
Design, Synthesis and Biological Characterization of Histone Deacetylase 8 (HDAC8) Proteolysis Targeting Chimeras (PROTACs) with Anti-Neuroblastoma Activity

 , , , and
, , , and 
Abstract
:1. Introduction
2. Results and Discussion
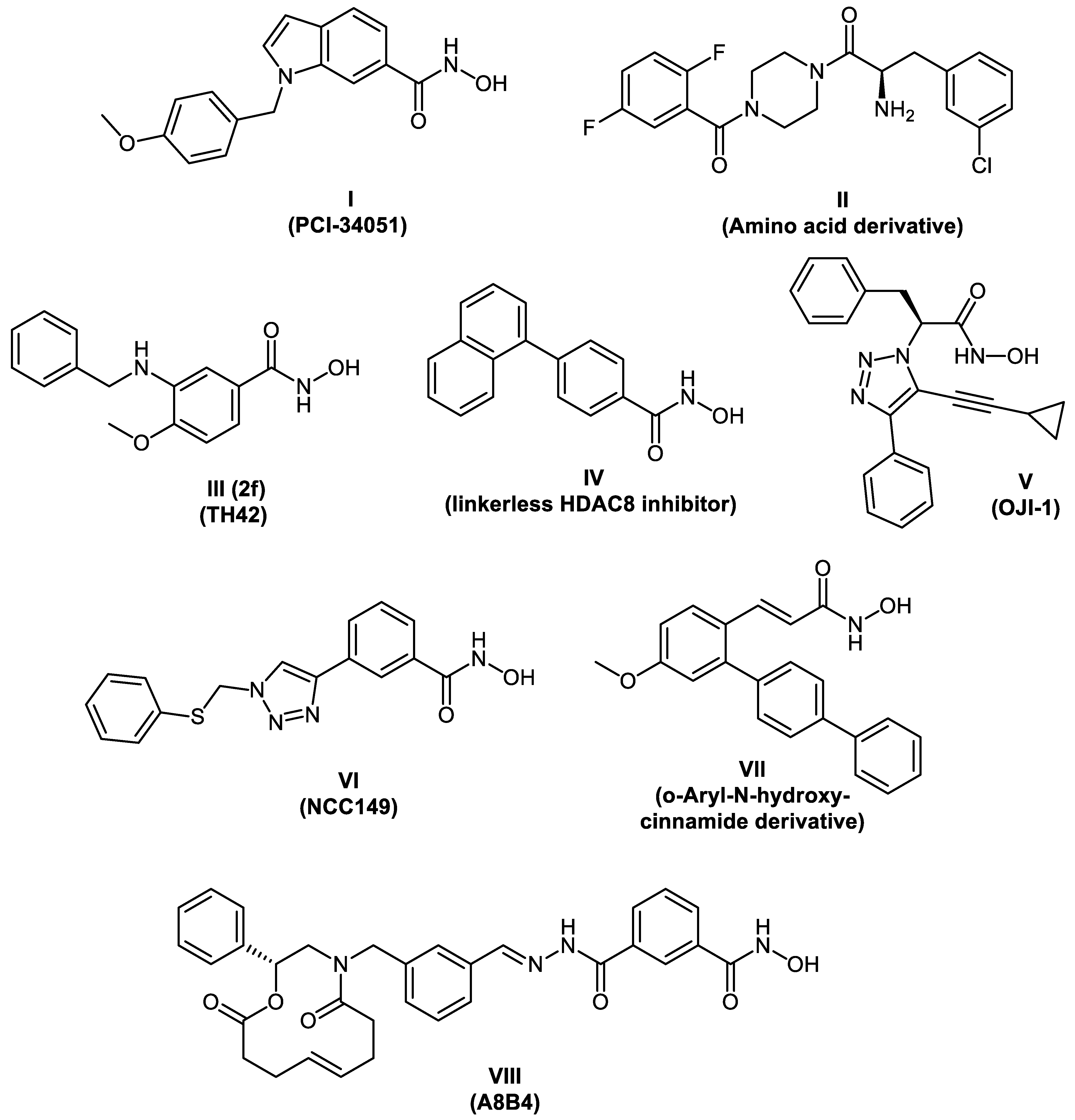
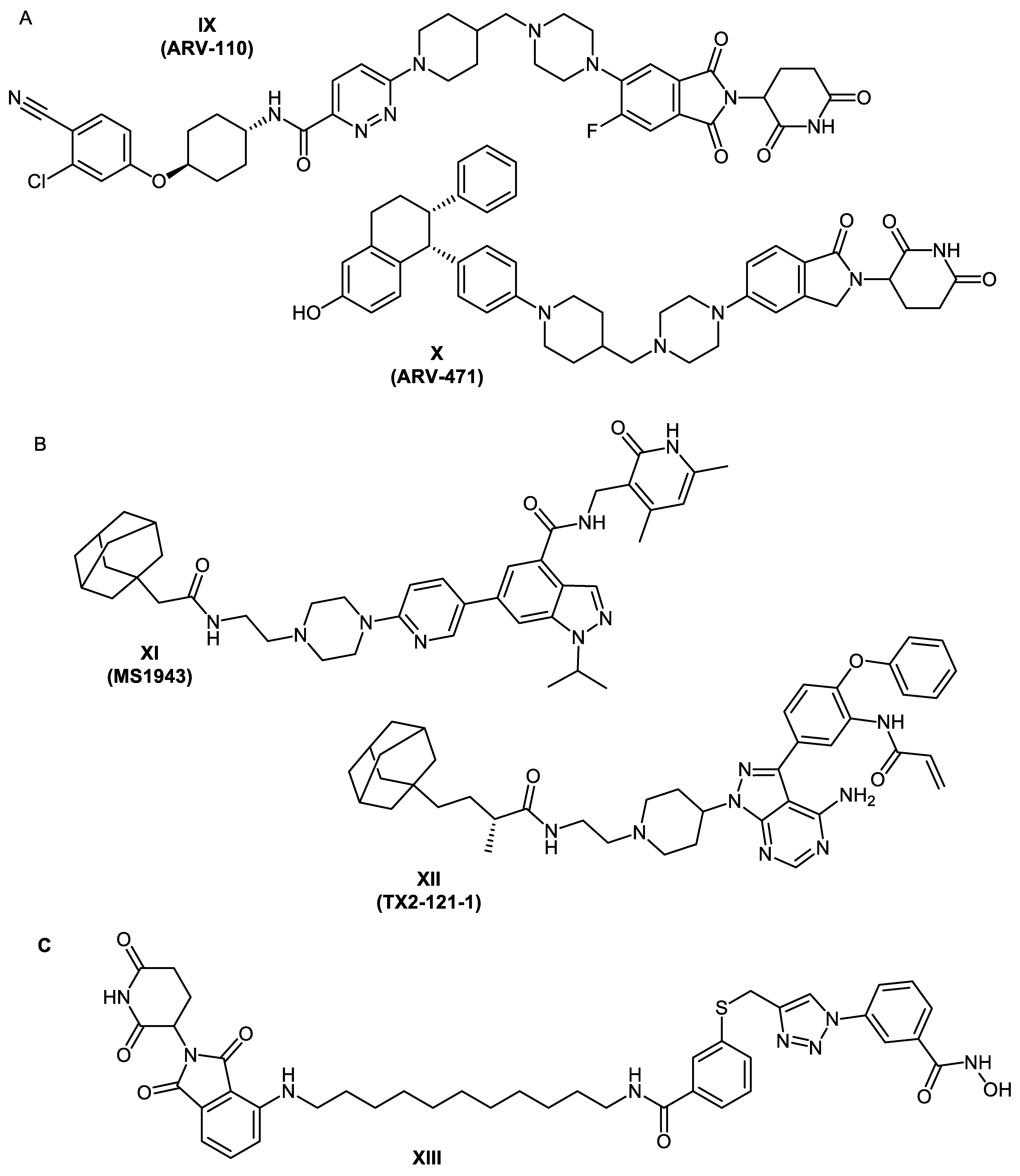
2.1. Design Concept
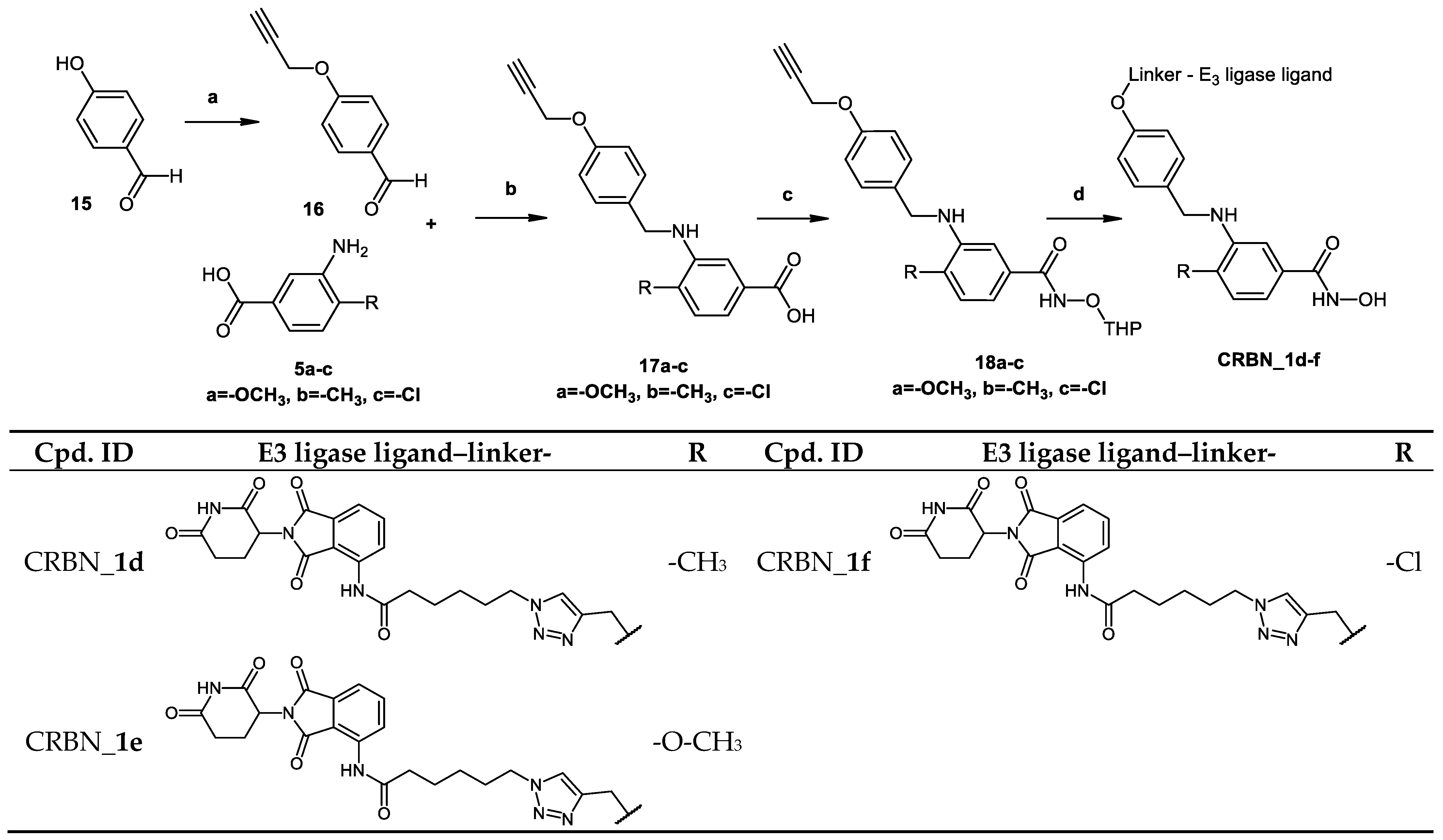
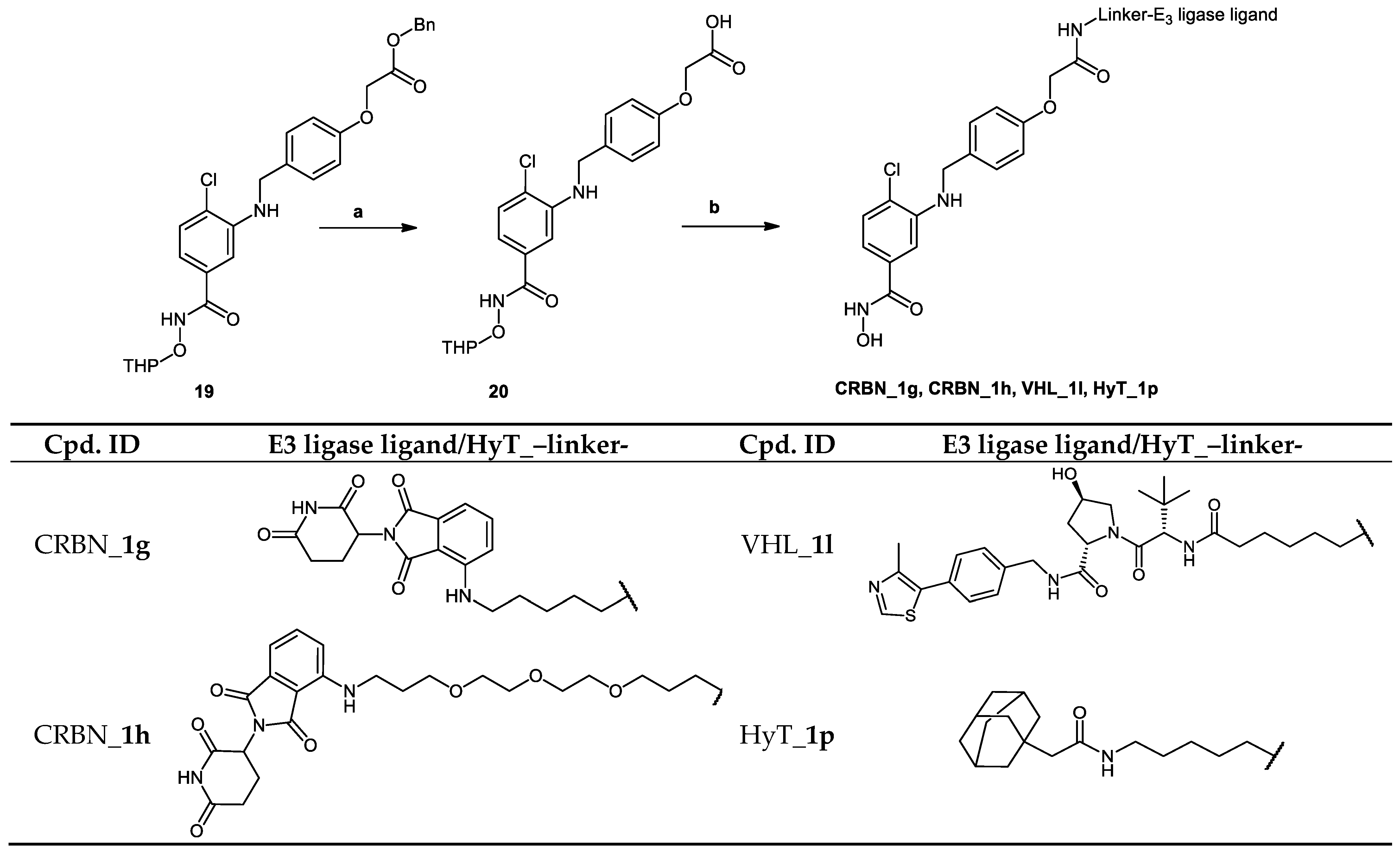
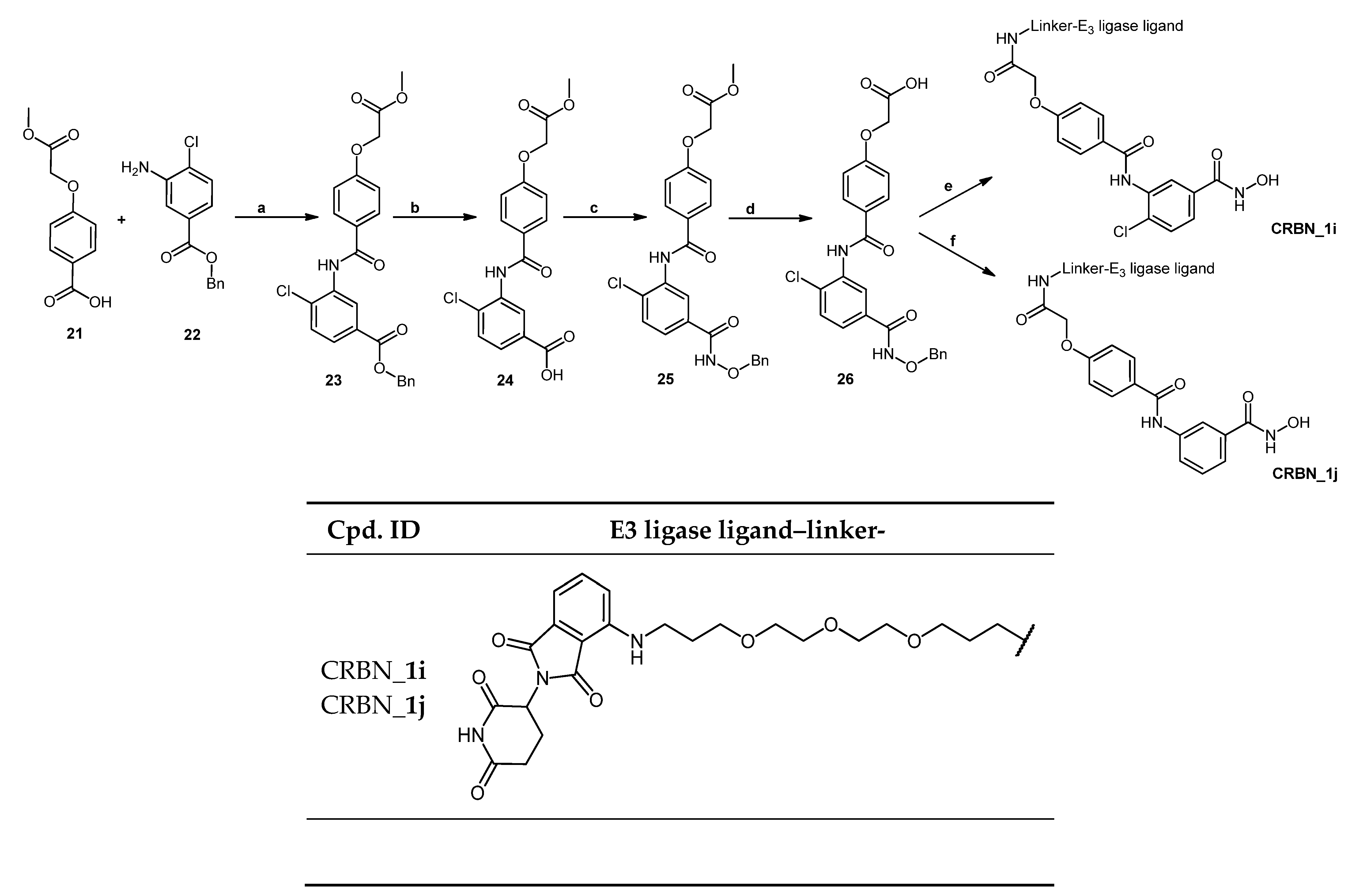
2.2. Chemical Synthesis of PROTACs and Inhibitors
2.3. In Vitro Testing Using Recombinant HDACs
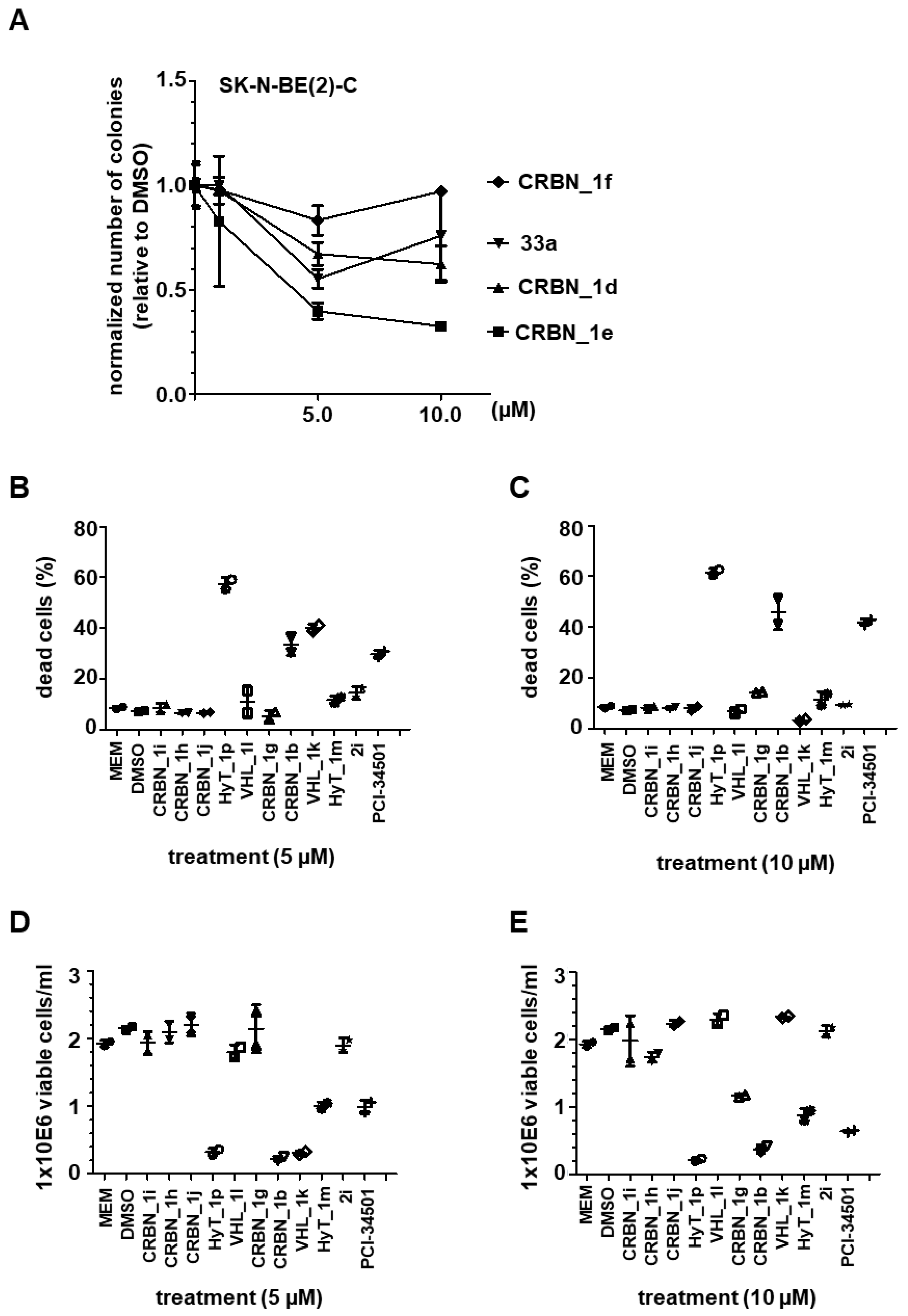
2.4. Cytotoxicity Assay
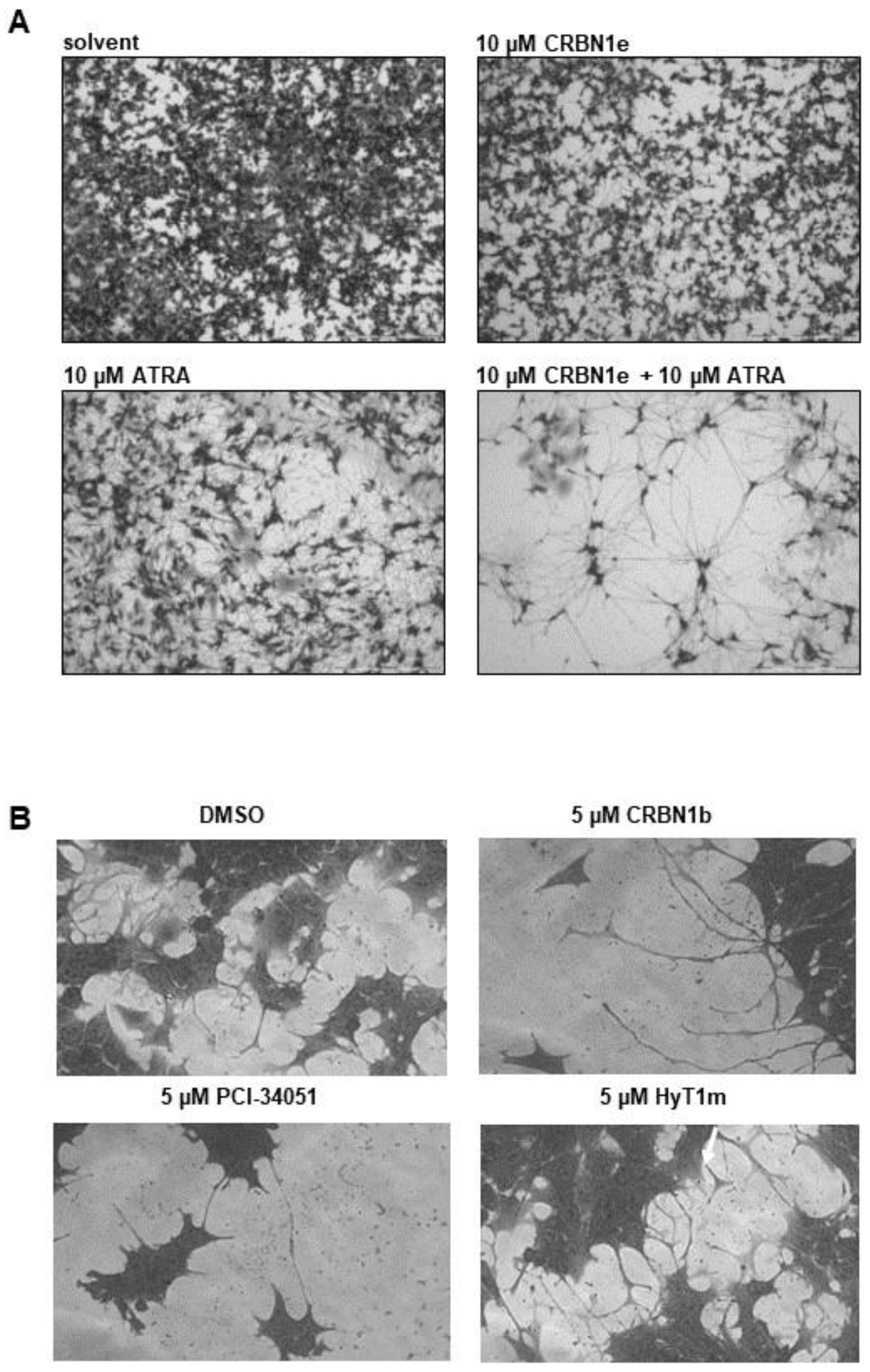
2.5. Testing on Neuroblastoma Cells
3. Conclusions
4. Materials and Methods
4.1. Chemistry
4.1.1. General
4.1.2. General Synthetic Methods
- Method I, reductive amination
- A mixture of the benzaldehyde (1 eq.) and the amine (5% molar excess) was dissolved in toluene and was heated under reflux using a water trap for 2 h. Afterwards, the solvent was removed under reduced pressure. The remaining residue was dissolved in dry tetrahydrofuran and the formed solution was cooled to 0 °C. Glacial acetic acid (2 eq.) was added followed by sodium triacetoxyborohydride (4 eq.) and the reaction mixture was stirred for 30 min at 0 °C. Afterwards, the ice bath was removed and stirring was continued for 24 h at room temperature. The reaction was then quenched by the addition of sodium bicarbonate and the product was extracted with ethyl acetate. The organic layer was washed with 1 M hydrochloric acid followed by brine and was dried over anhydrous sodium sulfate. Finally, it was filtered and evaporated under reduced pressure. The crude product was purified using the MPLC. The yields were in the 60–95% range.
- A mixture of benzaldehyde (1.1 eq.), the corresponding amine (1 eq.), trifluoroacetic acid (2 eq.), and sodium triacetoxyborohydride (1.2 eq.) was dissolved in a mixture of tetrahydrofuran and ethyl acetate (1:1). After stirring the reaction mixture at room temperature for 2 h, the reaction was quenched by adding water and the crude product was extracted with ethyl acetate. The combined organic layer was dried over anhydrous sodium sulfate, filtered, and concentrated in vacuo. The crude residue was purified using MPLC. The yield was around 50%.
- Method II, ester hydrolysis
- To a solution of the methyl ester (1 eq.) in methanol, 1 M aqueous sodium hydroxide (10 eq.) was added. The formed reaction mixture was refluxed for 2–4 h. After complete ester hydrolysis, the solvent was evaporated under reduced pressure to yield a crude residue that was dissolved in water. The aqueous solution was extracted using ethyl acetate to remove any organic impurities. In the next step, 1 M aqueous hydrochloric acid (10 eq.) was added to the aqueous solution to liberate the free acid which was extracted using ethyl acetate. The combined organic layer was washed with brine and dried over anhydrous sodium sulfate. It was then filtered, and the solvent was evaporated under reduced pressure to give the crude product which was purified using the MPLC. The yields were 70–96%.
- To the suspension of the methyl ester (1 eq.) in a mixture of tetrahydrofuran and water (1:1), lithium hydroxide (5 eq.) was added. The mixture was stirred at room temperature until complete hydrolysis of the ester then tetrahydrofuran was evaporated. Using aqueous 1 M hydrochloric acid, the pH of the remaining aqueous solution was adjusted to pH 6. The liberated free acid was extracted using a mixture of ethyl acetate and tetrahydrofuran. The combined organic layer was then dried over anhydrous sodium sulfate, filtered, and concentrated in vacuo to yield the product, which required no further purification. Crude yields were around 80–90%.
- Method III, amide bond formation
- A solution of the carboxylic acid (1–1.2 eq.) and N,N-diisopropylethylamine (3 eq.) in dimethylformamide was stirred for 15 min at room temperature then O-(7-Azabenzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium hexafluorophosphate (1.2–1.5 eq.) was added and stirring was continued for another 30 min. Next, the corresponding amine (1–1.5 eq.) was added to the solution. The formed reaction mixture was stirred at 0 °C or at room temperature or at 50 °C for 2–24 h. After completion of the reaction, water was added to the reaction mixture and the formed solution was extracted using ethyl acetate. The combined organic layer was washed with aqueous 1 M sodium bicarbonate solution followed by aqueous 1 M ammonium chloride solution and brine. After drying over anhydrous sodium sulfate, the organic layer was filtered then concentrated in vacuo to yield the crude compound which was purified using MPLC. The yields were around 27–100%.
- To a suspension of the carboxylic acid (1 eq.) in toluene, drops of dimethylformamide were added followed by pyridine then oxalyl chloride (2 eq.). The reaction mixture was stirred at room temperature for 6 h. The formed precipitate was then filtered and washed with toluene. Afterwards, the combined organic filtrates were concentrated under reduced pressure to give the acid chloride that was used directly without further purification. It was dissolved in pyridine and the amine (1 eq.) was added to the solution. The formed reaction mixture was stirred at room temperature for 24 h. After evaporation of the solvent the remaining residue was dissolved in chloroform and was successively washed with 10% hydrochloric acid, 1 M sodium bicarbonate, and brine. After drying the organic layer over anhydrous sodium sulfate, it was evaporated under reduced pressure to give the crude product which was purified using the MPLC. The yield was around 48%.
- After the dropwise addition of thionyl chloride (3 eq.) to the carboxylic acid (1 eq.) at 0 °C, the reaction mixture was heated under reflux for 2 h then the excess thionyl chloride was evaporated under vacuum. The formed acid chloride was dissolved in dry tetrahydrofuran and was added dropwise to a solution of the corresponding amine (0.9 eq.) and N,N-diisopropylethylamine (3 eq.) in tetrahydrofuran. The reaction mixture was stirred at room temperature until completion. Afterwards, it was diluted with ethyl acetate and was washed with a saturated aqueous solution of ammonium chloride followed by brine. Finally, the organic layer was dried over anhydrous sodium sulfate, filtered, and concentrated in vacuo to obtain the crude residue which was purified using MPLC. The yield was around 50–70%.
- A mixture of the carboxylic acid (3 eq.), N-methylimidazole (3.5 eq.), and chloro-N,N,N′,N′-tetramethylformamidinium-hexafluorophosphate (1.2 eq.) were stirred in acetonitrile for 15 min. The amine (1 eq.) was dissolved in some acetonitrile, then was added to the mixture. The formed reaction mixture was stirred at room temperature for 24 h. After completion of the reaction was confirmed by TLC, water was added, and the mixture was extracted using ethyl acetate. The combined organic layer was washed with water followed by brine. After drying over anhydrous sodium sulfate, the organic layer was filtered then concentrated in vacuo to yield the crude compound which was purified using MPLC. The yield was around 67%.
- Method IV, azide-alkyne Huisgen cycloaddition
- Method V, deprotection of tetrahydropyranyl ether
- Method VI, deprotection of tert-butyl protected carbamates and tert-butyl ester protected carboxylic acids
- Method VII, catalytic hydrogenation
4.2. In Vitro HDAC Inhibitory Activity Assay
4.3. Cellular Assay
- Cell Culture
- B.
- Western blot
- C.
- Cell viability assay (Trypan blue assay)
- D.
- Colony formation assay
- E.
- Cell differentiation assay
- F.
- Cytotoxicity Studies
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wolfson, N.A.; Pitcairn, C.A.; Fierke, C.A. HDAC8 substrates: Histones and beyond. Biopolymers 2013, 99, 112–126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Furdas, S.D.; Kannan, S.; Sippl, W.; Jung, M. Small Molecule Inhibitors of Histone Acetyltransferases as Epigenetic Tools and Drug Candidates. Arch. Pharm. 2012, 345, 7–21. [Google Scholar] [CrossRef] [PubMed]
- Seto, E.; Yoshida, M. Erasers of histone acetylation: The histone deacetylase enzymes. Cold Spring Harb. Perspect. Biol. 2014, 6, a018713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matthay, K.K.; Maris, J.M.; Schleiermacher, G.; Nakagawara, A.; Mackall, C.L.; Diller, L.; Weiss, W.A. Neuroblastoma. Nat. Rev. Dis. Primers 2016, 2, 16078. [Google Scholar] [CrossRef]
- Oehme, I.; Deubzer, H.E.; Wegener, D.; Pickert, D.; Linke, J.P.; Hero, B.; Kopp-Schneider, A.; Westermann, F.; Ulrich, S.M.; von Deimling, A.; et al. Histone deacetylase 8 in neuroblastoma tumorigenesis. Clin. Cancer Res. 2009, 15, 91–99. [Google Scholar] [CrossRef] [Green Version]
- Rettig, I.; Koeneke, E.; Trippel, F.; Mueller, W.C.; Burhenne, J.; Kopp-Schneider, A.; Fabian, J.; Schober, A.; Fernekorn, U.; von Deimling, A.; et al. Selective inhibition of HDAC8 decreases neuroblastoma growth in vitro and in vivo and enhances retinoic acid-mediated differentiation. Cell Death Dis. 2015, 6, e1657. [Google Scholar] [CrossRef]
- Zhao, G.; Wang, G.; Bai, H.; Li, T.; Gong, F.; Yang, H.; Wen, J.; Wang, W. Targeted inhibition of HDAC8 increases the doxorubicin sensitivity of neuroblastoma cells via up regulation of miR-137. Eur. J. Pharmacol. 2017, 802, 20–26. [Google Scholar] [CrossRef]
- Balasubramanian, S.; Ramos, J.; Luo, W.; Sirisawad, M.; Verner, E.; Buggy, J.J. A novel histone deacetylase 8 (HDAC8)-specific inhibitor PCI-34051 induces apoptosis in T-cell lymphomas. Leukemia 2008, 22, 1026–1034. [Google Scholar] [CrossRef]
- Krennhrubec, K.; Marshall, B.L.; Hedglin, M.; Verdin, E.; Ulrich, S.M. Design and evaluation of ‘Linkerless’ hydroxamic acids as selective HDAC8 inhibitors. Bioorg. Med. Chem. Lett. 2007, 17, 2874–2878. [Google Scholar] [CrossRef]
- Suzuki, T.; Ota, Y.; Ri, M.; Bando, M.; Gotoh, A.; Itoh, Y.; Tsumoto, H.; Tatum, P.R.; Mizukami, T.; Nakagawa, H.; et al. Rapid discovery of highly potent and selective inhibitors of histone deacetylase 8 using click chemistry to generate candidate libraries. J. Med. Chem. 2012, 55, 9562–9575. [Google Scholar] [CrossRef]
- Suzuki, T.; Muto, N.; Bando, M.; Itoh, Y.; Masaki, A.; Ri, M.; Ota, Y.; Nakagawa, H.; Iida, S.; Shirahige, K.; et al. Design, synthesis, and biological activity of NCC149 derivatives as histone deacetylase 8-selective inhibitors. Chem. Med. Chem. 2014, 9, 657–664. [Google Scholar] [CrossRef]
- Huang, W.J.; Wang, Y.C.; Chao, S.W.; Yang, C.Y.; Chen, L.C.; Lin, M.H.; Hou, W.C.; Chen, M.Y.; Lee, T.L.; Yang, P.; et al. Synthesis and biological evaluation of ortho-aryl N-hydroxycinnamides as potent histone deacetylase (HDAC) 8 isoform-selective inhibitors. Chem. Med. Chem. 2012, 7, 1815–1824. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.; Luo, T.; Greenberg, E.F.; Bradner, J.E.; Schreiber, S.L. Discovery of histone deacetylase 8 selective inhibitors. Bioorg. Med. Chem. Lett. 2011, 21, 2601–2605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ingham, O.J.; Paranal, R.M.; Smith, W.B.; Escobar, R.A.; Yueh, H.; Snyder, T.; Porco, J.A., Jr.; Bradner, J.E.; Beeler, A.B. Development of a Potent and Selective HDAC8 Inhibitor. ACS Med. Chem. Lett. 2016, 7, 929–932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whitehead, L.; Dobler, M.R.; Radetich, B.; Zhu, Y.; Atadja, P.W.; Claiborne, T.; Grob, J.E.; McRiner, A.; Pancost, M.R.; Patnaik, A.; et al. Human HDAC isoform selectivity achieved via exploitation of the acetate release channel with structurally unique small molecule inhibitors. Bioorg. Med. Chem. 2011, 19, 4626–4634. [Google Scholar] [CrossRef]
- Heimburg, T.; Kolbinger, F.R.; Zeyen, P.; Ghazy, E.; Herp, D.; Schmidtkunz, K.; Melesina, J.; Shaik, T.B.; Erdmann, F.; Schmidt, M.; et al. Structure-Based Design and Biological Characterization of Selective Histone Deacetylase 8 (HDAC8) Inhibitors with Anti-Neuroblastoma Activity. J. Med. Chem. 2017, 60, 10188–10204. [Google Scholar] [CrossRef]
- Lai, A.C.; Crews, C.M. Induced protein degradation: An emerging drug discovery paradigm. Nat. Rev. Drug Discov. 2017, 16, 101–114. [Google Scholar] [CrossRef] [Green Version]
- Cromm, P.M.; Crews, C.M. Targeted Protein Degradation: From Chemical Biology to Drug Discovery. Cell Chem. Biol. 2017, 24, 1181–1190. [Google Scholar] [CrossRef] [Green Version]
- Itoh, Y. Chemical Protein Degradation Approach and its Application to Epigenetic Targets. Chem. Rec. 2018, 18, 1681–1700. [Google Scholar] [CrossRef]
- Cecchini, C.; Pannilunghi, S.; Tardy, S.; Scapozza, L. From Conception to Development: Investigating PROTACs Features for Improved Cell Permeability and Successful Protein Degradation. Front. Chem. 2021, 9, 672267. [Google Scholar] [CrossRef]
- Yin, L.; Hu, Q. Chimera induced protein degradation: PROTACs and beyond. Eur. J. Med. Chem. 2020, 206, 112494. [Google Scholar] [CrossRef] [PubMed]
- Chamberlain, P.P.; Hamann, L.G. Development of targeted protein degradation therapeutics. Nat. Chem. Biol. 2019, 15, 937–944. [Google Scholar] [CrossRef] [PubMed]
- Ward, C.C.; Kleinman, J.I.; Brittain, S.M.; Lee, P.S.; Chung, C.Y.S.; Kim, K.; Petri, Y.; Thomas, J.R.; Tallarico, J.A.; McKenna, J.M.; et al. Covalent Ligand Screening Uncovers a RNF4 E3 Ligase Recruiter for Targeted Protein Degradation Applications. ACS Chem. Biol. 2019, 14, 2430–2440. [Google Scholar] [CrossRef]
- Zhang, X.; Crowley, V.M.; Wucherpfennig, T.G.; Dix, M.M.; Cravatt, B.F. Electrophilic PROTACs that degrade nuclear proteins by engaging DCAF16. Nat. Chem. Biol. 2019, 15, 737–746. [Google Scholar] [CrossRef] [PubMed]
- Tong, B.; Luo, M.; Xie, Y.; Spradlin, J.N.; Tallarico, J.A.; McKenna, J.M.; Schirle, M.; Maimone, T.J.; Nomura, D.K. Bardoxolone conjugation enables targeted protein degradation of BRD4. Sci. Rep. 2020, 10, 15543. [Google Scholar] [CrossRef]
- Burslem, G.M.; Schultz, A.R.; Bondeson, D.P.; Eide, C.A.; Savage Stevens, S.L.; Druker, B.J.; Crews, C.M. Targeting BCR-ABL1 in Chronic Myeloid Leukemia by PROTAC-Mediated Targeted Protein Degradation. Cancer Res. 2019, 79, 4744–4753. [Google Scholar] [CrossRef] [PubMed]
- Schiedel, M.; Herp, D.; Hammelmann, S.; Swyter, S.; Lehotzky, A.; Robaa, D.; Oláh, J.; Ovádi, J.; Sippl, W.; Jung, M. Chemically Induced Degradation of Sirtuin 2 (Sirt2) by a Proteolysis Targeting Chimera (PROTAC) Based on Sirtuin Rearranging Ligands (SirReals). J. Med. Chem. 2018, 61, 482–491. [Google Scholar] [CrossRef]
- Xue, G.; Chen, J.; Liu, L.; Zhou, D.; Zuo, Y.; Fu, T.; Pan, Z. Protein degradation through covalent inhibitor-based PROTACs. Chem. Commun. 2020, 56, 1521–1524. [Google Scholar] [CrossRef]
- Gabizon, R.; Shraga, A.; Gehrtz, P.; Livnah, E.; Shorer, Y.; Gurwicz, N.; Avram, L.; Unger, T.; Aharoni, H.; Albeck, S.; et al. Efficient Targeted Degradation via Reversible and Irreversible Covalent PROTACs. J. Am. Chem. Soc. 2020, 142, 11734–11742. [Google Scholar] [CrossRef]
- Neklesa, T.; Snyder, L.B.; Willard, R.R.; Vitale, N.; Raina, K.; Pizzano, J.; Gordon, D.; Bookbinder, M.; Macaluso, J.; Dong, H.; et al. Abstract 5236: ARV-110: An androgen receptor PROTAC degrader for prostate cancer. Cancer Res. 2018, 78, 5236. [Google Scholar] [CrossRef]
- Flanagan, J.J.; Qian, Y.; Gough, S.M.; Andreoli, M.; Bookbinder, M.; Cadelina, G.; Bradley, J.; Rousseau, E.; Willard, R.; Pizzano, J.; et al. Abstract P5-04-18: ARV-471, an oral estrogen receptor PROTAC degrader for breast cancer. Cancer Res. 2019, 79, P5-04-18. [Google Scholar] [CrossRef]
- Qi, S.-M.; Dong, J.; Xu, Z.-Y.; Cheng, X.-D.; Zhang, W.-D.; Qin, J.-J. PROTAC: An Effective Targeted Protein Degradation Strategy for Cancer Therapy. Front. Pharmacol. 2021, 12, 692574. [Google Scholar] [CrossRef] [PubMed]
- Tae, H.S.; Sundberg, T.B.; Neklesa, T.K.; Noblin, D.J.; Gustafson, J.L.; Roth, A.G.; Raina, K.; Crews, C.M. Identification of hydrophobic tags for the degradation of stabilized proteins. Chem. Bio. Chem. 2012, 13, 538–541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neklesa, T.K.; Tae, H.S.; Schneekloth, A.R.; Stulberg, M.J.; Corson, T.W.; Sundberg, T.B.; Raina, K.; Holley, S.A.; Crews, C.M. Small-molecule hydrophobic tagging–induced degradation of HaloTag fusion proteins. Nat. Chem. Biol. 2011, 7, 538–543. [Google Scholar] [CrossRef] [Green Version]
- Long, M.J.C.; Gollapalli, D.R.; Hedstrom, L. Inhibitor Mediated Protein Degradation. Chem. Biol. 2012, 19, 629–637. [Google Scholar] [CrossRef] [Green Version]
- Collins, I.; Wang, H.; Caldwell, J.J.; Chopra, R. Chemical approaches to targeted protein degradation through modulation of the ubiquitin-proteasome pathway. Biochem. J. 2017, 474, 1127–1147. [Google Scholar] [CrossRef]
- Neklesa, T.K.; Noblin, D.J.; Kuzin, A.; Lew, S.; Seetharaman, J.; Acton, T.B.; Kornhaber, G.; Xiao, R.; Montelione, G.T.; Tong, L.; et al. A Bidirectional System for the Dynamic Small Molecule Control of Intracellular Fusion Proteins. ACS Chem. Biol. 2013, 8, 2293–2300. [Google Scholar] [CrossRef] [Green Version]
- Shi, Y.; Long, M.J.C.; Rosenberg, M.M.; Li, S.; Kobjack, A.; Lessans, P.; Coffey, R.T.; Hedstrom, L. Boc3Arg-Linked Ligands Induce Degradation by Localizing Target Proteins to the 20S Proteasome. ACS Chem. Biol. 2016, 11, 3328–3337. [Google Scholar] [CrossRef] [Green Version]
- Gustafson, J.L.; Neklesa, T.K.; Cox, C.S.; Roth, A.G.; Buckley, D.L.; Tae, H.S.; Sundberg, T.B.; Stagg, D.B.; Hines, J.; McDonnell, D.P.; et al. Small-Molecule-Mediated Degradation of the Androgen Receptor through Hydrophobic Tagging. Angew. Chem. Int. Ed. 2015, 54, 9659–9662. [Google Scholar] [CrossRef]
- Ma, A.; Stratikopoulos, E.; Park, K.-S.; Wei, J.; Martin, T.C.; Yang, X.; Schwarz, M.; Leshchenko, V.; Rialdi, A.; Dale, B.; et al. Discovery of a first-in-class EZH2 selective degrader. Nat. Chem. Biol. 2020, 16, 214–222. [Google Scholar] [CrossRef]
- Xie, T.; Lim, S.M.; Westover, K.D.; Dodge, M.E.; Ercan, D.; Ficarro, S.B.; Udayakumar, D.; Gurbani, D.; Tae, H.S.; Riddle, S.M.; et al. Pharmacological targeting of the pseudokinase Her3. Nat. Chem. Biol. 2014, 10, 1006–1012. [Google Scholar] [CrossRef] [PubMed]
- Qiu, J.; Bai, X.; Zhang, W.; Ma, M.; Wang, W.; Liang, Y.; Wang, H.; Tian, J.; Yu, P. LPM3770277, a Potent Novel CDK4/6 Degrader, Exerts Antitumor Effect Against Triple-Negative Breast Cancer. Front. Pharmacol. 2022, 13, 853993. [Google Scholar] [CrossRef]
- Choi, S.R.; Wang, H.M.; Shin, M.H.; Lim, H.-S. Hydrophobic Tagging-Mediated Degradation of Transcription Coactivator SRC-1. Int. J. Mol. Sci. 2021, 22, 6407. [Google Scholar] [CrossRef] [PubMed]
- Long, X.; Nephew, K.P. Fulvestrant (ICI 182,780)-dependent Interacting Proteins Mediate Immobilization and Degradation of Estrogen Receptor-α*. J. Biol. Chem. 2006, 281, 9607–9615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, K.; Song, Y.; Xie, H.; Wu, H.; Wu, Y.-T.; Leisten, E.D.; Tang, W. Development of the first small molecule histone deacetylase 6 (HDAC6) degraders. Bioorg. Med. Chem. Lett. 2018, 28, 2493–2497. [Google Scholar] [CrossRef]
- Yang, K.; Wu, H.; Zhang, Z.; Leisten, E.D.; Nie, X.; Liu, B.; Wen, Z.; Zhang, J.; Cunningham, M.D.; Tang, W. Development of Selective Histone Deacetylase 6 (HDAC6) Degraders Recruiting Von Hippel–Lindau (VHL) E3 Ubiquitin Ligase. ACS Med. Chem. Lett. 2020, 11, 575–581. [Google Scholar] [CrossRef]
- Wu, H.; Yang, K.; Zhang, Z.; Leisten, E.D.; Li, Z.; Xie, H.; Liu, J.; Smith, K.A.; Novakova, Z.; Barinka, C.; et al. Development of Multifunctional Histone Deacetylase 6 Degraders with Potent Antimyeloma Activity. J. Med. Chem. 2019, 62, 7042–7057. [Google Scholar] [CrossRef]
- Xiao, Y.; Wang, J.; Zhao, L.Y.; Chen, X.; Zheng, G.; Zhang, X.; Liao, D. Discovery of histone deacetylase 3 (HDAC3)-specific PROTACs. Chem. Commun. 2020, 56, 9866–9869. [Google Scholar] [CrossRef]
- Chotitumnavee, J.; Yamashita, Y.; Takahashi, Y.; Takada, Y.; Iida, T.; Oba, M.; Itoh, Y.; Suzuki, T. Selective degradation of histone deacetylase 8 mediated by a proteolysis targeting chimera (PROTAC). Chem. Commun. 2022, 58, 4635–4638. [Google Scholar] [CrossRef]
- Heimburg, T.; Chakrabarti, A.; Lancelot, J.; Marek, M.; Melesina, J.; Hauser, A.-T.; Shaik, T.B.; Duclaud, S.; Robaa, D.; Erdmann, F.; et al. Structure-Based Design and Synthesis of Novel Inhibitors Targeting HDAC8 from Schistosoma mansoni for the Treatment of Schistosomiasis. J. Med. Chem. 2016, 59, 2423–2435. [Google Scholar] [CrossRef] [Green Version]
- Marek, M.; Shaik, T.B.; Heimburg, T.; Chakrabarti, A.; Lancelot, J.; Ramos-Morales, E.; Da Veiga, C.; Kalinin, D.; Melesina, J.; Robaa, D.; et al. Characterization of Histone Deacetylase 8 (HDAC8) Selective Inhibition Reveals Specific Active Site Structural and Functional Determinants. J. Med. Chem. 2018, 61, 10000–10016. [Google Scholar] [CrossRef] [PubMed]
- Lai, A.C.; Toure, M.; Hellerschmied, D.; Salami, J.; Jaime-Figueroa, S.; Ko, E.; Hines, J.; Crews, C.M. Modular PROTAC Design for the Degradation of Oncogenic BCR-ABL. Angew. Chem. Int. Ed. 2016, 55, 807–810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, B.E.; Wang, S.L.; Jaime-Figueroa, S.; Harbin, A.; Wang, J.; Hamman, B.D.; Crews, C.M. Differential PROTAC substrate specificity dictated by orientation of recruited E3 ligase. Nat. Commun. 2019, 10, 131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cyrus, K.; Wehenkel, M.; Choi, E.Y.; Han, H.J.; Lee, H.; Swanson, H.; Kim, K.B. Impact of linker length on the activity of PROTACs. Mol. Biosyst. 2011, 7, 359–364. [Google Scholar] [CrossRef] [Green Version]
- Zorba, A.; Nguyen, C.; Xu, Y.; Starr, J.; Borzilleri, K.; Smith, J.; Zhu, H.; Farley, K.A.; Ding, W.; Schiemer, J.; et al. Delineating the role of cooperativity in the design of potent PROTACs for BTK. Proc. Natl. Acad. Sci. USA 2018, 115, E7285–E7292. [Google Scholar] [CrossRef] [Green Version]
- Nowak, R.P.; DeAngelo, S.L.; Buckley, D.; He, Z.; Donovan, K.A.; An, J.; Safaee, N.; Jedrychowski, M.P.; Ponthier, C.M.; Ishoey, M.; et al. Plasticity in binding confers selectivity in ligand-induced protein degradation. Nat. Chem. Biol. 2018, 14, 706–714. [Google Scholar] [CrossRef]
- Cyrus, K.; Wehenkel, M.; Choi, E.-Y.; Lee, H.; Swanson, H.; Kim, K.-B. Jostling for Position: Optimizing Linker Location in the Design of Estrogen Receptor-Targeting PROTACs. Chem. Med. Chem. 2010, 5, 979–985. [Google Scholar] [CrossRef] [Green Version]
- Zoppi, V.; Hughes, S.J.; Maniaci, C.; Testa, A.; Gmaschitz, T.; Wieshofer, C.; Koegl, M.; Riching, K.M.; Daniels, D.L.; Spallarossa, A.; et al. Iterative Design and Optimization of Initially Inactive Proteolysis Targeting Chimeras (PROTACs) Identify VZ185 as a Potent, Fast, and Selective von Hippel–Lindau (VHL) Based Dual Degrader Probe of BRD9 and BRD7. J. Med. Chem. 2019, 62, 699–726. [Google Scholar] [CrossRef]
- Ghazy, E.; Zeyen, P.; Herp, D.; Hügle, M.; Schmidtkunz, K.; Erdmann, F.; Robaa, D.; Schmidt, M.; Morales, E.R.; Romier, C.; et al. Design, synthesis, and biological evaluation of dual targeting inhibitors of histone deacetylase 6/8 and bromodomain BRPF1. Eur. J. Med. Chem. 2020, 200, 112338. [Google Scholar] [CrossRef]
- Stolfa, D.A.; Stefanachi, A.; Gajer, J.M.; Nebbioso, A.; Altucci, L.; Cellamare, S.; Jung, M.; Carotti, A. Design, Synthesis, and Biological Evaluation of 2-Aminobenzanilide Derivatives as Potent and Selective HDAC Inhibitors. Chem. Med. Chem. 2012, 7, 1256–1266. [Google Scholar] [CrossRef]
- Marek, M.; Kannan, S.; Hauser, A.-T.; Moraes Mourão, M.; Caby, S.; Cura, V.; Stolfa, D.A.; Schmidtkunz, K.; Lancelot, J.; Andrade, L.; et al. Structural Basis for the Inhibition of Histone Deacetylase 8 (HDAC8), a Key Epigenetic Player in the Blood Fluke Schistosoma mansoni. PLoS Pathog. 2013, 9, e1003645. [Google Scholar] [CrossRef] [PubMed]
- Bensinger, D.; Stubba, D.; Cremer, A.; Kohl, V.; Waßmer, T.; Stuckert, J.; Engemann, V.; Stegmaier, K.; Schmitz, K.; Schmidt, B. Virtual Screening Identifies Irreversible FMS-like Tyrosine Kinase 3 Inhibitors with Activity toward Resistance-Conferring Mutations. J. Med. Chem. 2019, 62, 2428–2446. [Google Scholar] [CrossRef]
- Steinebach, C.; Lindner, S.; Udeshi, N.D.; Mani, D.C.; Kehm, H.; Köpff, S.; Carr, S.A.; Gütschow, M.; Krönke, J. Homo-PROTACs for the Chemical Knockdown of Cereblon. ACS Chem. Biol. 2018, 13, 2771–2782. [Google Scholar] [CrossRef] [PubMed]
- Sasmal, S.; Samajdar, S.; Mukherjee, S.; Abbineni, C. Pyridazine Derivatives as SMARCA2/4 Degraders. WO/2019/207538 A1, 26 April 2019. [Google Scholar]
- Nittoli, T.; Markotan, T. Mytansinoid derivatives, conjugates thereof and methods of use. WO 2016/160615 A1, 25 March 2016. [Google Scholar]
- Qiu, X.; Sun, N.; Kong, Y.; Li, Y.; Yang, X.; Jiang, B. Chemoselective Synthesis of Lenalidomide-Based PROTAC Library Using Alkylation Reaction. Org. Lett. 2019, 21, 3838–3841. [Google Scholar] [CrossRef]
- Kinberger, G.A.; Prakash, T.P.; Yu, J.; Vasquez, G.; Low, A.; Chappell, A.; Schmidt, K.; Murray, H.M.; Gaus, H.; Swayze, E.E.; et al. Conjugation of mono and di-GalNAc sugars enhances the potency of antisense oligonucleotides via ASGR mediated delivery to hepatocytes. Bioorg. Med. Chem. Lett. 2016, 26, 3690–3693. [Google Scholar] [CrossRef]
- Deane, F.M.; O’Sullivan, E.C.; Maguire, A.R.; Gilbert, J.; Sakoff, J.A.; McCluskey, A.; McCarthy, F.O. Synthesis and evaluation of novel ellipticines as potential anti-cancer agents. Org. Biomol. Chem. 2013, 11, 1334–1344. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Gao, C.; Zhao, L.; Yuan, Z.; Chen, Y.; Jiang, Y. Phthalimide conjugations for the degradation of oncogenic PI3K. Eur. J. Med. Chem. 2018, 151, 237–247. [Google Scholar] [CrossRef] [PubMed]
- Braun, M.; Hartnagel, U.; Ravanelli, E.; Schade, B.; Böttcher, C.; Vostrowsky, O.; Hirsch, A. Amphiphilic [5:1]- and [3:3]-Hexakisadducts of C60. Eur. J. Org. Chem. 2004, 2004, 1983–2001. [Google Scholar] [CrossRef]
- Peng, L.; Zhang, Z.; Lei, C.; Li, S.; Zhang, Z.; Ren, X.; Chang, Y.; Zhang, Y.; Xu, Y.; Ding, K. Identification of New Small-Molecule Inducers of Estrogen-related Receptor alpha (ERRalpha) Degradation. ACS Med. Chem. Lett. 2019, 10, 767–772. [Google Scholar] [CrossRef]
- Biraboneye, A.C.; Madonna, S.; Laras, Y.; Krantic, S.; Maher, P.; Kraus, J.L. Potential neuroprotective drugs in cerebral ischemia: New saturated and polyunsaturated lipids coupled to hydrophilic moieties: Synthesis and biological activity. J. Med. Chem. 2009, 52, 4358–4369. [Google Scholar] [CrossRef]
- Kaur, T.; Menon, A.; Garner, A.L. Synthesis of 7-benzylguanosine cap-analogue conjugates for eIF4E targeted degradation. Eur. J. Med. Chem. 2019, 166, 339–350. [Google Scholar] [CrossRef] [PubMed]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ID | Structure | HDAC1 IC50 (µM) or % inhib. at Given conc. | HDAC6 IC50 (µM) or % inhib. at Given conc. | HDAC8 IC50 (µM) |
|---|---|---|---|---|
| CRBN-based PROTACs | ||||
| CRBN_1a |  | 4.37 ± 0.65 | 0.22 ± 0.06 | 0.09 ± 0.03 |
| CRBN_1b |  | >20 | 0.56 ± 0.1 | 0.81 ± 0.17 |
| CRBN_1c |  | 10 µM: 40.1% 1 µM: 22.2% | 0.25 ± 0.02 | 0.15 ± 0.01 |
| CRBN_1d |  | 13.1 ± 0.6 | 6.7 ± 0.6 | 0.37 ± 0.05 |
| CRBN_1e |  | 16.2 ± 0.8 | 17.2 ± 2.4 | 0.25 ± 0.07 |
| CRBN_1f |  | 10.8 ± 0.7 | 1.3 ± 0.3 | 0.25 ± 0.03 |
| CRBN_1g |  | 10 µM: 69% 1µM: 10.1% | 10 µM: 72.6% 1µM: 24.8% | 0.59 ± 0.11 |
| CRBN_1h |  | 3.91 ± 0.48 | 10 µM: 66.8% 1µM: 33.5% | 0.33 ± 0.19 |
| CRBN_1i |  | 10 µM: 67.4% 1µM: 20.7% | 10 µM: 92.3% 1µM: 85.4% | 0.65 ± 0.14 |
| CRBN_1j |  | 10 µM: 60.0% 1µM: 13.9% | 10 µM: 80.9% 1µM: 46.2% | 4.84 ± 1.05 |
| VHL-based PROTACs | ||||
| VHL_1k |  | >20 | 0.31 ± 0.01 | 0.11 ± 0.01 |
| VHL_1l |  | 10 µM: 45.5% 1µM: 6.0% | 10 µM: 72.4% 1µM: 8.3% | 0.72 ± 0.15 |
| HyT-based PROTACs | ||||
| HyT_1m |  | 10 µM: 81.9% 1µM: 47.7% | 0.25 ± 0.08 | 0.57 ± 0.11 |
| HyT_1n |  | 10 µM: 30.0% 1 µM: 38.6% | 0.82 ± 0.05 | 0.39 ± 0.03 |
| HyT_1o |  | 10 µM: 52.6% 1 µM: 9.8% | 0.37 ± 0.03 | 0.09 ± 0.01 |
| HyT_1p |  | 10 µM: 40.0% 1µM: 2.3% | 10 µM: 72.8% 1µM: 12.4% | 0.75 ± 0.09 |
| ID | Inhibitor Structure | HDAC1 IC50 (µM) | HDAC6 IC50 (µM) | HDAC8 IC50 (µM) |
|---|---|---|---|---|
| 2a |  | 33.6 ± 1.8 | 3.0 ± 0.3 | 0.58 ± 0.05 |
| 2b |  | 2.3 ± 1.2 | 2.5 ± 1.1 | 0.09 ± 0.02 |
| 2c |  | 11.6 ± 3.9 | 0.12 ± 0.02 | 0.12 ± 0.04 |
| 2d |  | 2.3 ± 1.2 | 2.5 ± 1.1 | 0.14 ± 0.01 |
| 2e |  | 21.8 ± 2.1 | 5.1 ± 0.3 | 0.26 ± 0.04 |
| 2f |  | 14.5 ± 1.4 | 5.1 ± 0.8 | 0.07 ± 0.02 |
| 2g |  | 10.4 ± 1.2 | 4.0 ± 0.2 | 0.25 ± 0.04 |
| 2h |  | >20 | 0.15 ± 0.001 | 0.01 ± 0.001 |
| 2i |  | >20 | 7.4 ± 0.6 | 0.41 ± 0.05 |
| ID | HEK293 viability 50 µM | ID | HEK293 viability 50 µM | ID | HEK293 viability 50 µM |
|---|---|---|---|---|---|
| 2a | 72.0 ± 2.9 | CRBN_1a | 85.6 ± 2.4 | CRBN_1j | 83.7 ± 3.5 |
| 2b | 67.3 ± 3.9 | CRBN_1b | 64.1 ± 1.7 | VHL_1k | 78.2 ± 3.7 |
| 2c | 72.2 ± 3.5 | CRBN_1c | 70.1 ± 6.7 | VHL_1l | 90.4 ± 2.3 |
| 2d | 78.8 ± 6.1 | CRBN_1d | 80.2 ± 2.8 | HyT_1m | 51.5 ± 6.2 |
| 2e | 90.4 ± 1.7 | CRBN_1e | 65.1 ± 4.3 | HyT_1n | 60.0 ± 3.7 |
| 2f | 68.1 ± 1.2 | CRBN_1f | 100.5 ± 2.8 | HyT_1o | 69.3 ± 1.2 |
| 2g | 87.4 + 3.4 | CRBN_1g | 68.2 ± 2.5 | HyT_1p | 80.1 ± 1.9 |
| 2h | 70.4 ± 7.5 | CRBN_1h | 61.3 ± 0.9 | Daunorubicin | IC50 2.1 ± 0.2 μM |
| 2i | 88.1 ± 0.1 | CRBN_1i | 97.6 ± 7.0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Darwish, S.; Ghazy, E.; Heimburg, T.; Herp, D.; Zeyen, P.; Salem-Altintas, R.; Ridinger, J.; Robaa, D.; Schmidtkunz, K.; Erdmann, F.; et al. Design, Synthesis and Biological Characterization of Histone Deacetylase 8 (HDAC8) Proteolysis Targeting Chimeras (PROTACs) with Anti-Neuroblastoma Activity. Int. J. Mol. Sci. 2022, 23, 7535. https://doi.org/10.3390/ijms23147535
Darwish S, Ghazy E, Heimburg T, Herp D, Zeyen P, Salem-Altintas R, Ridinger J, Robaa D, Schmidtkunz K, Erdmann F, et al. Design, Synthesis and Biological Characterization of Histone Deacetylase 8 (HDAC8) Proteolysis Targeting Chimeras (PROTACs) with Anti-Neuroblastoma Activity. International Journal of Molecular Sciences. 2022; 23(14):7535. https://doi.org/10.3390/ijms23147535
Chicago/Turabian StyleDarwish, Salma, Ehab Ghazy, Tino Heimburg, Daniel Herp, Patrik Zeyen, Rabia Salem-Altintas, Johannes Ridinger, Dina Robaa, Karin Schmidtkunz, Frank Erdmann, and et al. 2022. "Design, Synthesis and Biological Characterization of Histone Deacetylase 8 (HDAC8) Proteolysis Targeting Chimeras (PROTACs) with Anti-Neuroblastoma Activity" International Journal of Molecular Sciences 23, no. 14: 7535. https://doi.org/10.3390/ijms23147535
APA StyleDarwish, S., Ghazy, E., Heimburg, T., Herp, D., Zeyen, P., Salem-Altintas, R., Ridinger, J., Robaa, D., Schmidtkunz, K., Erdmann, F., Schmidt, M., Romier, C., Jung, M., Oehme, I., & Sippl, W. (2022). Design, Synthesis and Biological Characterization of Histone Deacetylase 8 (HDAC8) Proteolysis Targeting Chimeras (PROTACs) with Anti-Neuroblastoma Activity. International Journal of Molecular Sciences, 23(14), 7535. https://doi.org/10.3390/ijms23147535