
Pathogenesis and Therapeutic Strategies Related to Non-Alcoholic Fatty Liver Disease


Abstract
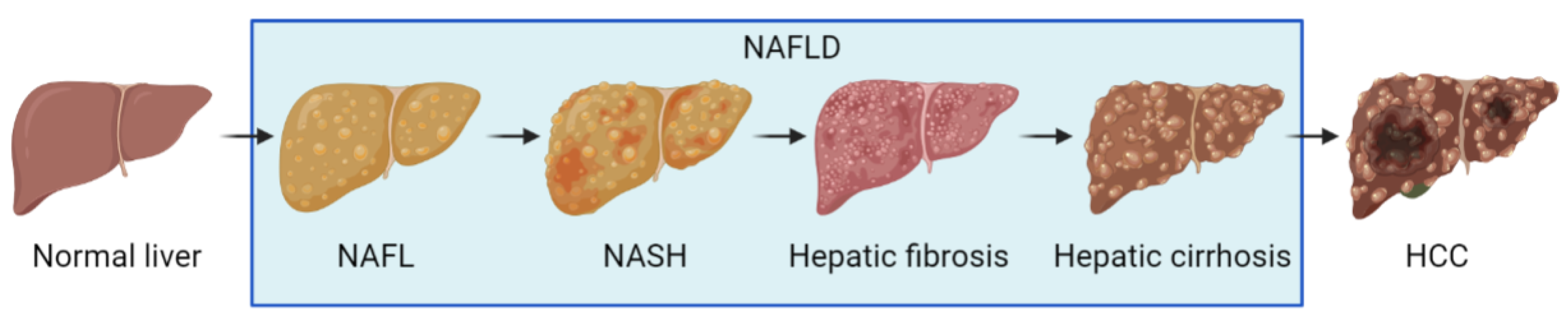
:1. Introduction
2. The Pathogenesis of NAFLD
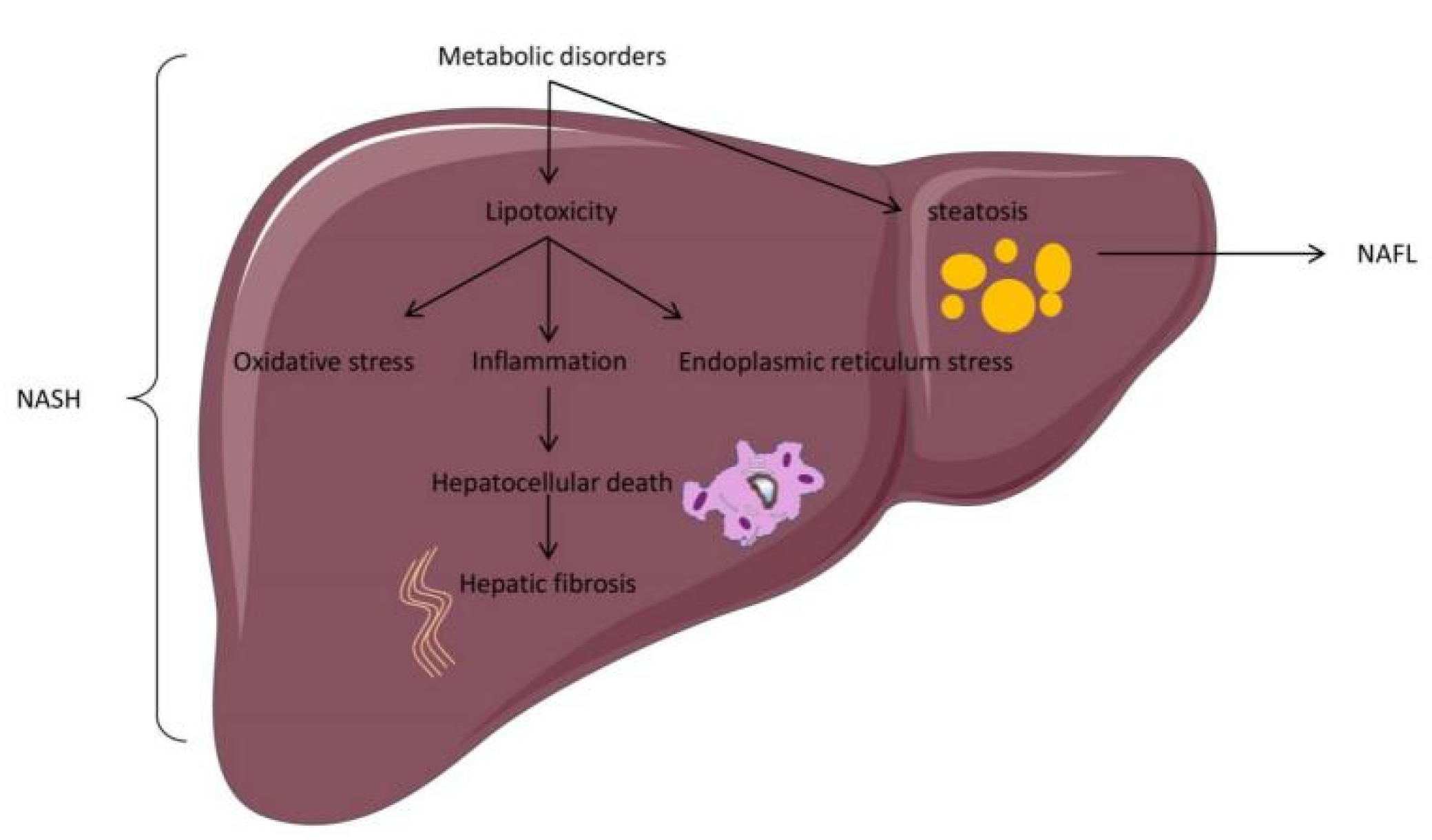
2.1. Two Hits Hypothesis
2.2. Multiple Hits Hypothesis
2.3. Progress on the Pathogenesis of NAFLD
3. Potential Targets for NAFLD Treatment
3.1. Potential Targets of Metabolic Disorders/Steatosis in NAFL
3.1.1. Sodium-Glucose Cotransporter 2 (SGLT2)
3.1.2. Perilipin Drip Protein-2 (PLIN2)
3.1.3. Liver X Receptor α (LXRα)
3.1.4. Acetyl CoA Carboxylase (ACC)
3.1.5. Fatty Acid Synthase (FASN)
3.1.6. AMP-Activated Protein Kinase (AMPK)
3.2. Potential Targets of Hepatic Inflammation in NASH
3.2.1. Apoptosis Signal-Regulated Kinase 1 (ASK1)
3.2.2. Sirtuin 1 (SIRT1)
3.2.3. NLPR3
3.3. Potential Targets of Hepatic Fibrosis in NASH
3.3.1. Lysyl Oxidase-like 2 (LOXL2)
3.3.2. Vascular Adhesion Protein-1 (VAP-1)
4. Potential Targeted Drugs for NAFLD
4.1. Hypoglycemic Drugs
4.2. Lipid-Lowering Drugs
4.3. THR-β Agonist
4.4. Vitamin E
4.5. FXR Agonist
4.6. PPAR Agonist
4.7. FGF19 Analogue
4.8. FGF21 Analogue
4.9. CCR2/CCR5 Inhibitor
4.10. Pan-Caspase Inhibitor
4.11. Galectin-3 Inhibitor

{kind=link}
{kind=link}
{kind=link}
| Drug | Mechanism | Therapeutic Benefits | Side Effects | Clinical No. | Ref. |
|---|---|---|---|---|---|
| Metformin | Activating AMPK and inhibiting ACC | Inhibiting adipogenesis and improving IR | Appetite suppression | NCT00736385 | [140] |
| Liraglutide | Activating GLP-1 | Improving insulin sensitivity and metabolic disorders | Appetite suppression | NCT01237119 | [141] |
| Statins | Inhibiting HMG-CoA | Reducing plasma total cholesterol and low density lipoprotein | Raised aminotransferases | NCT03434613 | [142] |
| GS-0976 | Inhibiting ACC | Reducing triglyceride accumulation in hepatocytes | Nausea and vomiting | NCT03987074 | [143] |
| MGL-3196 | Activating THR-β | Improving lipid metabolism and steatosis | Transient diarrhea | NCT04197479 | [144] |
| Vitamin E | Inhibiting ROS | Reducing oxidative stress and inflammation | |||
| Obeticholic acid | Activating FXR | Improving lipid metabolism | Pruritus | NCT01265498 | [145] |
| Cilofexor | Activating FXR | Improving inflammation and fibrosis | Pruritus | NCT02654002 | [146] |
| Tropifexor | Activating FXR | Improving adipogenesis, inflammation, and fibrosis | Pruritus and cholestatic disorders | NCT03681457 | [147] |
| Elafibranor | Activating PPARα/δ | Improving inflammation and fibrosis | Pruritus | NCT01694849 | [148] |
| Lanifibranor | Activating PPARα/δ/γ | Improving NASH and liver fibrosis | peripheral edema | NCT03459079 | [149] |
| NGM282 | Activating FGF19 | Reducing liver fat, liver injury, and inflammation | Nausea and abdominal pain | NCT02443116 | [150] |
| BMS-986036 | Activating FGF21 | Improving insulin sensitivity, liver fat content, and adiponectin content | Immunogenicity | NCT03486899 | [151] |
| Cenicriviroc | Inhibiting CCR2/CCR5 | Improving inflammation and fibrosis | Headache | NCT02330549 | [152] |
| IDN-6556 | Inhibiting pan-caspase | Improving apoptosis, inflammation, and fibrosis | NCT02077374 | [153] | |
| GR-MD-02 | Inhibiting galectin-3 | Improving fibrosis | NCT02077374 | [154] | |
| Empagliflozin | Inhibiting SGLT-2 | Reducing ALT and liver fat | Acute kidney injury | [155] | |
| Canagliflozin | Inhibiting SGLT-2 | Improving AST, FIB-4 index | Acute renal failure | [156] | |
| Rosiglitazone | Activating PPAR-γ | Improving steatosis and transaminase levels | Heart failure and peripheral edema | [157] | |
| Pioglitazone | Activating PPAR-γ | Improving steatosis, inflammation, and liver histology | Hypoglycemia and lower limb edema | NCT00063622 | [158] |
| Semaglutide | Activating GLP-1 | Reducing body weight and liver enzymes | Nausea and diarrhea | NCT02453711 | [159] |
| Pentoxifylline | Inhibiting TNF-a | Improving liver enzymes and insulin resistance | Nausea and vomiting | ||
| JKB-121 | Activating TLR-4 | Reducing liver fat content | Mild drug-related adverse events | NCT02442687 | [160] |
| Emricasan | Inhibiting caspase | Improving fibrosis | Chest pain and headache | NCT02686762 | [134] |
| Selonsertib | Inhibiting ASK-1 | Improving fibrosis and reduction in hepatic decompensation, hepatocellular carcinoma | Mild drug-related adverse events | NCT03053063 | [161] |
| Atorvastatin | Inhibiting HMG-CoA | Reducing steatosis and improving liver density | Autoimmune hepatitis | [162] | |
| Ezetimibe | Decreasing intestinal cholesterol absorption | Improving aminotransferases and hepatocyte ballooning | New-onset diabetes and increased HbA1c levels | [163] | |
| GS-9674 | Activating FXR | Reducing hepatic fat and improving liver biochemistry | NCT02854605 | [164] | |
| Aramchol | Inhibiting SCD-1 | Reducing liver fat, Ballooning, and AST | NCT04104321 | [165] | |
| Losartan | Activating TGF-β | Improving serum aminotransferases and histologic outcomes | Angioedema | [166] | |
| Telmisartan | Inhibiting CCR2 and CCR5 | Reducing serum ALT levels and improving insulin sensitivity steatosis | Angioedema | NCT01088295 | [167,168] |
| VK-2809 | Activating thyroid receptor β | Reducing fat in liver | [144] | ||
| Simtuzumab | Monoclonal antibody of LOXL2 | Improving liver cirrhosis | NCT01672866 | [152] |
5. Discussion
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Marchisello, S.; Di Pino, A.; Scicali, R.; Urbano, F.; Piro, S.; Purrello, F.; Rabuazzo, A.M. Pathophysiological, Molecular and Therapeutic Issues of Nonalcoholic Fatty Liver Disease: An Overview. Int. J. Mol. Sci. 2019, 20, 1948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Angulo, P. Nonalcoholic fatty liver disease. N. Engl. J. Med. 2002, 346, 1221–1231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Younossi, Z.; Anstee, Q.M.; Marietti, M.; Hardy, T.; Henry, L.; Eslam, M.; George, J.; Bugianesi, E. Global burden of NAFLD and NASH: Trends, predictions, risk factors and prevention. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, C.S.; Graubard, B.I.; Thistle, J.E.; Petrick, J.L.; McGlynn, K.A. Attributable Fractions of NAFLD for Mortality in the United States: Results From NHANES III With 27 Years of Follow-up. Hepatology 2019, 72, 430–440. [Google Scholar] [CrossRef] [Green Version]
- Younossi, Z.; Tacke, F.; Arrese, M.; Chander Sharma, B.; Mostafa, I.; Bugianesi, E.; Wai-Sun Wong, V.; Yilmaz, Y.; George, J.; Fan, J.; et al. Global Perspectives on Nonalcoholic Fatty Liver Disease and Nonalcoholic Steatohepatitis. Hepatology 2019, 69, 2672–2682. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Zou, B.; Yeo, Y.H.; Feng, Y.; Xie, X.; Lee, D.H.; Fujii, H.; Wu, Y.; Kam, L.Y.; Ji, F.; et al. Prevalence, incidence, and outcome of non-alcoholic fatty liver disease in Asia, 1999-2019: A systematic review and meta-analysis. Lancet Gastroenterol. Hepatol. 2019, 4, 389–398. [Google Scholar] [CrossRef]
- Rehm, J.; Mathers, C.; Popova, S.; Thavorncharoensap, M.; Teerawattananon, Y.; Patra, J. Global burden of disease and injury and economic cost attributable to alcohol use and alcohol-use disorders. Lancet 2009, 373, 2223–2233. [Google Scholar] [CrossRef]
- Calzadilla Bertot, L.; Adams, L.A. The Natural Course of Non-Alcoholic Fatty Liver Disease. Int. J. Mol. Sci. 2016, 17, 774. [Google Scholar] [CrossRef] [Green Version]
- Eslam, M.; Valenti, L.; Romeo, S. Genetics and epigenetics of NAFLD and NASH: Clinical impact. J. Hepatol. 2018, 68, 268–279. [Google Scholar] [CrossRef]
- Cohen, J.C.; Horton, J.D.; Hobbs, H.H. Human fatty liver disease: Old questions and new insights. Science 2011, 332, 1519–1523. [Google Scholar] [CrossRef] [Green Version]
- Targher, G.; Byrne, C.D.; Lonardo, A.; Zoppini, G.; Barbui, C. Non-alcoholic fatty liver disease and risk of incident cardiovascular disease: A meta-analysis. J. Hepatol. 2016, 65, 589–600. [Google Scholar] [CrossRef] [Green Version]
- Lonardo, A.; Nascimbeni, F.; Mantovani, A.; Targher, G. Hypertension, diabetes, atherosclerosis and NASH: Cause or consequence? J. Hepatol. 2018, 68, 335–352. [Google Scholar] [CrossRef] [PubMed]
- Day, C.P.; James, O.F. Steatohepatitis: A tale of two “hits”? Gastroenterology 1998, 114, 842–845. [Google Scholar] [CrossRef]
- Friedman, S.L.; Neuschwander-Tetri, B.A.; Rinella, M.; Sanyal, A.J. Mechanisms of NAFLD development and therapeutic strategies. Nat. Med. 2018, 24, 908–922. [Google Scholar] [CrossRef] [PubMed]
- Buzzetti, E.; Pinzani, M.; Tsochatzis, E.A. The multiple-hit pathogenesis of non-alcoholic fatty liver disease (NAFLD). Metabolism 2016, 65, 1038–1048. [Google Scholar] [CrossRef]
- Marra, F.; Svegliati-Baroni, G. Lipotoxicity and the gut-liver axis in NASH pathogenesis. J. Hepatol. 2018, 68, 280–295. [Google Scholar] [CrossRef]
- Byrne, C.D.; Targher, G. What’s new in NAFLD pathogenesis, biomarkers and treatment? Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 70–71. [Google Scholar] [CrossRef]
- Tsuchida, T.; Friedman, S.L. Mechanisms of hepatic stellate cell activation. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 397–411. [Google Scholar] [CrossRef]
- Tacke, F.; Weiskirchen, R. Non-alcoholic fatty liver disease (NAFLD)/non-alcoholic steatohepatitis (NASH)-related liver fibrosis: Mechanisms, treatment and prevention. Ann. Transl. Med. 2021, 9, 729. [Google Scholar] [CrossRef]
- Zhou, W.C.; Zhang, Q.B.; Qiao, L. Pathogenesis of liver cirrhosis. World J. Gastroenterol. 2014, 20, 7312–7324. [Google Scholar] [CrossRef]
- Tilg, H.; Adolph, T.E.; Moschen, A.R. Multiple Parallel Hits Hypothesis in Nonalcoholic Fatty Liver Disease: Revisited After a Decade. Hepatology 2021, 73, 833–842. [Google Scholar] [CrossRef]
- Makri, E.; Goulas, A.; Polyzos, S.A. Epidemiology, Pathogenesis, Diagnosis and Emerging Treatment of Nonalcoholic Fatty Liver Disease. Arch. Med. Res. 2021, 52, 25–37. [Google Scholar] [CrossRef]
- Fujii, H.; Kawada, N.; Japan Study Group Of Nafld, J.-N. The Role of Insulin Resistance and Diabetes in Nonalcoholic Fatty Liver Disease. Int. J. Mol. Sci. 2020, 21, 3863. [Google Scholar] [CrossRef]
- Koh, A.; Molinaro, A.; Ståhlman, M.; Khan, M.T.; Schmidt, C.; Mannerås-Holm, L.; Wu, H.; Carreras, A.; Jeong, H.; Olofsson, L.E.; et al. Microbially Produced Imidazole Propionate Impairs Insulin Signaling through mTORC1. Cell 2018, 175, 947–961.e17. [Google Scholar] [CrossRef] [Green Version]
- Zhao, M.; Zhao, L.; Xiong, X.; He, Y.; Huang, W.; Liu, Z.; Ji, L.; Pan, B.; Guo, X.; Wang, L.; et al. TMAVA, a Metabolite of Intestinal Microbes, Is Increased in Plasma From Patients With Liver Steatosis, Inhibits γ-Butyrobetaine Hydroxylase, and Exacerbates Fatty Liver in Mice. Gastroenterology 2020, 158, 2266–2281.e27. [Google Scholar] [CrossRef]
- Nati, M.; Chung, K.J.; Chavakis, T. The Role of Innate Immune Cells in Nonalcoholic Fatty Liver Disease. J. Innate Immun. 2022, 14, 31–41. [Google Scholar] [CrossRef]
- Ala, M. SGLT2 Inhibition for Cardiovascular Diseases, Chronic Kidney Disease, and NAFLD. Endocrinology 2021, 162, bqab157. [Google Scholar] [CrossRef]
- Sumida, Y.; Yoneda, M. Current and future pharmacological therapies for NAFLD/NASH. J. Gastroenterol. 2018, 53, 362–376. [Google Scholar] [CrossRef] [Green Version]
- Chiang, H.; Lee, J.C.; Huang, H.C.; Huang, H.; Liu, H.K.; Huang, C. Delayed intervention with a novel SGLT2 inhibitor NGI001 suppresses diet-induced metabolic dysfunction and non-alcoholic fatty liver disease in mice. Br. J. Pharm. 2020, 177, 239–253. [Google Scholar] [CrossRef] [Green Version]
- Xu, L.; Nagata, N.; Nagashimada, M.; Zhuge, F.; Ni, Y.; Chen, G.; Mayoux, E.; Kaneko, S.; Ota, T. SGLT2 Inhibition by Empagliflozin Promotes Fat Utilization and Browning and Attenuates Inflammation and Insulin Resistance by Polarizing M2 Macrophages in Diet-induced Obese Mice. EBioMedicine 2017, 20, 137–149. [Google Scholar] [CrossRef] [Green Version]
- Trauner, M.; Fuchs, C.D. Novel therapeutic targets for cholestatic and fatty liver disease. Gut 2022, 71, 194–209. [Google Scholar] [CrossRef]
- Libby, A.E.; Bales, E.; Orlicky, D.J.; McManaman, J.L. Perilipin-2 Deletion Impairs Hepatic Lipid Accumulation by Interfering with Sterol Regulatory Element-binding Protein (SREBP) Activation and Altering the Hepatic Lipidome. J. Biol. Chem. 2016, 291, 24231–24246. [Google Scholar] [CrossRef] [Green Version]
- Najt, C.P.; Senthivinayagam, S.; Aljazi, M.B.; Fader, K.A.; Olenic, S.D.; Brock, J.R.; Lydic, T.A.; Jones, A.D.; Atshaves, B.P. Liver-specific loss of Perilipin 2 alleviates diet-induced hepatic steatosis, inflammation, and fibrosis. Am. J. Physiol. Gastrointest. Liver Physiol. 2016, 310, G726–G738. [Google Scholar] [CrossRef] [Green Version]
- Mitro, N.; Mak, P.A.; Vargas, L.; Godio, C.; Hampton, E.; Molteni, V.; Kreusch, A.; Saez, E. The nuclear receptor LXR is a glucose sensor. Nature 2007, 445, 219–223. [Google Scholar] [CrossRef]
- Zhou, J.; Febbraio, M.; Wada, T.; Zhai, Y.; Kuruba, R.; He, J.; Lee, J.H.; Khadem, S.; Ren, S.; Li, S.; et al. Hepatic fatty acid transporter Cd36 is a common target of LXR, PXR, and PPARgamma in promoting steatosis. Gastroenterology 2008, 134, 556–567. [Google Scholar] [CrossRef]
- Huang, P.; Kaluba, B.; Jiang, X.L.; Chang, S.; Tang, X.F.; Mao, L.F.; Zhang, Z.P.; Huang, F.Z. Liver X Receptor Inverse Agonist SR9243 Suppresses Nonalcoholic Steatohepatitis Intrahepatic Inflammation and Fibrosis. Biomed. Res. Int. 2018, 2018, 8071093. [Google Scholar] [CrossRef] [Green Version]
- Kim, W.; Kim, B.G.; Lee, J.S.; Lee, C.K.; Yeon, J.E.; Chang, M.S.; Kim, J.H.; Kim, H.; Yi, S.; Lee, J.; et al. Randomised clinical trial: The efficacy and safety of oltipraz, a liver X receptor alpha-inhibitory dithiolethione in patients with non-alcoholic fatty liver disease. Aliment. Pharm. Ther. 2017, 45, 1073–1083. [Google Scholar] [CrossRef] [Green Version]
- Harriman, G.; Greenwood, J.; Bhat, S.; Huang, X.; Wang, R.; Paul, D.; Tong, L.; Saha, A.K.; Westlin, W.F.; Kapeller, R.; et al. Acetyl-CoA carboxylase inhibition by ND-630 reduces hepatic steatosis, improves insulin sensitivity, and modulates dyslipidemia in rats. Proc. Natl. Acad. Sci. USA 2016, 113, E1796–E1805. [Google Scholar] [CrossRef] [Green Version]
- Alkhouri, N.; Lawitz, E.; Noureddin, M.; DeFronzo, R.; Shulman, G.I. GS-0976 (Firsocostat): An investigational liver-directed acetyl-CoA carboxylase (ACC) inhibitor for the treatment of non-alcoholic steatohepatitis (NASH). Expert Opin. Investig. Drugs 2020, 29, 135–141. [Google Scholar] [CrossRef]
- Lawitz, E.J.; Coste, A.; Poordad, F.; Alkhouri, N.; Loo, N.; McColgan, B.J.; Tarrant, J.M.; Nguyen, T.; Han, L.; Chung, C.; et al. Acetyl-CoA Carboxylase Inhibitor GS-0976 for 12 Weeks Reduces Hepatic De Novo Lipogenesis and Steatosis in Patients With Nonalcoholic Steatohepatitis. Clin. Gastroenterol. Hepatol. 2018, 16, 1983–1991.e3. [Google Scholar] [CrossRef]
- Beysen, C.; Schroeder, P.; Wu, E.; Brevard, J.; Ribadeneira, M.; Lu, W.; Dole, K.; O’Reilly, T.; Morrow, L.; Hompesch, M.; et al. Inhibition of fatty acid synthase with FT-4101 safely reduces hepatic de novo lipogenesis and steatosis in obese subjects with non-alcoholic fatty liver disease: Results from two early-phase randomized trials. Diabetes Obes. Metab. 2021, 23, 700–710. [Google Scholar] [CrossRef]
- Loomba, R.; Mohseni, R.; Lucas, K.J.; Gutierrez, J.A.; Perry, R.G.; Trotter, J.F.; Rahimi, R.S.; Harrison, S.A.; Ajmera, V.; Wayne, J.D.; et al. TVB-2640 (FASN Inhibitor) for the Treatment of Nonalcoholic Steatohepatitis: FASCINATE-1, a Randomized, Placebo-Controlled Phase 2a Trial. Gastroenterology 2021, 161, 1475–1486. [Google Scholar] [CrossRef]
- Gaballah, A.H.; Bingham, K.; Hammoud, G.M.; Kemble, G.; Buckley, D.; McCulloch, W.; Manrique-Acevedo, C. Fatty Acid Synthase Inhibitor TVB-2640 Reduces Hepatic de Novo Lipogenesis in Males With Metabolic Abnormalities. Hepatology 2020, 72, 103–118. [Google Scholar]
- Zhang, M.; Tang, Y.; Tang, E.; Lu, W. MicroRNA-103 represses hepatic de novo lipogenesis and alleviates NAFLD via targeting FASN and SCD1. Biochem. Biophys. Res. Commun. 2020, 524, 716–722. [Google Scholar] [CrossRef]
- Day, E.A.; Ford, R.J.; Steinberg, G.R. AMPK as a Therapeutic Target for Treating Metabolic Diseases. Trends Endocrinol. Metab. 2017, 28, 545–560. [Google Scholar] [CrossRef]
- Boudaba, N.; Marion, A.; Huet, C.; Pierre, R.; Viollet, B.; Foretz, M. AMPK Re-Activation Suppresses Hepatic Steatosis but its Downregulation Does Not Promote Fatty Liver Development. EBioMedicine 2018, 28, 194–209. [Google Scholar] [CrossRef] [Green Version]
- Zhu, X.; Bian, H.; Wang, L.; Sun, X.; Xu, X.; Yan, H.; Xia, M.; Chang, X.; Lu, Y.; Li, Y.; et al. Berberine attenuates nonalcoholic hepatic steatosis through the AMPK-SREBP-1c-SCD1 pathway. Free Radic. Biol. Med. 2019, 141, 192–204. [Google Scholar] [CrossRef]
- Shang, J.; Chen, L.L.; Xiao, F.X.; Sun, H.; Ding, H.C.; Xiao, H. Resveratrol improves non-alcoholic fatty liver disease by activating AMP-activated protein kinase. Acta Pharm. Sin. 2008, 29, 698–706. [Google Scholar] [CrossRef] [Green Version]
- Qiang, X.; Xu, L.; Zhang, M.; Zhang, P.; Wang, Y.; Wang, Y.; Zhao, Z.; Chen, H.; Liu, X.; Zhang, Y. Demethyleneberberine attenuates non-alcoholic fatty liver disease with activation of AMPK and inhibition of oxidative stress. Biochem. Biophys. Res. Commun. 2016, 472, 603–609. [Google Scholar] [CrossRef]
- Sakauchi, C.; Wakatsuki, H.; Ichijo, H.; Hattori, K. Pleiotropic properties of ASK1. Biochim. Biophys. Acta Gen. Subj. 2017, 1861, 3030–3038. [Google Scholar] [CrossRef]
- Challa, T.D.; Wueest, S.; Lucchini, F.C.; Dedual, M.; Modica, S.; Borsigova, M.; Wolfrum, C.; Blüher, M.; Konrad, D. Liver ASK1 protects from non-alcoholic fatty liver disease and fibrosis. EMBO Mol. Med. 2019, 11, e10124. [Google Scholar] [CrossRef] [PubMed]
- Ye, P.; Xiang, M.; Liao, H.; Liu, J.; Luo, H.; Wang, Y.; Huang, L.; Chen, M.; Xia, J. Dual-Specificity Phosphatase 9 Protects Against Nonalcoholic Fatty Liver Disease in Mice Through ASK1 Suppression. Hepatology 2019, 69, 76–93. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Peng, J.; Kong, D.; Wang, X.; Wang, Z.; Liu, J.; Yu, W.; Wu, H.; Long, Z.; Zhang, W.; et al. Silent information regulator 1 suppresses epithelial-to-mesenchymal transition in lung cancer cells via its regulation of mitochondria status. Life Sci. 2021, 280, 119716. [Google Scholar] [CrossRef] [PubMed]
- Silva, J.P.; Wahlestedt, C. Role of Sirtuin 1 in metabolic regulation. Drug Discov. Today 2010, 15, 781–791. [Google Scholar] [CrossRef] [PubMed]
- You, M.; Jogasuria, A.; Taylor, C.; Wu, J. Sirtuin 1 signaling and alcoholic fatty liver disease. Hepatobiliary Surg. Nutr. 2015, 4, 88–100. [Google Scholar]
- Wu, T.; Liu, Y.H.; Fu, Y.C.; Liu, X.M.; Zhou, X.H. Direct evidence of sirtuin downregulation in the liver of non-alcoholic fatty liver disease patients. Ann. Clin. Lab. Sci. 2014, 44, 410–418. [Google Scholar]
- Haohao, Z.; Guijun, Q.; Juan, Z.; Wen, K.; Lulu, C. Resveratrol improves high-fat diet induced insulin resistance by rebalancing subsarcolemmal mitochondrial oxidation and antioxidantion. J. Physiol. Biochem. 2015, 71, 121–131. [Google Scholar] [CrossRef]
- Côté, C.D.; Rasmussen, B.A.; Duca, F.A.; Zadeh-Tahmasebi, M.; Baur, J.A.; Daljeet, M.; Breen, D.M.; Filippi, B.M.; Lam, T.K. Resveratrol activates duodenal Sirt1 to reverse insulin resistance in rats through a neuronal network. Nat. Med. 2015, 21, 498–505. [Google Scholar] [CrossRef]
- Xin, F.Z.; Zhao, Z.H.; Zhang, R.N.; Pan, Q.; Gong, Z.Z.; Sun, C.; Fan, J.G. Folic acid attenuates high-fat diet-induced steatohepatitis via deacetylase SIRT1-dependent restoration of PPARα. World J. Gastroenterol. 2020, 26, 2203–2220. [Google Scholar] [CrossRef]
- Leclerc, D.; Jelinek, J.; Christensen, K.E.; Issa, J.J.; Rozen, R. High folic acid intake increases methylation-dependent expression of Lsr and dysregulates hepatic cholesterol homeostasis. J. Nutr. Biochem. 2021, 88, 108554. [Google Scholar] [CrossRef]
- Salman, M.; Kamel, M.A.; El-Nabi, S.E.H.; Ismail, A.H.A.; Ullah, S.; Al-Ghamdi, A.; Hathout, H.M.R.; El-Garawani, I.M. The regulation of HBP1, SIRT1, and SREBP-1c genes and the related microRNAs in non-alcoholic fatty liver rats: The association with the folic acid anti-steatosis. PLoS ONE 2022, 17, e0265455. [Google Scholar] [CrossRef]
- Yu, L.; Hong, W.; Lu, S.; Li, Y.; Guan, Y.; Weng, X.; Feng, Z. The NLRP3 Inflammasome in Non-Alcoholic Fatty Liver Disease and Steatohepatitis: Therapeutic Targets and Treatment. Front. Pharm. 2022, 13, 780496. [Google Scholar] [CrossRef]
- Mridha, A.R.; Wree, A.; Robertson, A.A.B.; Yeh, M.M.; Johnson, C.D.; Van Rooyen, D.M.; Haczeyni, F.; Teoh, N.C.; Savard, C.; Ioannou, G.N.; et al. NLRP3 inflammasome blockade reduces liver inflammation and fibrosis in experimental NASH in mice. J. Hepatol. 2017, 66, 1037–1046. [Google Scholar] [CrossRef] [PubMed]
- Qu, J.; Yuan, Z.; Wang, G.; Wang, X.; Li, K. The selective NLRP3 inflammasome inhibitor MCC950 alleviates cholestatic liver injury and fibrosis in mice. Int. Immunopharmacol. 2019, 70, 147–155. [Google Scholar] [CrossRef]
- Wang, Q.; Ou, Y.; Hu, G.; Wen, C.; Yue, S.; Chen, C.; Xu, L.; Xie, J.; Dai, H.; Xiao, H.; et al. Naringenin attenuates non-alcoholic fatty liver disease by down-regulating the NLRP3/NF-κB pathway in mice. Br. J. Pharm. 2020, 177, 1806–1821. [Google Scholar] [CrossRef]
- Barry-Hamilton, V.; Spangler, R.; Marshall, D.; McCauley, S.; Rodriguez, H.M.; Oyasu, M.; Mikels, A.; Vaysberg, M.; Ghermazien, H.; Wai, C.; et al. Allosteric inhibition of lysyl oxidase-like-2 impedes the development of a pathologic microenvironment. Nat. Med. 2010, 16, 1009–1017. [Google Scholar] [CrossRef]
- Puente, A.; Fortea, J.I.; Cabezas, J.; Arias Loste, M.T.; Iruzubieta, P.; Llerena, S.; Huelin, P.; Fábrega, E.; Crespo, J. LOXL2-A New Target in Antifibrogenic Therapy? Int. J. Mol. Sci. 2019, 20, 1634. [Google Scholar] [CrossRef] [Green Version]
- Dongiovanni, P.; Meroni, M.; Baselli, G.A.; Bassani, G.A.; Rametta, R.; Pietrelli, A.; Maggioni, M.; Facciotti, F.; Trunzo, V.; Badiali, S.; et al. Insulin resistance promotes Lysyl Oxidase Like 2 induction and fibrosis accumulation in non-alcoholic fatty liver disease. Clin. Sci. 2017, 131, 1301–1315. [Google Scholar] [CrossRef] [Green Version]
- Moon, H.J.; Finney, J.; Ronnebaum, T.; Mure, M. Human lysyl oxidase-like 2. Bioorganic Chem. 2014, 57, 231–241. [Google Scholar] [CrossRef]
- Schilter, H.; Findlay, A.D.; Perryman, L.; Yow, T.T.; Moses, J.; Zahoor, A.; Turner, C.I.; Deodhar, M.; Foot, J.S.; Zhou, W.; et al. The lysyl oxidase like 2/3 enzymatic inhibitor, PXS-5153A, reduces crosslinks and ameliorates fibrosis. J. Cell Mol. Med. 2019, 23, 1759–1770. [Google Scholar] [CrossRef] [Green Version]
- Weston, C.J.; Shepherd, E.L.; Claridge, L.C.; Rantakari, P.; Curbishley, S.M.; Tomlinson, J.W.; Hubscher, S.G.; Reynolds, G.M.; Aalto, K.; Anstee, Q.M.; et al. Vascular adhesion protein-1 promotes liver inflammation and drives hepatic fibrosis. J. Clin. Investig. 2015, 125, 501–520. [Google Scholar] [CrossRef] [Green Version]
- Öksüz, Z.; Üçbilek, E.; Serin, M.S.; Yaraş, S.; Temel, G.O.; Sezgin, O. Circulating vascular adhesion protein-1(VAP-1): A possible biomarker for liver fibrosis associated with chronic hepatitis B and C. Braz. J. Microbiol. Publ. Braz. Soc. Microbiol. 2020, 51, 1757–1763. [Google Scholar] [CrossRef] [PubMed]
- Shepherd, E.L.; Karim, S.; Newsome, P.N.; Lalor, P.F. Inhibition of vascular adhesion protein-1 modifies hepatic steatosis in vitro and in vivo. World J. Hepatol. 2020, 12, 931–948. [Google Scholar] [CrossRef]
- Trivedi, P.J.; Tickle, J.; Vesterhus, M.N.; Eddowes, P.J.; Bruns, T.; Vainio, J.; Parker, R.; Smith, D.; Liaskou, E.; Thorbjørnsen, L.W.; et al. Vascular adhesion protein-1 is elevated in primary sclerosing cholangitis, is predictive of clinical outcome and facilitates recruitment of gut-tropic lymphocytes to liver in a substrate-dependent manner. Gut 2018, 67, 1135–1145. [Google Scholar] [CrossRef]
- Holst, J.J.; Jepsen, S.L.; Modvig, I. GLP-1—Incretin and pleiotropic hormone with pharmacotherapy potential. Increasing secretion of endogenous GLP-1 for diabetes and obesity therapy. Curr. Opin. Pharmacol. 2022, 63, 102189. [Google Scholar] [CrossRef]
- Mathiesen, D.S.; Bagger, J.I.; Bergmann, N.C.; Lund, A.; Christensen, M.B.; Vilsbøll, T.; Knop, F.K. The Effects of Dual GLP-1/GIP Receptor Agonism on Glucagon Secretion-A Review. Int. J. Mol. Sci. 2019, 20, 4092. [Google Scholar] [CrossRef] [Green Version]
- Hartman, M.L.; Sanyal, A.J.; Loomba, R.; Wilson, J.M.; Nikooienejad, A.; Bray, R.; Karanikas, C.A.; Duffin, K.L.; Robins, D.A.; Haupt, A. Effects of Novel Dual GIP and GLP-1 Receptor Agonist Tirzepatide on Biomarkers of Nonalcoholic Steatohepatitis in Patients With Type 2 Diabetes. Diabetes Care 2020, 43, 1352–1355. [Google Scholar] [CrossRef]
- Qin, Y.; Chen, M.; Yang, Y.; Zhou, X.R.; Shao, S.Y.; Wang, D.W.; Yuan, G. Liraglutide improves hepatic insulin resistance via the canonical Wnt signaling pathway. Mol. Med. Rep. 2018, 17, 7372–7380. [Google Scholar] [CrossRef] [Green Version]
- McCrimmon, R.J.; Catarig, A.M.; Frias, J.P.; Lausvig, N.L.; le Roux, C.W.; Thielke, D.; Lingvay, I. Effects of once-weekly semaglutide vs once-daily canagliflozin on body composition in type 2 diabetes: A substudy of the SUSTAIN 8 randomised controlled clinical trial. Diabetologia 2020, 63, 473–485. [Google Scholar] [CrossRef] [Green Version]
- Sirtori, C.R. The pharmacology of statins. Pharm. Res. 2014, 88, 3–11. [Google Scholar] [CrossRef]
- Nascimbeni, F.; Pellegrini, E.; Lugari, S.; Mondelli, A.; Bursi, S.; Onfiani, G.; Carubbi, F.; Lonardo, A. Statins and nonalcoholic fatty liver disease in the era of precision medicine: More friends than foes. Atherosclerosis 2019, 284, 66–74. [Google Scholar] [CrossRef] [Green Version]
- Bril, F.; Portillo Sanchez, P.; Lomonaco, R.; Orsak, B.; Hecht, J.; Tio, F.; Cusi, K. Liver Safety of Statins in Prediabetes or T2DM and Nonalcoholic Steatohepatitis: Post Hoc Analysis of a Randomized Trial. J. Clin. Endocrinol. Metab. 2017, 102, 2950–2961. [Google Scholar] [CrossRef]
- Kannt, A.; Wohlfart, P.; Madsen, A.N.; Veidal, S.S.; Feigh, M.; Schmoll, D. Activation of thyroid hormone receptor-β improved disease activity and metabolism independent of body weight in a mouse model of non-alcoholic steatohepatitis and fibrosis. Br. J. Pharm. 2021, 178, 2412–2423. [Google Scholar] [CrossRef]
- Harrison, S.A.; Bashir, M.R.; Guy, C.D.; Zhou, R.; Moylan, C.A.; Frias, J.P.; Alkhouri, N.; Bansal, M.B.; Baum, S.; Neuschwander-Tetri, B.A.; et al. Resmetirom (MGL-3196) for the treatment of non-alcoholic steatohepatitis: A multicentre, randomised, double-blind, placebo-controlled, phase 2 trial. Lancet 2019, 394, 2012–2024. [Google Scholar] [CrossRef]
- Zhou, J.; Waskowicz, L.R.; Lim, A.; Liao, X.H.; Lian, B.; Masamune, H.; Refetoff, S.; Tran, B.; Koeberl, D.D.; Yen, P.M. A Liver-Specific Thyromimetic, VK2809, Decreases Hepatosteatosis in Glycogen Storage Disease Type Ia. Thyroid Off. J. Am. Thyroid Assoc. 2019, 29, 1158–1167. [Google Scholar] [CrossRef]
- Luong, X.G.; Stevens, S.K.; Jekle, A.; Lin, T.I.; Gupta, K.; Misner, D.; Chanda, S.; Mukherjee, S.; Williams, C.; Stoycheva, A.; et al. Regulation of gene transcription by thyroid hormone receptor β agonists in clinical development for the treatment of non-alcoholic steatohepatitis (NASH). PLoS ONE 2020, 15, e0240338. [Google Scholar] [CrossRef]
- Lee, G.Y.; Han, S.N. The Role of Vitamin E in Immunity. Nutrients 2018, 10, 1614. [Google Scholar] [CrossRef] [Green Version]
- Peh, H.Y.; Tan, W.S.; Liao, W.; Wong, W.S. Vitamin E therapy beyond cancer: Tocopherol versus tocotrienol. Pharmacol. Ther. 2016, 162, 152–169. [Google Scholar] [CrossRef]
- Bril, F.; Biernacki, D.M.; Kalavalapalli, S.; Lomonaco, R.; Subbarayan, S.K.; Lai, J.; Tio, F.; Suman, A.; Orsak, B.K.; Hecht, J.; et al. Role of Vitamin E for Nonalcoholic Steatohepatitis in Patients With Type 2 Diabetes: A Randomized Controlled Trial. Diabetes Care 2019, 42, 1481–1488. [Google Scholar] [CrossRef]
- Kawanaka, M.; Nishino, K.; Nakamura, J.; Suehiro, M.; Goto, D.; Urata, N.; Oka, T.; Kawamoto, H.; Nakamura, H.; Yodoi, J.; et al. Treatment of nonalcoholic steatohepatitis with vitamins E and C: A pilot study. Hepatic Med. Evid. Res. 2013, 5, 11–16. [Google Scholar] [CrossRef] [Green Version]
- Murer, S.B.; Aeberli, I.; Braegger, C.P.; Gittermann, M.; Hersberger, M.; Leonard, S.W.; Taylor, A.W.; Traber, M.G.; Zimmermann, M.B. Antioxidant supplements reduced oxidative stress and stabilized liver function tests but did not reduce inflammation in a randomized controlled trial in obese children and adolescents. J. Nutr. 2014, 144, 193–201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perumpail, B.J.; Li, A.A.; John, N.; Sallam, S.; Shah, N.D.; Kwong, W.; Cholankeril, G.; Kim, D.; Ahmed, A. The Role of Vitamin E in the Treatment of NAFLD. Diseases 2018, 6, 86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schürks, M.; Glynn, R.J.; Rist, P.M.; Tzourio, C.; Kurth, T. Effects of vitamin E on stroke subtypes: Meta-analysis of randomised controlled trials. BMJ Clin. Res. Ed. 2010, 341, c5702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oliver, C.J.; Myers, S.P. Validity of a Cochrane Systematic Review and meta-analysis for determining the safety of vitamin E. BMC Complementary Altern. Med. 2017, 17, 408. [Google Scholar] [CrossRef] [Green Version]
- Massafra, V.; van Mil, S.W.C. Farnesoid X receptor: A “homeostat” for hepatic nutrient metabolism. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 45–59. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Pang, Y.; Wang, X.; Wu, Q.; Liu, H.; Liu, B.; Liu, G.; Ye, M.; Kong, W.; Jiang, C. Ablation of gut microbiota alleviates obesity-induced hepatic steatosis and glucose intolerance by modulating bile acid metabolism in hamsters. Acta Pharm. Sin. B 2019, 9, 702–710. [Google Scholar] [CrossRef] [PubMed]
- Pierre, J.F.; Li, Y.; Gomes, C.K.; Rao, P.; Chang, E.B.; Yin, D.P. Bile Diversion Improves Metabolic Phenotype Dependent on Farnesoid X Receptor (FXR). Obesity 2019, 27, 803–812. [Google Scholar] [CrossRef]
- Hernandez, E.D.; Zheng, L.; Kim, Y.; Fang, B.; Liu, B.; Valdez, R.A.; Dietrich, W.F.; Rucker, P.V.; Chianelli, D.; Schmeits, J.; et al. Tropifexor-Mediated Abrogation of Steatohepatitis and Fibrosis Is Associated With the Antioxidative Gene Expression Profile in Rodents. Hepatol. Commun. 2019, 3, 1085–1097. [Google Scholar] [CrossRef] [Green Version]
- Tølbøl, K.S.; Kristiansen, M.N.; Hansen, H.H.; Veidal, S.S.; Rigbolt, K.T.; Gillum, M.P.; Jelsing, J.; Vrang, N.; Feigh, M. Metabolic and hepatic effects of liraglutide, obeticholic acid and elafibranor in diet-induced obese mouse models of biopsy-confirmed nonalcoholic steatohepatitis. World J. Gastroenterol. 2018, 24, 179–194. [Google Scholar] [CrossRef]
- Mudaliar, S.; Henry, R.R.; Sanyal, A.J.; Morrow, L.; Marschall, H.U.; Kipnes, M.; Adorini, L.; Sciacca, C.I.; Clopton, P.; Castelloe, E.; et al. Efficacy and safety of the farnesoid X receptor agonist obeticholic acid in patients with type 2 diabetes and nonalcoholic fatty liver disease. Gastroenterology 2013, 145, 574–582.e1. [Google Scholar] [CrossRef]
- Patel, K.; Harrison, S.A.; Elkhashab, M.; Trotter, J.F.; Herring, R.; Rojter, S.E.; Kayali, Z.; Wong, V.W.; Greenbloom, S.; Jayakumar, S.; et al. Cilofexor, a Nonsteroidal FXR Agonist, in Patients With Noncirrhotic NASH: A Phase 2 Randomized Controlled Trial. Hepatology 2020, 72, 58–71. [Google Scholar] [CrossRef] [PubMed]
- Fiorucci, S.; Biagioli, M.; Sepe, V.; Zampella, A.; Distrutti, E. Bile acid modulators for the treatment of nonalcoholic steatohepatitis (NASH). Expert Opin. Investig. Drugs 2020, 29, 623–632. [Google Scholar] [CrossRef] [PubMed]
- Broeders, N.; Abramowicz, D. Peroxisome proliferator-activated receptors (PPARs): Novel therapeutic targets in renal disease. Kidney Int. 2002, 61, 354–355. [Google Scholar] [CrossRef] [PubMed]
- Schoonjans, K.; Staels, B.; Auwerx, J. The peroxisome proliferator activated receptors (PPARS) and their effects on lipid metabolism and adipocyte differentiation. Biochim. Biophys. Acta 1996, 1302, 93–109. [Google Scholar] [CrossRef]
- Francque, S.; Verrijken, A.; Caron, S.; Prawitt, J.; Paumelle, R.; Derudas, B.; Lefebvre, P.; Taskinen, M.R.; Van Hul, W.; Mertens, I.; et al. PPARα gene expression correlates with severity and histological treatment response in patients with non-alcoholic steatohepatitis. J. Hepatol. 2015, 63, 164–173. [Google Scholar] [CrossRef] [PubMed]
- Sairyo, M.; Kobayashi, T.; Masuda, D.; Kanno, K.; Zhu, Y.; Okada, T.; Koseki, M.; Ohama, T.; Nishida, M.; Sakata, Y.; et al. A Novel Selective PPARα Modulator (SPPARMα), K-877 (Pemafibrate), Attenuates Postprandial Hypertriglyceridemia in Mice. J. Atheroscler. Thromb. 2018, 25, 142–152. [Google Scholar] [CrossRef] [Green Version]
- Lim, H.J.; Park, J.H.; Lee, S.; Choi, H.E.; Lee, K.S.; Park, H.Y. PPARdelta ligand L-165041 ameliorates Western diet-induced hepatic lipid accumulation and inflammation in LDLR-/- mice. Eur. J. Pharm. 2009, 622, 45–51. [Google Scholar] [CrossRef] [Green Version]
- Gan, Z.; Burkart-Hartman, E.M.; Han, D.H.; Finck, B.; Leone, T.C.; Smith, E.Y.; Ayala, J.E.; Holloszy, J.; Kelly, D.P. The nuclear receptor PPARβ/δ programs muscle glucose metabolism in cooperation with AMPK and MEF2. Genes Dev. 2011, 25, 2619–2630. [Google Scholar] [CrossRef] [Green Version]
- Haczeyni, F.; Wang, H.; Barn, V.; Mridha, A.R.; Yeh, M.M.; Haigh, W.G.; Ioannou, G.N.; Choi, Y.J.; McWherter, C.A.; Teoh, N.C.; et al. The selective peroxisome proliferator-activated receptor-delta agonist seladelpar reverses nonalcoholic steatohepatitis pathology by abrogating lipotoxicity in diabetic obese mice. Hepatol. Commun 2017, 1, 663–674. [Google Scholar] [CrossRef] [Green Version]
- Westerouen Van Meeteren, M.J.; Drenth, J.P.H.; Tjwa, E. Elafibranor: A potential drug for the treatment of nonalcoholic steatohepatitis (NASH). Expert Opin. Investig. Drugs 2020, 29, 117–123. [Google Scholar] [CrossRef]
- Pascual, G.; Fong, A.L.; Ogawa, S.; Gamliel, A.; Li, A.C.; Perissi, V.; Rose, D.W.; Willson, T.M.; Rosenfeld, M.G.; Glass, C.K. A SUMOylation-dependent pathway mediates transrepression of inflammatory response genes by PPAR-gamma. Nature 2005, 437, 759–763. [Google Scholar] [CrossRef] [PubMed]
- Ricote, M.; Huang, J.T.; Welch, J.S.; Glass, C.K. The peroxisome proliferator-activated receptor(PPARgamma) as a regulator of monocyte/macrophage function. J. Leukoc. Biol. 1999, 66, 733–739. [Google Scholar] [CrossRef] [PubMed]
- Konstantinopoulos, P.A.; Vandoros, G.P.; Sotiropoulou-Bonikou, G.; Kominea, A.; Papavassiliou, A.G. NF-kappaB/PPAR gamma and/or AP-1/PPAR gamma ‘on/off’ switches and induction of CBP in colon adenocarcinomas: Correlation with COX-2 expression. Int. J. Colorectal Dis. 2007, 22, 57–68. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.W.; Chu, E.S.; Lam, C.N.; Cheng, A.S.; Lee, C.W.; Wong, V.W.; Sung, J.J.; Yu, J. PPARgamma is essential for protection against nonalcoholic steatohepatitis. Gene Ther. 2010, 17, 790–798. [Google Scholar] [CrossRef] [Green Version]
- Luo, W.; Xu, Q.; Wang, Q.; Wu, H.; Hua, J. Effect of modulation of PPAR-γ activity on Kupffer cells M1/M2 polarization in the development of non-alcoholic fatty liver disease. Sci. Rep. 2017, 7, 44612. [Google Scholar] [CrossRef] [Green Version]
- Goyal, O.; Nohria, S.; Goyal, P.; Kaur, J.; Sharma, S.; Sood, A.; Chhina, R.S. Saroglitazar in patients with non-alcoholic fatty liver disease and diabetic dyslipidemia: A prospective, observational, real world study. Sci. Rep. 2020, 10, 21117. [Google Scholar] [CrossRef]
- Krishnappa, M.; Patil, K.; Parmar, K.; Trivedi, P.; Mody, N.; Shah, C.; Faldu, K.; Maroo, S.; Parmar, D. Effect of saroglitazar 2 mg and 4 mg on glycemic control, lipid profile and cardiovascular disease risk in patients with type 2 diabetes mellitus: A 56-week, randomized, double blind, phase 3 study (PRESS XII study). Cardiovasc. Diabetol. 2020, 19, 93. [Google Scholar] [CrossRef]
- Boyer-Diaz, Z.; Aristu-Zabalza, P.; Andrés-Rozas, M.; Robert, C.; Ortega-Ribera, M.; Fernández-Iglesias, A.; Broqua, P.; Junien, J.L.; Wettstein, G.; Bosch, J.; et al. Pan-PPAR agonist lanifibranor improves portal hypertension and hepatic fibrosis in experimental advanced chronic liver disease. J. Hepatol. 2021, 74, 1188–1199. [Google Scholar] [CrossRef]
- Liu, W.Y.; Xie, D.M.; Zhu, G.Q.; Huang, G.Q.; Lin, Y.Q.; Wang, L.R.; Shi, K.Q.; Hu, B.; Braddock, M.; Chen, Y.P.; et al. Targeting fibroblast growth factor 19 in liver disease: A potential biomarker and therapeutic target. Expert Opin. Ther. Targets 2015, 19, 675–685. [Google Scholar] [CrossRef]
- Roberts, S.K.; Majeed, A. A short report on NGM282/aldafermin for the treatment of nonalcoholic steatohepatitis (NASH). Expert Opin. Ther. Targets 2021, 25, 889–895. [Google Scholar] [CrossRef]
- Harrison, S.A.; Rinella, M.E.; Abdelmalek, M.F.; Trotter, J.F.; Paredes, A.H.; Arnold, H.L.; Kugelmas, M.; Bashir, M.R.; Jaros, M.J.; Ling, L.; et al. NGM282 for treatment of non-alcoholic steatohepatitis: A multicentre, randomised, double-blind, placebo-controlled, phase 2 trial. Lancet 2018, 391, 1174–1185. [Google Scholar] [CrossRef]
- Fisher, F.M.; Maratos-Flier, E. Understanding the Physiology of FGF21. Annu. Rev. Physiol. 2016, 78, 223–241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Babaknejad, N.; Nayeri, H.; Hemmati, R.; Bahrami, S.; Esmaillzadeh, A. An Overview of FGF19 and FGF21: The Therapeutic Role in the Treatment of the Metabolic Disorders and Obesity. Horm. Metab. Res. Horm.-Und Stoffwechs. Horm. Metab. 2018, 50, 441–452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kliewer, S.A.; Mangelsdorf, D.J. A Dozen Years of Discovery: Insights into the Physiology and Pharmacology of FGF21. Cell Metab 2019, 29, 246–253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jimenez, V.; Jambrina, C.; Casana, E.; Sacristan, V.; Muñoz, S.; Darriba, S.; Rodó, J.; Mallol, C.; Garcia, M.; León, X.; et al. FGF21 gene therapy as treatment for obesity and insulin resistance. EMBO Mol. Med. 2018, 10, e8791. [Google Scholar] [CrossRef]
- Verzijl, C.R.C.; Van De Peppel, I.P.; Struik, D.; Jonker, J.W. Pegbelfermin (BMS-986036): An investigational PEGylated fibroblast growth factor 21 analogue for the treatment of nonalcoholic steatohepatitis. Expert Opin. Investig. Drugs 2020, 29, 125–133. [Google Scholar] [CrossRef]
- Sanyal, A.; Charles, E.D.; Neuschwander-Tetri, B.A.; Loomba, R.; Harrison, S.A.; Abdelmalek, M.F.; Lawitz, E.J.; Halegoua-DeMarzio, D.; Kundu, S.; Noviello, S.; et al. Pegbelfermin (BMS-986036), a PEGylated fibroblast growth factor 21 analogue, in patients with non-alcoholic steatohepatitis: A randomised, double-blind, placebo-controlled, phase 2a trial. Lancet 2019, 392, 2705–2717. [Google Scholar] [CrossRef]
- Kaufman, A.; Abuqayyas, L.; Denney, W.S.; Tillman, E.J.; Rolph, T. AKR-001, an Fc-FGF21 Analog, Showed Sustained Pharmacodynamic Effects on Insulin Sensitivity and Lipid Metabolism in Type 2 Diabetes Patients. Cell Rep. Med. 2020, 1, 100057. [Google Scholar] [CrossRef]
- Seki, E.; De Minicis, S.; Gwak, G.Y.; Kluwe, J.; Inokuchi, S.; Bursill, C.A.; Llovet, J.M.; Brenner, D.A.; Schwabe, R.F. CCR1 and CCR5 promote hepatic fibrosis in mice. J. Clin. Investig. 2009, 119, 1858–1870. [Google Scholar] [CrossRef] [Green Version]
- Saiman, Y.; Friedman, S.L. The role of chemokines in acute liver injury. Front. Physiol. 2012, 3, 213. [Google Scholar] [CrossRef] [Green Version]
- Ortiz Zacarías, N.V.; van Veldhoven, J.P.D.; den Hollander, L.S.; Dogan, B.; Openy, J.; Hsiao, Y.Y.; Lenselink, E.B.; Heitman, L.H.; AP, I.J. Synthesis and Pharmacological Evaluation of Triazolopyrimidinone Derivatives as Noncompetitive, Intracellular Antagonists for CC Chemokine Receptors 2 and 5. J. Med. Chem. 2019, 62, 11035–11053. [Google Scholar] [CrossRef] [PubMed]
- Carter, P.H.; Brown, G.D.; Cherney, R.J.; Batt, D.G.; Chen, J.; Clark, C.M.; Cvijic, M.E.; Duncia, J.V.; Ko, S.S.; Mandlekar, S.; et al. Discovery of a Potent and Orally Bioavailable Dual Antagonist of CC Chemokine Receptors 2 and 5. ACS Med. Chem. Lett. 2015, 6, 439–444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barreyro, F.J.; Holod, S.; Finocchietto, P.V.; Camino, A.M.; Aquino, J.B.; Avagnina, A.; Carreras, M.C.; Poderoso, J.J.; Gores, G.J. The pan-caspase inhibitor Emricasan (IDN-6556) decreases liver injury and fibrosis in a murine model of non-alcoholic steatohepatitis. Liver Int. 2015, 35, 953–966. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shiffman, M.; Freilich, B.; Vuppalanchi, R.; Watt, K.; Chan, J.L.; Spada, A.; Hagerty, D.T.; Schiff, E. Randomised clinical trial: Emricasan versus placebo significantly decreases ALT and caspase 3/7 activation in subjects with non-alcoholic fatty liver disease. Aliment. Pharmacol. Ther. 2019, 49, 64–73. [Google Scholar] [CrossRef] [Green Version]
- Bral, M.; Pawlick, R.; Marfil-Garza, B.; Dadheech, N.; Hefler, J.; Thiesen, A.; Shapiro, A.M.J. Pan-caspase inhibitor F573 mitigates liver ischemia reperfusion injury in a murine model. PLoS ONE 2019, 14, e0224567. [Google Scholar] [CrossRef]
- Pejnovic, N.; Jeftic, I.; Jovicic, N.; Arsenijevic, N.; Lukic, M.L. Galectin-3 and IL-33/ST2 axis roles and interplay in diet-induced steatohepatitis. World J. Gastroenterol. 2016, 22, 9706–9717. [Google Scholar] [CrossRef] [PubMed]
- Iacobini, C.; Menini, S.; Ricci, C.; Blasetti Fantauzzi, C.; Scipioni, A.; Salvi, L.; Cordone, S.; Delucchi, F.; Serino, M.; Federici, M.; et al. Galectin-3 ablation protects mice from diet-induced NASH: A major scavenging role for galectin-3 in liver. J. Hepatol. 2011, 54, 975–983. [Google Scholar] [CrossRef]
- Weiskirchen, R. Hepatoprotective and Anti-fibrotic Agents: It’s Time to Take the Next Step. Front. Pharm. 2015, 6, 303. [Google Scholar] [CrossRef] [Green Version]
- Rotman, Y.; Sanyal, A.J. Current and upcoming pharmacotherapy for non-alcoholic fatty liver disease. Gut 2017, 66, 180–190. [Google Scholar] [CrossRef] [Green Version]
- Tarry-Adkins, J.L.; Grant, I.D.; Ozanne, S.E.; Reynolds, R.M.; Aiken, C.E. Efficacy and Side Effect Profile of Different Formulations of Metformin: A Systematic Review and Meta-Analysis. Diabetes Ther. Res. Treat. Educ. Diabetes Relat. Disord. 2021, 12, 1901–1914. [Google Scholar] [CrossRef]
- Mehta, A.; Marso, S.P.; Neeland, I.J. Liraglutide for weight management: A critical review of the evidence. Obes. Sci. Pract. 2017, 3, 3–14. [Google Scholar] [CrossRef]
- Pose, E.; Trebicka, J.; Mookerjee, R.P.; Angeli, P.; Gines, P. Statins: Old drugs as new therapy for liver diseases? J. Hepatol. 2019, 70, 194–202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwabl, P.; Hambruch, E.; Budas, G.R.; Supper, P.; Burnet, M.; Liles, J.T.; Birkel, M.; Brusilovskaya, K.; Konigshofer, P.; Peck-Radosavljevic, M.; et al. The Non-Steroidal FXR Agonist Cilofexor Improves Portal Hypertension and Reduces Hepatic Fibrosis in a Rat NASH Model. Biomedicines 2021, 9, 60. [Google Scholar] [CrossRef] [PubMed]
- Sinakos, E.; Liava, C.; Loomba, R. Emerging advances in the pharmacologic treatment of nonalcoholic steatohepatitis and related cirrhosis. Ann. Gastroenterol. 2022, 35, 213–225. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.M.; Cai, J.J.; Yu, Y.; She, Z.G.; Li, H. Current and Emerging Approaches for Nonalcoholic Steatohepatitis Treatment. Gene Expr. 2019, 19, 175–185. [Google Scholar] [CrossRef]
- Polyzos, S.A.; Katsiki, N. Semaglutide, cilofexor, and firsocostat for nonalcoholic steatohepatitis: A dance that may need more than one dancer. Hormones 2022, 2, 1–2. [Google Scholar] [CrossRef]
- Jiang, L.; Xiao, D.; Li, Y.; Dai, S.; Qu, L.; Chen, X.; Guo, M.; Wei, H.; Chen, Y. Structural basis of tropifexor as a potent and selective agonist of farnesoid X receptor. Biochem. Biophys. Res. Commun. 2021, 534, 1047–1052. [Google Scholar] [CrossRef]
- Schattenberg, J.M.; Pares, A.; Kowdley, K.V.; Heneghan, M.A.; Caldwell, S.; Pratt, D.; Bonder, A.; Hirschfield, G.M.; Levy, C.; Vierling, J.; et al. A randomized placebo-controlled trial of elafibranor in patients with primary biliary cholangitis and incomplete response to UDCA. J. Hepatol. 2021, 74, 1344–1354. [Google Scholar] [CrossRef]
- Sven, M.F.; Pierre, B.; Manal, F.A.; Quentin, M.A.; Elisabetta, B.; Vlad, R.; Philippe, H.M.; Bruno, S.; Jean-Louis, J.; Pierre, B.; et al. A randomised, double-blind, placebo-controlled, multi-centre, dose-range, proof-of-concept, 24-week treatment study of lanifibranor in adult subjects with non-alcoholic steatohepatitis: Design of the NATIVE study. Contemp. Clin. Trials 2020, 98, 106170. [Google Scholar] [CrossRef]
- Mayo, M.J.; Wigg, A.J.; Leggett, B.A.; Arnold, H.; Thompson, A.J.; Weltman, M.; Carey, E.J.; Muir, A.J.; Ling, L.; Rossi, S.J.; et al. NGM282 for Treatment of Patients With Primary Biliary Cholangitis: A Multicenter, Randomized, Double-Blind, Placebo-Controlled Trial. Hepatol. Commun. 2018, 2, 1037–1050. [Google Scholar] [CrossRef]
- Shao, W.; Jin, T. Hepatic hormone FGF21 and its analogues in clinical trials. Chronic Dis. Transl. Med. 2022, 8, 19–25. [Google Scholar] [CrossRef] [PubMed]
- Attia, S.L.; Softic, S.; Mouzaki, M. Evolving Role for Pharmacotherapy in NAFLD/NASH. Clin. Transl. Sci. 2021, 14, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Huang, N.; Guo, Y.; Cui, S.; Ge, C.; He, Q.; Pan, X.; Wang, G.; Wang, H.; Hao, H. Combined obeticholic acid and apoptosis inhibitor treatment alleviates liver fibrosis. Acta Pharm. Sinica. B 2019, 9, 526–536. [Google Scholar] [CrossRef]
- Harrison, S.A.; Marri, S.R.; Chalasani, N.; Kohli, R.; Aronstein, W.; Thompson, G.A.; Irish, W.; Miles, M.V.; Xanthakos, S.A.; Lawitz, E.; et al. Randomised clinical study: GR-MD-02, a galectin-3 inhibitor, vs. placebo in patients having non-alcoholic steatohepatitis with advanced fibrosis. Aliment. Pharmacol. Ther. 2016, 44, 1183–1198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cehic, M.G.; Muir, C.A.; Greenfield, J.R.; Hayward, C.; Jabbour, A.; Keogh, A.; Kotlyar, E.; Muthiah, K.; Macdonald, P.S. Efficacy and Safety of Empagliflozin in the Management of Diabetes Mellitus in Heart Transplant Recipients. Transplant. Direct 2019, 5, e450. [Google Scholar] [CrossRef] [PubMed]
- Hsiang, J.C.; Wong, V.W. SGLT2 Inhibitors in Liver Patients. Clin. Gastroenterol. Hepatol. 2020, 18, 2168–2172.e2. [Google Scholar] [CrossRef]
- Tacelli, M.; Celsa, C.; Magro, B.; Giannetti, A.; Pennisi, G.; Spatola, F.; Petta, S. Antidiabetic Drugs in NAFLD: The Accomplishment of Two Goals at Once? Pharmaceuticals 2018, 11, 121. [Google Scholar] [CrossRef] [Green Version]
- Lian, J.; Fu, J. Pioglitazone for NAFLD Patients With Prediabetes or Type 2 Diabetes Mellitus: A Meta-Analysis. Front. Endocrinol. 2021, 12, 615409. [Google Scholar] [CrossRef]
- Tilinca, M.C.; Tiuca, R.A.; Niculas, C.; Varga, A.; Tilea, I. Future perspectives in diabesity treatment: Semaglutide, a glucagon-like peptide 1 receptor agonist (Review). Exp. Ther. Med. 2021, 22, 1167. [Google Scholar] [CrossRef]
- Dibba, P.; Li, A.A.; Perumpail, B.J.; John, N.; Sallam, S.; Shah, N.D.; Kwong, W.; Cholankeril, G.; Kim, D.; Ahmed, A. Emerging Therapeutic Targets and Experimental Drugs for the Treatment of NAFLD. Diseases 2018, 6, 83. [Google Scholar] [CrossRef] [Green Version]
- Yu, Y.; Cai, J.; She, Z.; Li, H. Insights into the Epidemiology, Pathogenesis, and Therapeutics of Nonalcoholic Fatty Liver Diseases. Adv. Sci. 2019, 6, 1801585. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Dominguez, E.; Gisbert, J.P.; Moreno-Monteagudo, J.A.; Garcia-Buey, L.; Moreno-Otero, R. A pilot study of atorvastatin treatment in dyslipemid, non-alcoholic fatty liver patients. Aliment. Pharmacol. Ther. 2006, 23, 1643–1647. [Google Scholar] [CrossRef] [PubMed]
- Jun, B.G.; Cheon, G.J. The utility of ezetimibe therapy in nonalcoholic fatty liver disease. Korean J. Intern. Med. 2019, 34, 284–285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, B.; Lu, L.G. Efficacy and safety of drugs for nonalcoholic steatohepatitis. J. Dig. Dis. 2021, 22, 72–82. [Google Scholar] [CrossRef] [PubMed]
- Brodosi, L.; Marchignoli, F.; Petroni, M.L.; Marchesini, G. NASH: A glance at the landscape of pharmacological treatment. Ann. Hepatol. 2016, 15, 673–681. [Google Scholar]
- Boner, G.; Cooper, M.E.; McCarroll, K.; Brenner, B.M.; de Zeeuw, D.; Kowey, P.R.; Shahinfar, S.; Dickson, T.; Crow, R.S.; Parving, H.H.; et al. Adverse effects of left ventricular hypertrophy in the reduction of endpoints in NIDDM with the angiotensin II antagonist losartan (RENAAL) study. Diabetologia 2005, 48, 1980–1987. [Google Scholar] [CrossRef] [Green Version]
- Mancia, G.; Schumacher, H. Incidence of adverse events with telmisartan compared with ACE inhibitors: Evidence from a pooled analysis of clinical trials. Patient Prefer. Adherence 2012, 6, 1–9. [Google Scholar] [CrossRef]
- Lake, J.E.; Tseng, C.H.; Currier, J.S. A pilot study of telmisartan for visceral adiposity in HIV infection: The metabolic abnormalities, telmisartan, and HIV infection (MATH) trial. PLoS ONE 2013, 8, e58135. [Google Scholar] [CrossRef] [Green Version]
- Peng, C.; Stewart, A.G.; Woodman, O.L.; Ritchie, R.H.; Qin, C.X. Non-Alcoholic Steatohepatitis: A Review of Its Mechanism, Models and Medical Treatments. Front. Pharmacol. 2020, 11, 603926. [Google Scholar] [CrossRef]
- Ganguli, S.; DeLeeuw, P.; Satapathy, S.K. A Review Of Current And Upcoming Treatment Modalities In Non-Alcoholic Fatty Liver Disease And Non-Alcoholic Steatohepatitis. Hepatic Med. Evid. Res. 2019, 11, 159–178. [Google Scholar] [CrossRef] [Green Version]
- Roeb, E. Diagnostic and Therapy of Nonalcoholic Fatty Liver Disease: A Narrative Review. Visc. Med. 2022, 38, 126–132. [Google Scholar] [CrossRef] [PubMed]
- Pydyn, N.; Miekus, K.; Jura, J.; Kotlinowski, J. New therapeutic strategies in nonalcoholic fatty liver disease: A focus on promising drugs for nonalcoholic steatohepatitis. Pharmacol. Rep. 2020, 72, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Teng, T.; Qiu, S.; Zhao, Y.; Zhao, S.; Sun, D.; Hou, L.; Li, Y.; Zhou, K.; Yu, X.; Yang, C.; et al. Pathogenesis and Therapeutic Strategies Related to Non-Alcoholic Fatty Liver Disease. Int. J. Mol. Sci. 2022, 23, 7841. https://doi.org/10.3390/ijms23147841
Teng T, Qiu S, Zhao Y, Zhao S, Sun D, Hou L, Li Y, Zhou K, Yu X, Yang C, et al. Pathogenesis and Therapeutic Strategies Related to Non-Alcoholic Fatty Liver Disease. International Journal of Molecular Sciences. 2022; 23(14):7841. https://doi.org/10.3390/ijms23147841
Chicago/Turabian StyleTeng, Tieshan, Shuai Qiu, Yiming Zhao, Siyuan Zhao, Dequan Sun, Lingzhu Hou, Yihang Li, Ke Zhou, Xixi Yu, Changyong Yang, and et al. 2022. "Pathogenesis and Therapeutic Strategies Related to Non-Alcoholic Fatty Liver Disease" International Journal of Molecular Sciences 23, no. 14: 7841. https://doi.org/10.3390/ijms23147841
APA StyleTeng, T., Qiu, S., Zhao, Y., Zhao, S., Sun, D., Hou, L., Li, Y., Zhou, K., Yu, X., Yang, C., & Li, Y. (2022). Pathogenesis and Therapeutic Strategies Related to Non-Alcoholic Fatty Liver Disease. International Journal of Molecular Sciences, 23(14), 7841. https://doi.org/10.3390/ijms23147841