ohiwa, N.; Fukata, S.; Nakashima, N.; Takeuchi, J. Immunohistochemical localization of chondroitin sulphate and dermatan sulphate proteoglycans in tumour tissues. Br. J. Cancer 1988, 57, 74–78. [Google Scholar] [CrossRef] [Green Version]
- Ten Dam, G.B.; Yamada, S.; Kobayashi, F.; Purushothaman, A.; van de Westerlo, E.M.; Bulten, J.; Malmström, A.; Sugahara, K.; Massuger, L.F.; van Kuppevelt, T.H. Dermatan sulfate domains defined by the novel antibody GD3A12, in normal tissues and ovarian adenocarcinomas. Histochem. Cell. Biol. 2009, 132, 117–127. [Google Scholar] [CrossRef] [PubMed]
- Thelin, M.A.; Svensson, K.J.; Shi, X.; Bagher, M.; Axelsson, J.; Isinger-Ekstrand, A.; van Kuppevelt, T.H.; Johansson, J.; Nilbert, M.; Zaia, J.; et al. Dermatan sulfate is involved in the tumorigenic properties of esophagus squamous cell carcinoma. Cancer Res. 2012, 72, 1943–1952. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakai, S.; Kim, W.S.; Lee, I.S.; Kim, Y.S.; Nakamura, A.; Toida, T.; Imanari, T. Purification and characterization of dermatan sulfate from the skin of the eel, Anguilla japonica. Carbohydr. Res. 2003, 338, 263–269. [Google Scholar] [CrossRef]
- Pavão, M.S.; Mourão, P.A.; Mulloy, B.; Tollefsen, D.M. A unique dermatan sulfate-like glycosaminoglycan from ascidian. Its structure and the effect of its unusual sulfation pattern on anticoagulant activity. J. Biol. Chem. 1995, 270, 31027–31036. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pavão, M.S.; Aiello, K.R.; Werneck, C.C.; Silva, L.C.; Valente, A.P.; Mulloy, B.; Colwell, N.S.; Tollefsen, D.M.; Mourão, P.A. Highly sulfated dermatan sulfates from Ascidians. Structure versus anticoagulant activity of these glycosaminoglycans. J. Biol. Chem. 1998, 273, 27848–27857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vilela-Silva, A.C.; Werneck, C.C.; Valente, A.P.; Vacquier, V.D.; Mourão, P.A. Embryos of the sea urchin Strongylocentrotus purpuratus synthesize a dermatan sulfate enriched in 4-O- and 6-O-disulfated galactosamine units. Glycobiology 2001, 11, 433–440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, F.; Yamada, S.; Basappa; Shetty, A.K.; Sugiura, M.; Sugahara, K. Determination of iduronic acid and glucuronic acid in sulfated chondroitin/dermatan hybrid chains by (1)H-nuclear magnetic resonance spectroscopy. Glycoconj. J. 2008, 25, 603–610. [Google Scholar] [CrossRef]
- Ogura, C.; Hirano, K.; Mizumoto, S.; Yamada, S.; Nishihara, S. Dermatan sulphate promotes neuronal differentiation in mouse and human stem cells. J. Biochem. 2021, 169, 55–64. [Google Scholar] [CrossRef]
- Ogura, C.; Nishihara, S. Dermatan-4-O-Sulfotransferase-1 Contributes to the Undifferentiated State of Mouse Embryonic Stem Cells. Front. Cell Dev. Biol. 2021, 9, 733964. [Google Scholar] [CrossRef]
- Sahu, S.; Li, R.; Loers, G.; Schachner, M. Knockdown of chondroitin-4-sulfotransferase-1, but not of dermatan-4-sulfotransferase-1, accelerates regeneration of zebrafish after spinal cord injury. FASEB J. 2019, 33, 2252–2262. [Google Scholar] [CrossRef] [Green Version]
- Danielson, K.G.; Baribault, H.; Holmes, D.F.; Graham, H.; Kadler, K.E.; Iozzo, R.V. Targeted disruption of decorin leads to abnormal collagen fibril morphology and skin fragility. J. Cell Biol. 1997, 136, 729–743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goossens, D.; Van Gestel, S.; Claes, S.; De Rijk, P.; Souery, D.; Massat, I.; Van den Bossche, D.; Backhovens, H.; Mendlewicz, J.; Van Broeckhoven, C.; et al. A novel CpG-associated brain-expressed candidate gene for chromosome 18q-linked bipolar disorder. Mol. Psychiatry 2003, 8, 83–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakao, M.; Shichijo, S.; Imaizumi, T.; Inoue, Y.; Matsunaga, K.; Yamada, A.; Kikuchi, M.; Tsuda, N.; Ohta, K.; Takamori, S.; et al. Identification of a gene coding for a new squamous cell carcinoma antigen recognized by the CTL. J. Immunol. 2000, 164, 2565–2574. [Google Scholar] [CrossRef] [Green Version]
- Stachtea, X.N.; Tykesson, E.; van Kuppevelt, T.H.; Feinstein, R.; Malmström, A.; Reijmers, R.M.; Maccarana, M. Dermatan sulfate-free mice display embryological defects and are neonatal lethal despite normal lymphoid and non-lymphoid organogenesis. PLoS ONE 2015, 10, e0140279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartolini, B.; Thelin, M.A.; Rauch, U.; Feinstein, R.; Oldberg, A.; Malmström, A.; Maccarana, M. Mouse development is not obviously affected by the absence of dermatan sulfate epimerase 2 in spite of a modified brain dermatan sulfate composition. Glycobiology 2021, 22, 1007–1016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bian, S.; Akyüz, N.; Bernreuther, C.; Loers, G.; Laczynska, E.; Jakovcevski, I.; Schachner, M. Dermatan sulfotransferase Chst14/D4st1, but not chondroitin sulfotransferase Chst11/C4st1, regulates proliferation and neurogenesis of neural progenitor cells. J. Cell Sci. 2011, 124, 4051–4063. [Google Scholar] [CrossRef] [Green Version]
- Yoshizawa, T.; Mizumoto, S.; Takahashi, Y.; Shimada, S.; Sugahara, K.; Nakayama, J.; Takeda, S.; Nomura, Y.; Nitahara-Kasahara, Y.; Okada, T.; et al. Vascular abnormalities in the placenta of Chst14-/- fetuses: Implications in the pathophysiology of perinatal lethality of the murine model and vascular lesions in human CHST14/D4ST1 deficiency. Glycobiology 2018, 28, 80–89. [Google Scholar] [CrossRef] [Green Version]
- Syx, D.; Van Damme, T.; Symoens, S.; Maiburg, M.C.; van de Laar, I.; Morton, J.; Suri, M.; Del Campo, M.; Hausser, I.; Hermanns-Lê, T.; et al. Genetic heterogeneity and clinical variability in musculocontractural Ehlers-Danlos syndrome caused by impaired dermatan sulfate biosynthesis. Hum. Mutat. 2015, 36, 535–547. [Google Scholar] [CrossRef]
- Schirwani, S.; Metcalfe, K.; Wagner, B.; Berry, I.; Sobey, G.; Jewell, R. DSE associated musculocontractural EDS, a milder phenotype or phenotypic variability. Eur. J. Med. Genet. 2020, 63, 103798. [Google Scholar] [CrossRef]
- Lautrup, C.K.; Teik, K.W.; Unzaki, A.; Mizumoto, S.; Syx, D.; Sin, H.H.; Nielsen, I.K.; Markholt, S.; Yamada, S.; Malfait, F.; et al. Delineation of musculocontractural Ehlers-Danlos syndrome caused by dermatan sulfate epimerase deficiency. Mol. Genet. Genom. Med. 2020, 8, e1197. [Google Scholar] [CrossRef] [Green Version]
- Ullah, I.; Aamir, M.; Ilyas, M.; Ahmed, A.; Jelani, M.; Ullah, W.; Abbas, M.; Ishfaq, M.; Ali, F.; Yip, J.; et al. A novel variant in the DSE gene leads to Ehlers-Danlos musculocontractural type 2 in a Pakistani family. Congenit. Anom. 2021, 61, 177–182. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Potash, J.B.; Knowles, J.A.; Weissman, M.M.; Coryell, W.; Scheftner, W.A.; Lawson, W.B.; DePaulo, J.R., Jr.; Gejman, P.V.; Sanders, A.R.; et al. Genome-wide association study of recurrent early-onset major depressive disorder. Mol. Psychiatry 2011, 16, 193–201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zayed, H.; Chao, R.; Moshrefi, A.; Lopezjimenez, N.; Delaney, A.; Chen, J.; Shaw, G.M.; Slavotinek, A.M. A maternally inherited chromosome 18q22.1 deletion in a male with late-presenting diaphragmatic hernia and microphthalmia-evaluation of DSEL as a candidate gene for the diaphragmatic defect. Am. J. Med. Genet. 2010, 152A, 916–923. [Google Scholar] [CrossRef] [Green Version]
- Kosho, T.; Miyake, N.; Hatamochi, A.; Takahashi, J.; Kato, H.; Miyahara, T.; Igawa, Y.; Yasui, H.; Ishida, T.; Ono, K.; et al. A new Ehlers-Danlos syndrome with craniofacial characteristics, multiple congenital contractures, progressive joint and skin laxity, and multisystem fragility-related manifestations. Am. J. Med. Genet. 2010, 152A, 1333–1346. [Google Scholar] [CrossRef]
- Voermans, N.C.; Kempers, M.; Lammens, M.; van Alfen, N.; Janssen, M.C.; Bönnemann, C.; van Engelen, B.G.; Hamel, B.C. Myopathy in a 20-year-old female patient with D4ST-1 deficient Ehlers-Danlos syndrome due to a homozygous CHST14 mutation. Am. J. Med. Genet. A 2012, 158A, 850–855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winters, K.A.; Jiang, Z.; Xu, W.; Li, S.; Ammous, Z.; Jayakar, P.; Wierenga, K.J. Re-assigned diagnosis of D4ST1-deficient Ehlers-Danlos syndrome (adducted thumb-clubfoot syndrome) after initial diagnosis of Marden-Walker syndrome. Am. J. Med. Genet. A. 2012, 158A, 2935–2940. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, K.; Okamoto, N.; Miyake, N.; Taira, K.; Sato, Y.; Matsuda, K.; Akimaru, N.; Ohashi, H.; Wakui, K.; Fukushima, Y.; et al. Delineation of dermatan 4-O-sulfotransferase 1 deficient Ehlers-Danlos syndrome: Observation of two additional patients and comprehensive review of 20 reported patients. Am. J. Med. Genet. A. 2011, 155A, 1949–1958. [Google Scholar] [CrossRef]
- Mendoza-Londono, R.; Chitayat, D.; Kahr, W.H.; Hinek, A.; Blaser, S.; Dupuis, L.; Goh, E.; Badilla-Porras, R.; Howard, A.; Mittaz, L.; et al. Extracellular matrix and platelet function in patients with musculocontractural Ehlers-Danlos syndrome caused by mutations in the CHST14 gene. Am. J. Med. Genet. A 2012, 158A, 1344–1354. [Google Scholar] [CrossRef]
- Janecke, A.R.; Li, B.; Boehm, M.; Krabichler, B.; Rohrbach, M.; Müller, T.; Fuchs, I.; Golas, G.; Katagiri, Y.; Ziegler, S.G.; et al. The phenotype of the musculocontractural type of Ehlers-Danlos syndrome due to CHST14 mutations. Am. J. Med. Genet. A 2016, 170A, 103–115. [Google Scholar] [CrossRef] [Green Version]
- Alazami, A.M.; Al-Qattan, S.M.; Faqeih, E.; Alhashem, A.; Alshammari, M.; Alzahrani, F.; Al-Dosari, M.S.; Patel, N.; Alsagheir, A.; Binabbas, B.; et al. Expanding the clinical and genetic heterogeneity of hereditary disorders of connective tissue. Hum. Genet. 2016, 135, 525–540. [Google Scholar] [CrossRef]
- Sandal, S.; Kaur, A.; Panigrahi, I. Novel mutation in the CHST14 gene causing musculocontractural type of Ehlers-Danlos syndrome. BMJ Case Rep. 2018, 2018, bcr2018226165. [Google Scholar] [CrossRef] [PubMed]
- Uehara, M.; Kosho, T.; Yamamoto, N.; Takahashi, H.E.; Shimakura, T.; Nakayama, J.; Kato, H.; Takahashi, J. Spinal manifestations in 12 patients with musculocontractural Ehlers-Danlos syndrome caused by CHST14/D4ST1 deficiency (mcEDS-CHST14). Am. J. Med. Genet. A 2018, 176, 2331–2341. [Google Scholar] [CrossRef] [PubMed]
- Minatogawa, M.; Unzaki, A.; Morisaki, H.; Syx, D.; Sonoda, T.; Janecke, A.R.; Slavotinek, A.; Voermans, N.C.; Lacassie, Y.; Mendoza-Londono, R.; et al. Clinical and molecular features of 66 patients with musculocontractural Ehlers-Danlos syndrome caused by pathogenic variants in CHST14 (mcEDS-CHST14). J. Med. Genet. 2021, in press. [CrossRef] [PubMed]
- Akatsu, C.; Mizumoto, S.; Kaneiwa, T.; Maccarana, M.; Malmström, A.; Yamada, S.; Sugahara, K. Dermatan sulfate epimerase 2 is the predominant isozyme in the formation of the chondroitin sulfate/dermatan sulfate hybrid structure in postnatal developing mouse brain. Glycobiology 2011, 21, 565–574. [Google Scholar] [CrossRef] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mizumoto, S.; Yamada, S. The Specific Role of Dermatan Sulfate as an Instructive Glycosaminoglycan in Tissue Development. Int. J. Mol. Sci. 2022, 23, 7485. https://doi.org/10.3390/ijms23137485
Mizumoto S, Yamada S. The Specific Role of Dermatan Sulfate as an Instructive Glycosaminoglycan in Tissue Development. International Journal of Molecular Sciences. 2022; 23(13):7485. https://doi.org/10.3390/ijms23137485
Chicago/Turabian StyleMizumoto, Shuji, and Shuhei Yamada. 2022. "The Specific Role of Dermatan Sulfate as an Instructive Glycosaminoglycan in Tissue Development" International Journal of Molecular Sciences 23, no. 13: 7485. https://doi.org/10.3390/ijms23137485
APA StyleMizumoto, S., & Yamada, S. (2022). The Specific Role of Dermatan Sulfate as an Instructive Glycosaminoglycan in Tissue Development. International Journal of Molecular Sciences, 23(13), 7485. https://doi.org/10.3390/ijms23137485