
Tumor-Localized Administration of α-GalCer to Recruit Invariant Natural Killer T Cells and Enhance Their Antitumor Activity against Solid Tumors

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Generation and Characterization of Healthy Donor Peripheral Blood Mononuclear (PBMC)-Derived iNKT (PBMC-iNKT) Cells
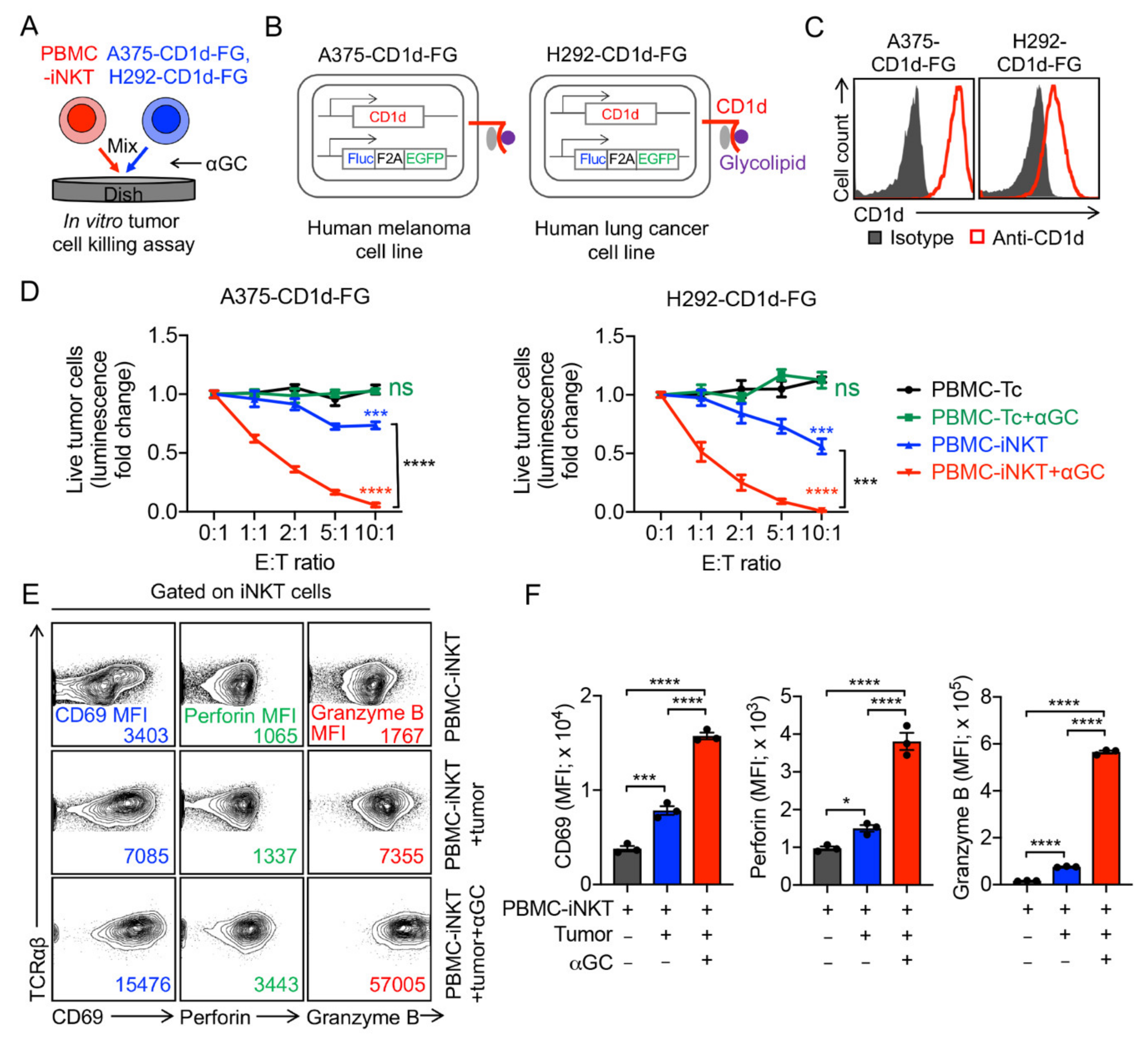
2.2. Tumor Targeting of PBMC-iNKT Cells
2.3. Pharmacokinetics/Pharmacodynamics (PK/PD) Study of PBMC-iNKT Cells
3. Discussion
4. Materials and Methods
4.1. Lentiviral Vectors and Transduction
4.2. Cell Culture Media and Reagents
4.3. Tumor Cell Lines
4.4. Mice
4.5. Antibodies and Flow Cytometry
4.6. Enzyme-Linked Immunosorbent Assay (ELISA)
4.7. In Vitro Generation of Peripheral Blood Mononuclear (PBMC)-Derived Conventional αß T (PBMC-Tc) Cells
4.8. In Vitro Generation and Activation of PBMC-Derived iNKT (PBMC-iNKT) Cells
4.9. Cell Phenotype Study
4.10. In Vitro Tumor Cell Killing Assay
4.11. Bioluminescence Live Animal Imaging (BLI)
4.12. In Vivo Antitumor Efficacy Study: H292 Human Lung Cancer Xenograft NSG Mouse Model
4.13. In Vivo Antitumor Efficacy Study: A375 Human Melanoma Xenograft NSG Mouse Model
4.14. In Vivo Pharmacokinetics/Pharmacodynamics (PK/PD) Study of PBMC-iNKT Cells: A375 Human Melanoma Xenograft NSG Mouse Model
4.15. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- King, L.A.; Lameris, R.; de Gruijl, T.D.; van der Vliet, H.J. CD1d-Invariant Natural Killer T Cell-Based Cancer Immunotherapy: α-Galactosylceramide and Beyond. Front. Immunol. 2018, 9, 1519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bendelac, A.; Savage, P.B.; Teyton, L. The biology of NKT cells. Annu. Rev. Immunol. 2007, 25, 297–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McEwen-Smith, R.M.; Salio, M.; Cerundolo, V. The regulatory role of invariant NKT cells in tumor immunity. Cancer Immunol. Res. 2015, 3, 425–435. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.-R.; Dunn, Z.S.; Zhou, Y.; Lee, D.; Yang, L. Development of Stem Cell-Derived Immune Cells for Off-the-Shelf Cancer Immunotherapies. Cells 2021, 10, 3479. [Google Scholar] [CrossRef]
- Li, Y.-R.; Zhou, Y.; Kramer, A.; Yang, L. Engineering stem cells for cancer immunotherapy. Trends Cancer 2021, 7, 1059–1073. [Google Scholar] [CrossRef]
- Van Kaer, L.; Parekh, V.V.; Wu, L. Invariant natural killer T cells: Bridging innate and adaptive immunity. Cell Tissue Res. 2011, 343, 43–55. [Google Scholar] [CrossRef] [Green Version]
- Kronenberg, M.; Gapin, L. The unconventional lifestyle of NKT cells. Nat. Rev. Immunol. 2002, 2, 557–568. [Google Scholar] [CrossRef]
- Bae, E.A.; Seo, H.; Kim, I.K.; Jeon, I.; Kang, C.Y. Roles of NKT cells in cancer immunotherapy. Arch. Pharm. Res. 2019, 42, 543–548. [Google Scholar] [CrossRef]
- Brennan, P.J.; Brigl, M.; Brenner, M.B. Invariant natural killer T cells: An innate activation scheme linked to diverse effector functions. Nat. Rev. Immunol. 2013, 13, 101–117. [Google Scholar] [CrossRef]
- Snyder-Cappione, J.E.; Nixon, D.F.; Chi, J.C.; Nguyen, M.-L.T.; Kirby, C.K.; Milush, J.M.; Koth, L.L. Invariant natural killer T (iNKT) cell exhaustion in sarcoidosis. Eur. J. Immunol. 2013, 43, 2194–2205. [Google Scholar] [CrossRef] [Green Version]
- Díaz-Basabe, A.; Strati, F.; Facciotti, F. License to Kill: When iNKT Cells Are Granted the Use of Lethal Cytotoxicity. Int. J. Mol. Sci. 2020, 21, 3909. [Google Scholar] [CrossRef]
- Godfrey, D.I.; Berzins, S.P. Control points in NKT-cell development. Nat. Rev. Immunol. 2007, 7, 505–518. [Google Scholar] [CrossRef]
- Rudak, P.T.; Haeryfar, S.M.M. In Vivo Cytotoxicity by α-GalCer-Transactivated NK Cells BT—Invariant Natural Killer T-Cells: Methods and Protocols; Liu, C., Ed.; Springer: New York, NY, USA, 2021; pp. 157–174. ISBN 978-1-0716-1775-5. [Google Scholar]
- Waldowska, M.; Bojarska-Junak, A.; Roliński, J. A brief review of clinical trials involving manipulation of invariant NKT cells as a promising approach in future cancer therapies. Cent. J. Immunol. 2017, 42, 181–195. [Google Scholar] [CrossRef]
- Zhang, Y.; Springfield, R.; Chen, S.; Li, X.; Feng, X.; Moshirian, R.; Yang, R.; Yuan, W. α-GalCer and iNKT Cell-Based Cancer Immunotherapy: Realizing the Therapeutic Potentials. Front. Immunol. 2019, 10, 1126. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Y.; Smith, D.J.; Zhou, Y.; Li, Y.R.; Yu, J.; Lee, D.; Wang, Y.C.; Di Biase, S.; Wang, X.; Hardoy, C.; et al. Development of Hematopoietic Stem Cell-Engineered Invariant Natural Killer T Cell Therapy for Cancer. Cell Stem Cell 2019, 25, 542–557.e9. [Google Scholar] [CrossRef]
- Kunii, N.; Horiguchi, S.; Motohashi, S.; Yamamoto, H.; Ueno, N.; Yamamoto, S.; Sakurai, D.; Taniguchi, M.; Nakayama, T.; Okamoto, Y. Combination therapy of in vitro-expanded natural killer T cells and α-galactosylceramide-pulsed antigen-presenting cells in patients with recurrent head and neck carcinoma. Cancer Sci. 2009, 100, 1092–1098. [Google Scholar] [CrossRef]
- Li, Y.-R.; Zhou, Y.; Kim, Y.J.; Zhu, Y.; Ma, F.; Yu, J.; Wang, Y.-C.; Chen, X.; Li, Z.; Zeng, S.; et al. Development of allogeneic HSC-engineered iNKT cells for off-the-shelf cancer immunotherapy. Cell Rep. Med. 2021, 2, 100449. [Google Scholar] [CrossRef]
- Li, Y.R.; Dunn, Z.S.; Garcia, G., Jr.; Carmona, C.; Zhou, Y.; Lee, D.; Yu, J.; Huang, J.; Kim, J.T.; Arumugaswami, V.; et al. Development of off-the-shelf hematopoietic stem cell—Engineered invariant natural killer T cells for COVID-19 therapeutic intervention. Stem Cell Res. Ther. 2022, 13, 112. [Google Scholar] [CrossRef]
- Fu, S.; He, K.; Tian, C.; Sun, H.; Zhu, C.; Bai, S.; Liu, J.; Wu, Q.; Xie, D.; Yue, T.; et al. Impaired lipid biosynthesis hinders anti-tumor efficacy of intratumoral iNKT cells. Nat. Commun. 2020, 11, 438. [Google Scholar] [CrossRef] [Green Version]
- Park, J.-Y.; Keller, H.; Sato, N.; Park, J.-H. NKT cells require IL-7, not IL-15, for survival and homeostasis (LYM4P.761). J. Immunol. 2014, 192 (Suppl. S1), 65.18. [Google Scholar]
- Uchida, T.; Horiguchi, S.; Tanaka, Y.; Yamamoto, H.; Kunii, N.; Motohashi, S.; Taniguchi, M.; Nakayama, T.; Okamoto, Y. Phase I study of alpha-galactosylceramide-pulsed antigen presenting cells administration to the nasal submucosa in unresectable or recurrent head and neck cancer. Cancer Immunol. Immunother. 2008, 57, 337–345. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, A.; Motohashi, S.; Ishikawa, E.; Fuchida, H.; Higashino, K.; Otsuji, M.; Iizasa, T.; Nakayama, T.; Taniguchi, M.; Fujisawa, T. A phase I study of alpha-galactosylceramide (KRN7000)-pulsed dendritic cells in patients with advanced and recurrent non-small cell lung cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2005, 11, 1910–1917. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toyoda, T.; Kamata, T.; Tanaka, K.; Ihara, F.; Takami, M.; Suzuki, H.; Nakajima, T.; Ikeuchi, T.; Kawasaki, Y.; Hanaoka, H.; et al. Phase II study of α-galactosylceramide-pulsed antigen-presenting cells in patients with advanced or recurrent non-small cell lung cancer. J. Immunother. Cancer 2020, 8, e000316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagato, K.; Motohashi, S.; Ishibashi, F.; Okita, K.; Yamasaki, K.; Moriya, Y.; Hoshino, H.; Yoshida, S.; Hanaoka, H.; Fujii, S.I.; et al. Accumulation of activated invariant natural killer T cells in the tumor microenvironment after α-galactosylceramide-pulsed antigen presenting cells. J. Clin. Immunol. 2012, 32, 1071–1081. [Google Scholar] [CrossRef]
- Takami, M.; Ihara, F.; Motohashi, S. Clinical application of iNKT cell-mediated anti-tumor activity against lung cancer and head and neck cancer. Front. Immunol. 2018, 9, 2021. [Google Scholar] [CrossRef]
- Yamasaki, K.; Horiguchi, S.; Kurosaki, M.; Kunii, N.; Nagato, K.; Hanaoka, H.; Shimizu, N.; Ueno, N.; Yamamoto, S.; Taniguchi, M.; et al. Induction of NKT cell-specific immune responses in cancer tissues after NKT cell-targeted adoptive immunotherapy. Clin. Immunol. 2011, 138, 255–265. [Google Scholar] [CrossRef]
- Giaccone, G.; Punt, C.J.A.; Ando, Y.; Ruijter, R.; Nishi, N.; Peters, M.; Von Blomberg, B.M.E.; Scheper, R.J.; van der Vliet, H.J.J.; van den Eertwegh, A.J.M.; et al. A phase I study of the natural killer T-cell ligand α-galactosylceramide (KRN7000) in patients with solid tumors. Clin. Cancer Res. 2002, 8, 3702–3709. [Google Scholar]
- Nicol, A.J.; Tazbirkova, A.; Nieda, M. Comparison of clinical and immunological effects of intravenous and intradermal administration of α-galactosylceramide (KRN7000)-pulsed dendritic cells. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2011, 17, 5140–5151. [Google Scholar] [CrossRef] [Green Version]
- Chang, D.H.; Osman, K.; Connolly, J.; Kukreja, A.; Krasovsky, J.; Pack, M.; Hutchinson, A.; Geller, M.; Liu, N.; Annable, R.; et al. Sustained expansion of NKT cells and antigen-specific T cells after injection of α-galactosyl-ceramide loaded mature dendritic cells in cancer patients. J. Exp. Med. 2005, 201, 1503–1517. [Google Scholar] [CrossRef]
- Nieda, M.; Okai, M.; Tazbirkova, A.; Lin, H.; Yamaura, A.; Ide, K.; Abraham, R.; Juji, T.; Macfarlane, D.J.; Nicol, A.J. Therapeutic activation of Vα24+Vβ11+ NKT cells in human subjects results in highly coordinated secondary activation of acquired and innate immunity. Blood 2004, 103, 383–389. [Google Scholar] [CrossRef]
- Gao, Y.; Guo, J.; Bao, X.; Xiong, F.; Ma, Y.; Tan, B.; Yu, L.; Zhao, Y.; Lu, J. Adoptive Transfer of Autologous Invariant Natural Killer T Cells as Immunotherapy for Advanced Hepatocellular Carcinoma: A Phase I Clinical Trial. Oncologist 2021, 26, e1919–e1930. [Google Scholar] [CrossRef]
- Motohashi, S.; Ishikawa, A.; Ishikawa, E.; Otsuji, M.; Iizasa, T.; Hanaoka, H.; Shimizu, N.; Horiguchi, S.; Okamoto, Y.; Fujii, S.; et al. A phase I study of in vitro expanded natural killer T cells in patients with advanced and recurrent non-small cell lung cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2006, 12, 6079–6086. [Google Scholar] [CrossRef] [Green Version]
- Exley, M.A.; Friedlander, P.; Alatrakchi, N.; Vriend, L.; Yue, S.; Sasada, T.; Zeng, W.; Mizukami, Y.; Clark, J.; Nemer, D.; et al. Adoptive transfer of invariant NKT cells as immunotherapy for advanced melanoma: A phase I clinical trial. Clin. Cancer Res. 2017, 23, 3510–3519. [Google Scholar] [CrossRef] [Green Version]
- Cheng, X.; Wang, J.; Qiu, C.; Jin, Y.; Xia, B.; Qin, R.; Hu, H.; Yan, J.; Zhang, X.; Xu, J. Feasibility of iNKT cell and PD-1+CD8+ T cell-based immunotherapy in patients with lung adenocarcinoma: Preliminary results of a phase I/II clinical trial. Clin. Immunol. 2022, 238, 108992. [Google Scholar] [CrossRef]
- Nelson, A.; Lukacs, J.D.; Johnston, B. The Current Landscape of NKT Cell Immunotherapy and the Hills Ahead. Cancers 2021, 13, 5174. [Google Scholar] [CrossRef]
- Muhammad Ali Tahir, S.; Cheng, O.; Shaulov, A.; Koezuka, Y.; Bubley, G.J.; Wilson, S.B.; Balk, S.P.; Exley, M.A. Loss of IFN-γ Production by Invariant NK T Cells in Advanced Cancer. J. Immunol. 2001, 167, 4046–4050. [Google Scholar] [CrossRef] [Green Version]
- Kawano, T.; Nakayama, T.; Kamada, N.; Kaneko, Y.; Harada, M.; Ogura, N.; Akutsu, Y.; Motohashi, S.; Iizasa, T.; Endo, H.; et al. Antitumor cytotoxicity mediated by ligand-activated human V alpha24 NKT cells. Cancer Res. 1999, 59, 5102–5105. [Google Scholar]
- Uchida, T.; Nakashima, H.; Yamagata, A.; Ito, S.; Ishikiriyama, T.; Nakashima, M.; Seki, S.; Kumagai, H.; Oshima, N. Repeated administration of alpha-galactosylceramide ameliorates experimental lupus nephritis in mice. Sci. Rep. 2018, 8, 8225. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, G.; Chai, D.; Dang, Y.; Zheng, J.; Li, H. iNKT: A new avenue for CAR-based cancer immunotherapy. Transl. Oncol. 2022, 17, 101342. [Google Scholar] [CrossRef]
- Wolf, B.J.; Choi, J.E.; Exley, M.A. Novel Approaches to Exploiting Invariant NKT Cells in Cancer Immunotherapy. Front. Immunol. 2018, 9, 384. [Google Scholar] [CrossRef]
- Metelitsa, L.S. Anti-tumor potential of type-I NKT cells against CD1d-positive and CD1d-negative tumors in humans. Clin. Immunol. 2011, 140, 119–129. [Google Scholar] [CrossRef] [Green Version]
- Dai, H.; Wang, Y.; Lu, X.; Han, W. Chimeric Antigen Receptors Modified T-Cells for Cancer Therapy. JNCI J. Natl. Cancer Inst. 2016, 108, djv439. [Google Scholar] [CrossRef] [Green Version]
- Rotolo, A.; Caputo, V.S.; Holubova, M.; Baxan, N.; Dubois, O.; Chaudhry, M.S.; Xiao, X.; Goudevenou, K.; Pitcher, D.S.; Petevi, K.; et al. Enhanced Anti-lymphoma Activity of CAR19-iNKT Cells Underpinned by Dual CD19 and CD1d Targeting. Cancer Cell 2018, 34, 596–610.e11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poels, R.; Drent, E.; Lameris, R.; Katsarou, A.; Themeli, M.; van der Vliet, H.J.; de Gruijl, T.D.; van de Donk, N.W.C.J.; Mutis, T. Preclinical Evaluation of Invariant Natural Killer T Cells Modified with CD38 or BCMA Chimeric Antigen Receptors for Multiple Myeloma. Int. J. Mol. Sci. 2021, 22, 1096. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Huang, W.; Heczey, A.; Liu, D.; Guo, L.; Wood, M.; Jin, J.; Courtney, A.N.; Liu, B.; Di Pierro, E.J.; et al. NKT cells coexpressing a GD2-specific chimeric antigen receptor and IL15 show enhanced in vivo persistence and antitumor activity against neuroblastoma. Clin. Cancer Res. 2019, 25, 7126–7138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krijgsman, D.; Hokland, M.; Kuppen, P.J.K. The Role of Natural Killer T Cells in Cancer-A Phenotypical and Functional Approach. Front. Immunol. 2018, 9, 367. [Google Scholar] [CrossRef]
- Zhou, Y.; Li, Y.-R.; Zeng, S.; Yang, L. Methods for Studying Mouse and Human Invariant Natural Killer T Cells. Methods Mol. Biol. 2021, 2388, 35–57. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Y.-R.; Zhou, Y.; Wilson, M.; Kramer, A.; Hon, R.; Zhu, Y.; Fang, Y.; Yang, L. Tumor-Localized Administration of α-GalCer to Recruit Invariant Natural Killer T Cells and Enhance Their Antitumor Activity against Solid Tumors. Int. J. Mol. Sci. 2022, 23, 7547. https://doi.org/10.3390/ijms23147547
Li Y-R, Zhou Y, Wilson M, Kramer A, Hon R, Zhu Y, Fang Y, Yang L. Tumor-Localized Administration of α-GalCer to Recruit Invariant Natural Killer T Cells and Enhance Their Antitumor Activity against Solid Tumors. International Journal of Molecular Sciences. 2022; 23(14):7547. https://doi.org/10.3390/ijms23147547
Chicago/Turabian StyleLi, Yan-Ruide, Yang Zhou, Matthew Wilson, Adam Kramer, Ryan Hon, Yichen Zhu, Ying Fang, and Lili Yang. 2022. "Tumor-Localized Administration of α-GalCer to Recruit Invariant Natural Killer T Cells and Enhance Their Antitumor Activity against Solid Tumors" International Journal of Molecular Sciences 23, no. 14: 7547. https://doi.org/10.3390/ijms23147547
APA StyleLi, Y. -R., Zhou, Y., Wilson, M., Kramer, A., Hon, R., Zhu, Y., Fang, Y., & Yang, L. (2022). Tumor-Localized Administration of α-GalCer to Recruit Invariant Natural Killer T Cells and Enhance Their Antitumor Activity against Solid Tumors. International Journal of Molecular Sciences, 23(14), 7547. https://doi.org/10.3390/ijms23147547