
Integrated Analysis of Single-Molecule Real-Time Sequencing and Next-Generation Sequencing Eveals Insights into Drought Tolerance Mechanism of Lolium multiflorum



Abstract
:1. Introduction
2. Results
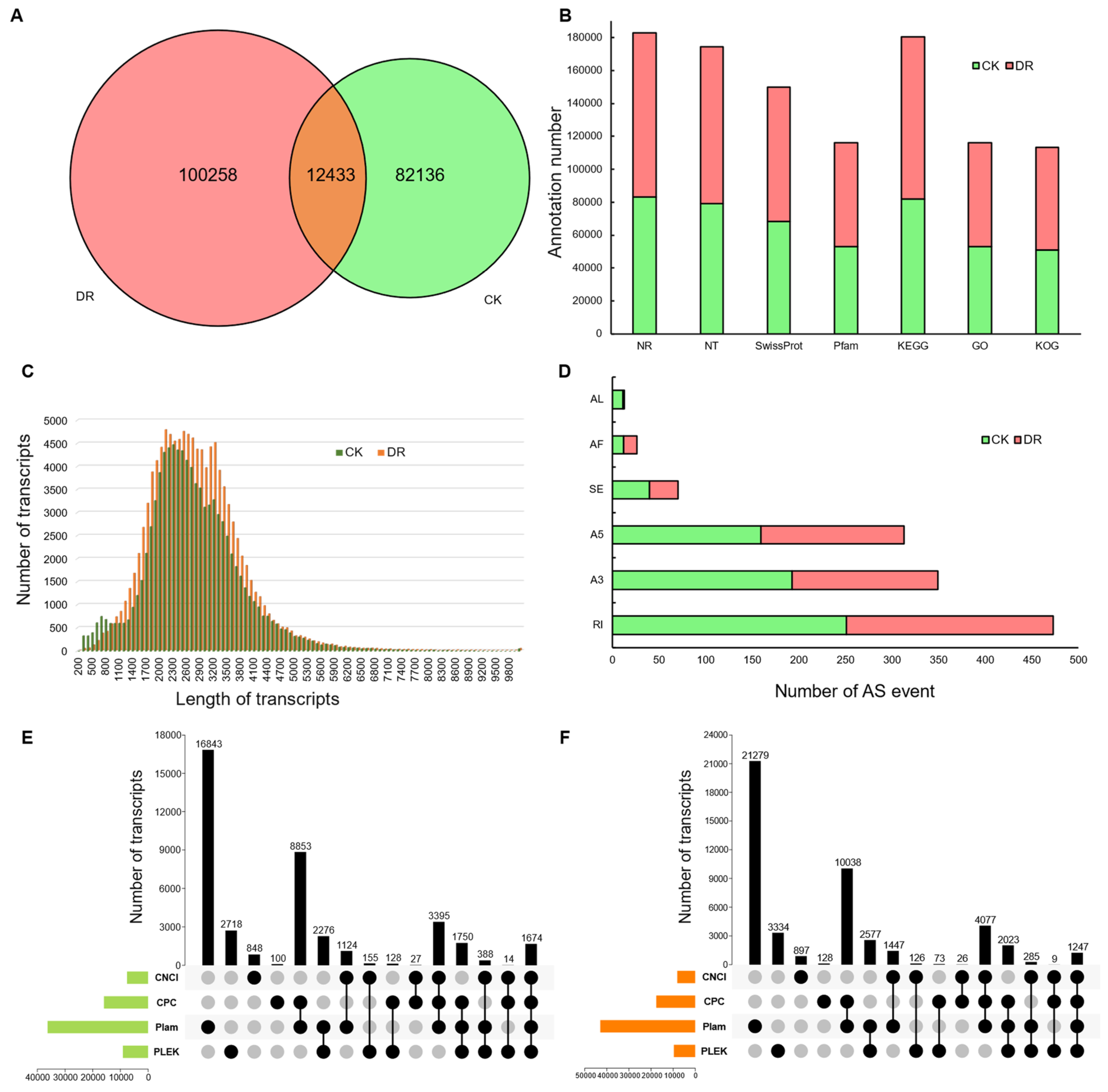
2.1. Assembly of the Sequence Datasets and Functional Annotation
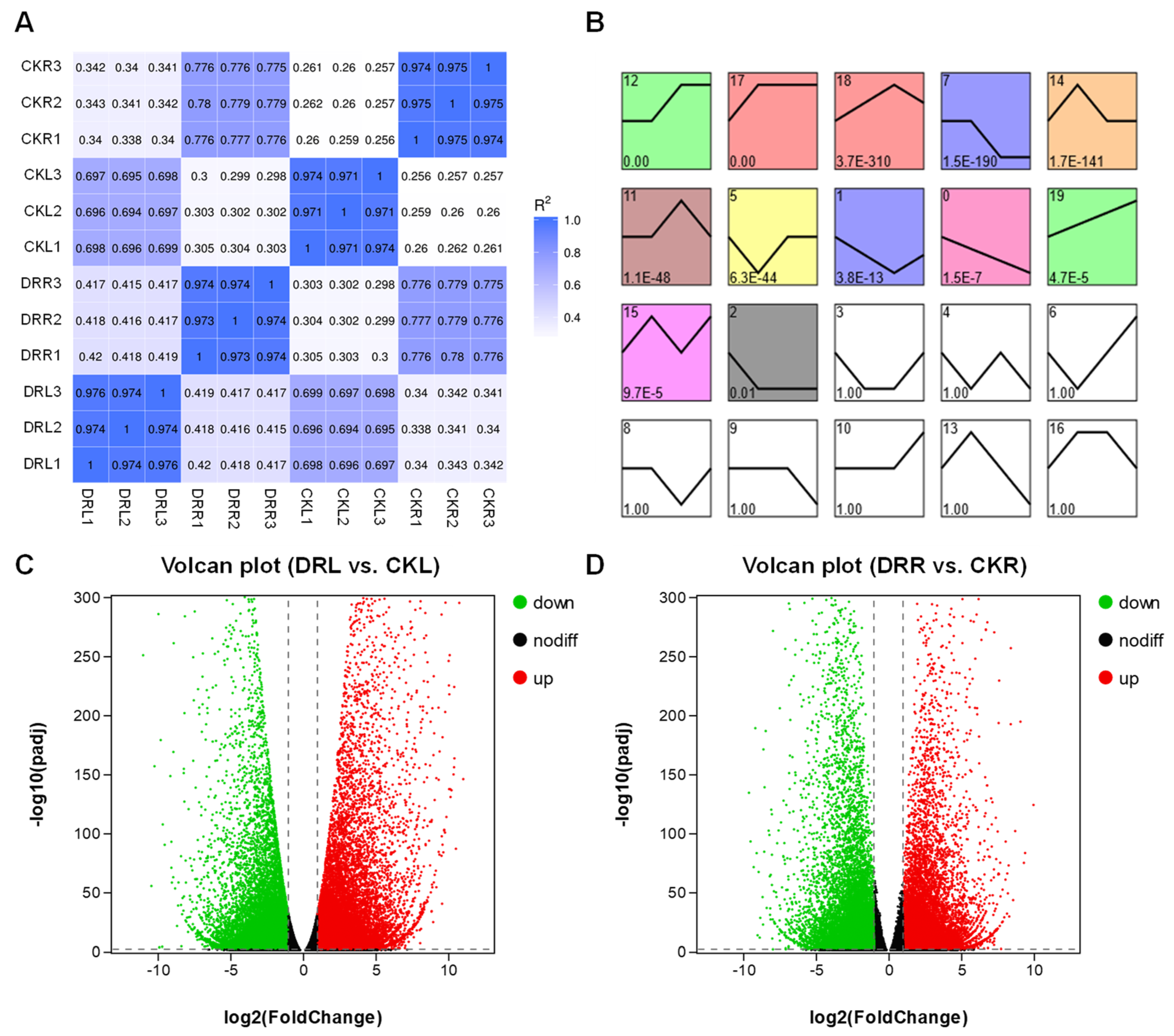
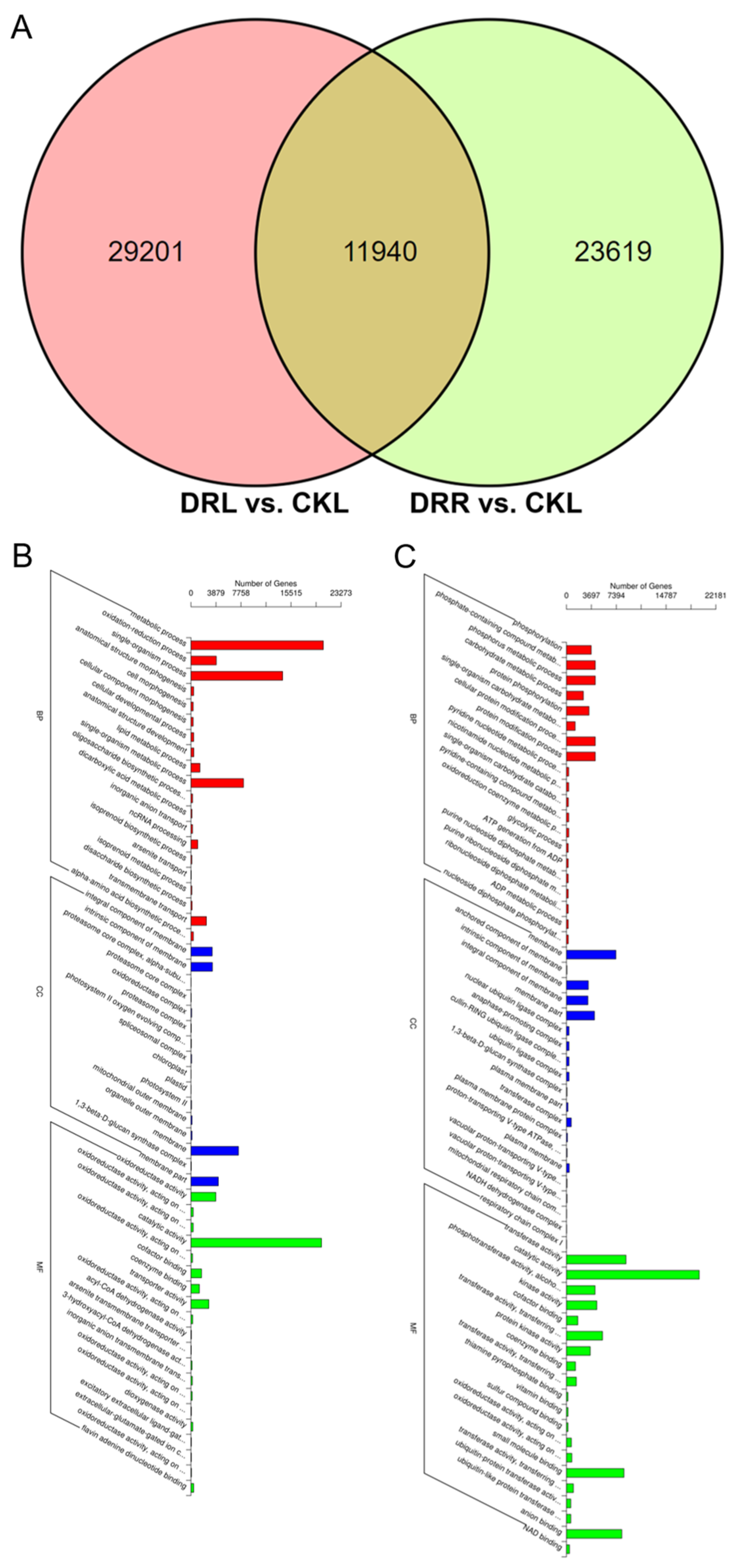
2.2. Differentially Expressed Genes in L. multiflorum Leaves and Roots under Drought
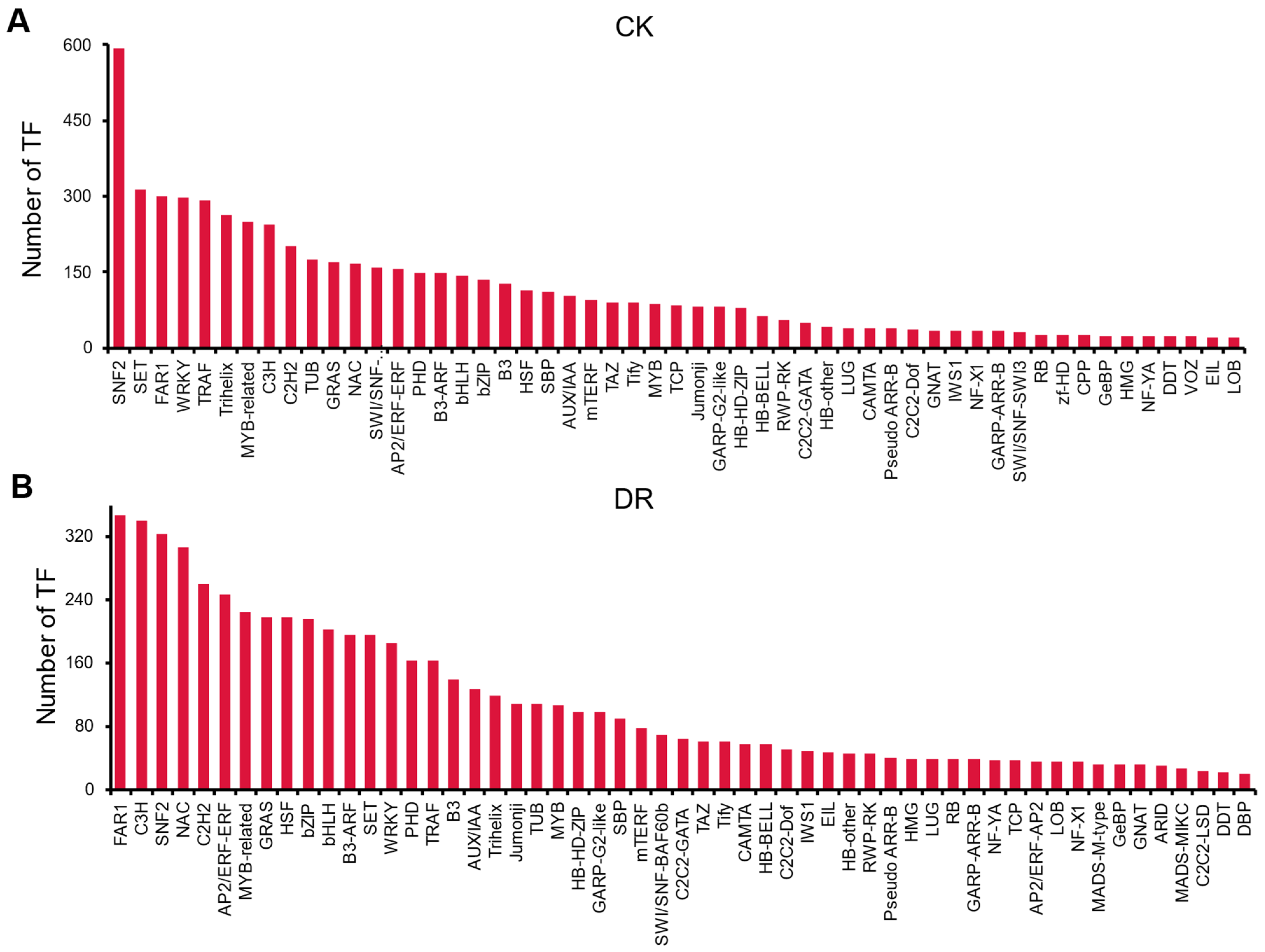
2.3. Transcription Factor Statistics and Identification of R2R3-MYB Family Members
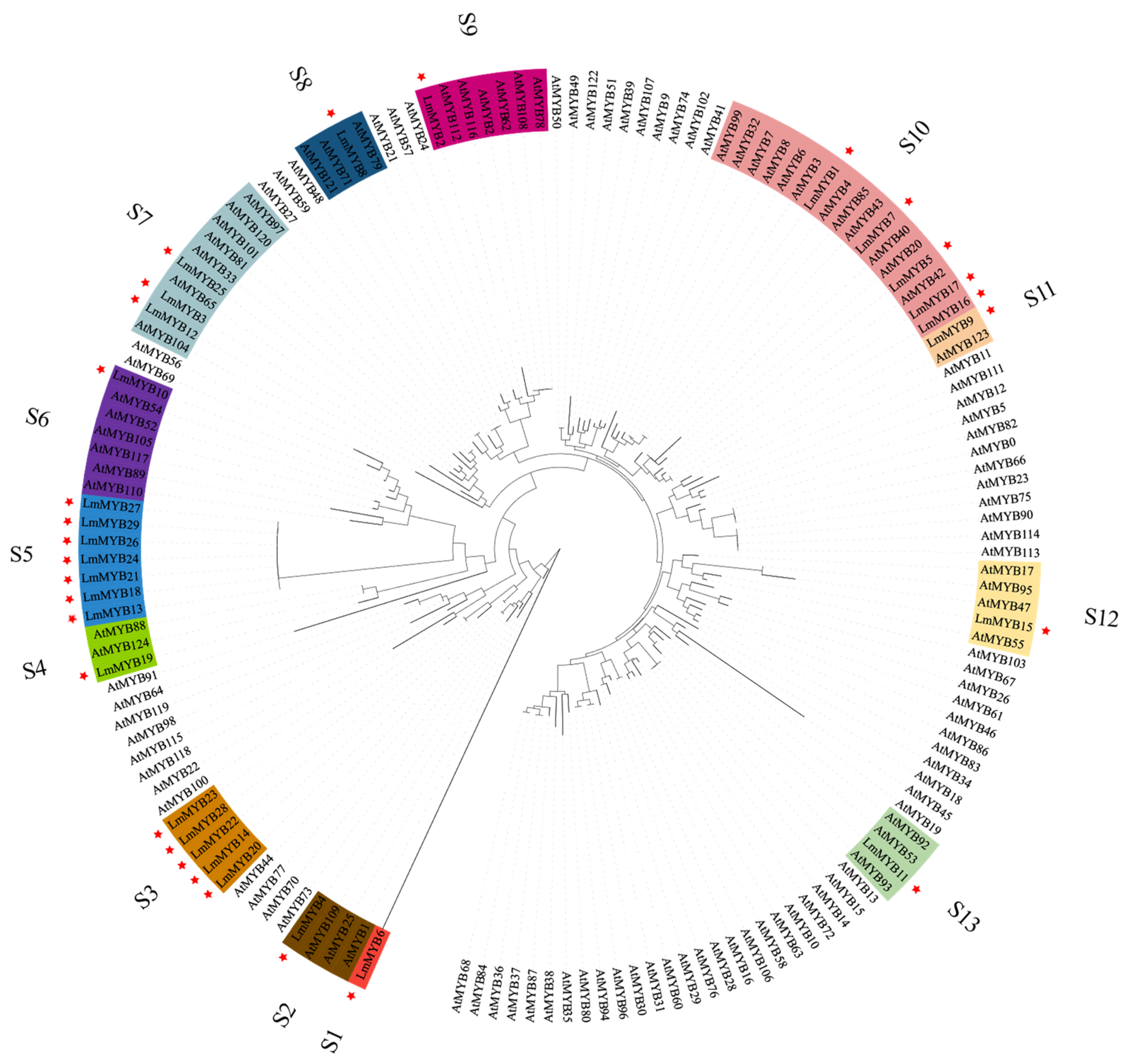
2.4. Genetic Analysis of Members of the R2R3-MYB Gene Family
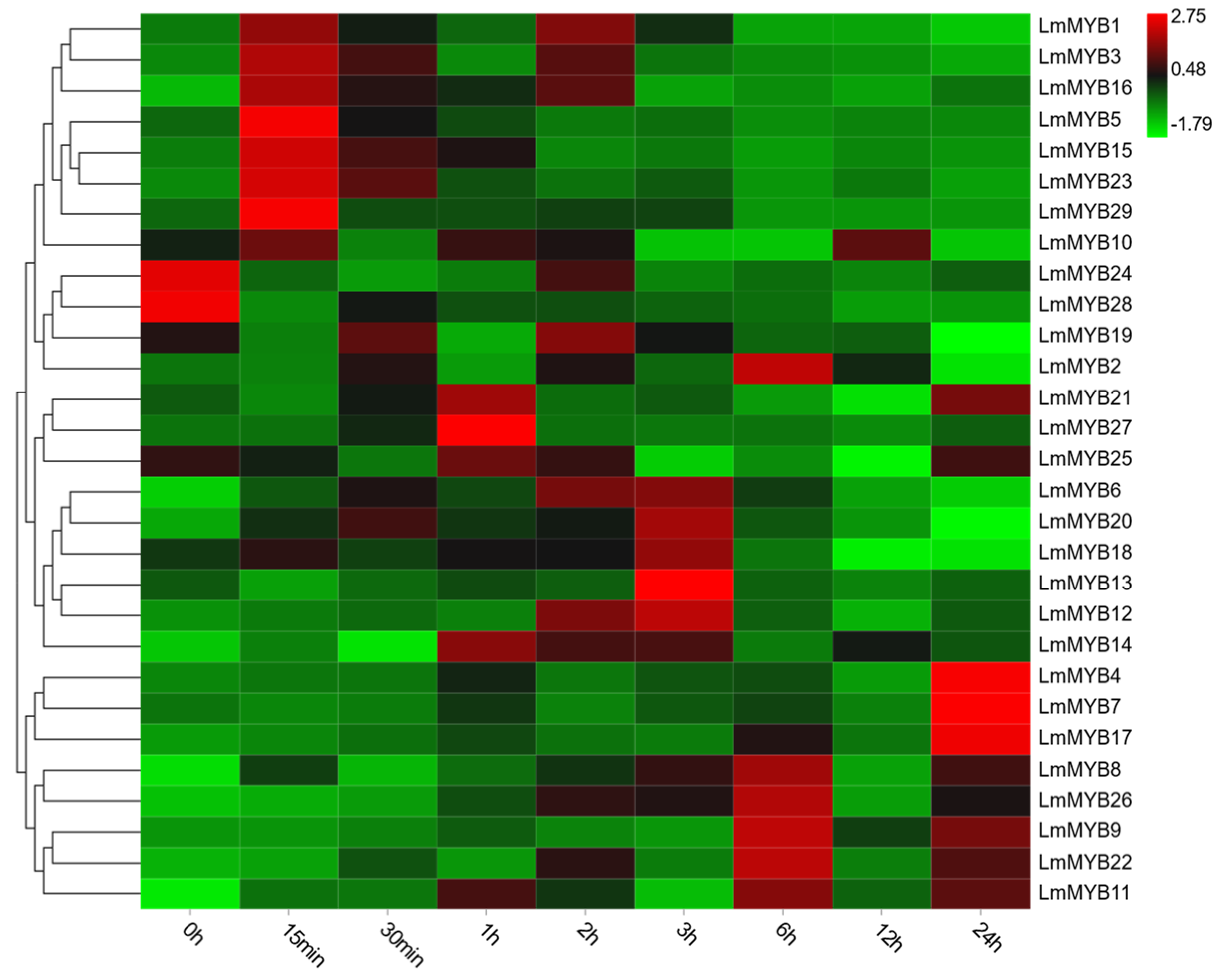
2.5. The Expression Patterns of R2R3-MYB Family Members under Drought
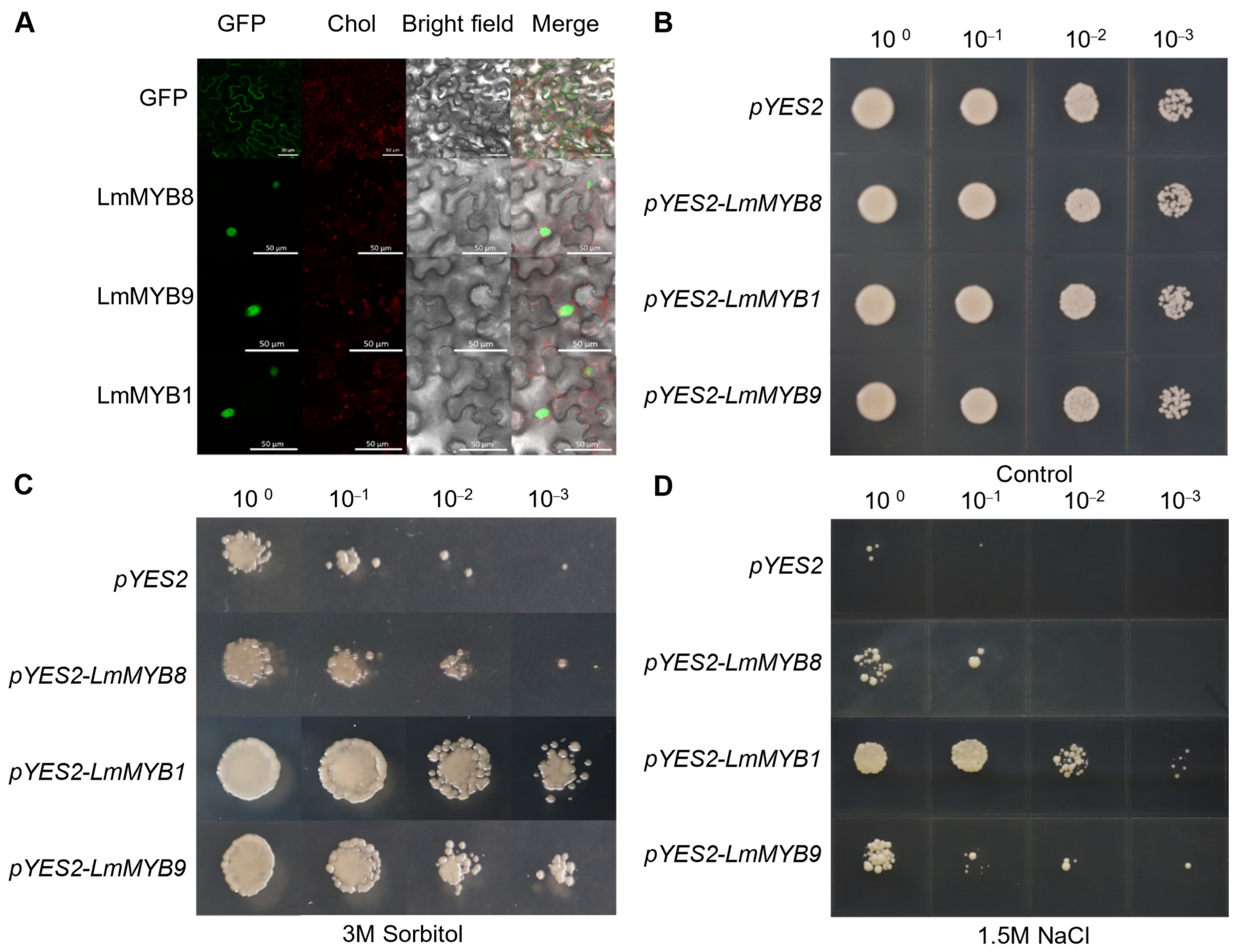
2.6. LmMYB Transcripts Localized to the Nucleus Enhanced Abiotic Stress Tolerance in Yeast
3. Discussion
3.1. A More Extensive and Complete Transcriptome Dataset
3.2. Alternative Splicing Plays an Important Role in Complex Transcriptional Regulation
3.3. Key Distinctive Candidate Genes Involved in the L. multiflorum Drought Stress Response
4. Materials and Methods
4.1. Material Cultivation and Sample Collection
4.2. Illumina cDNA Library Construction and Next-Generation Sequencing
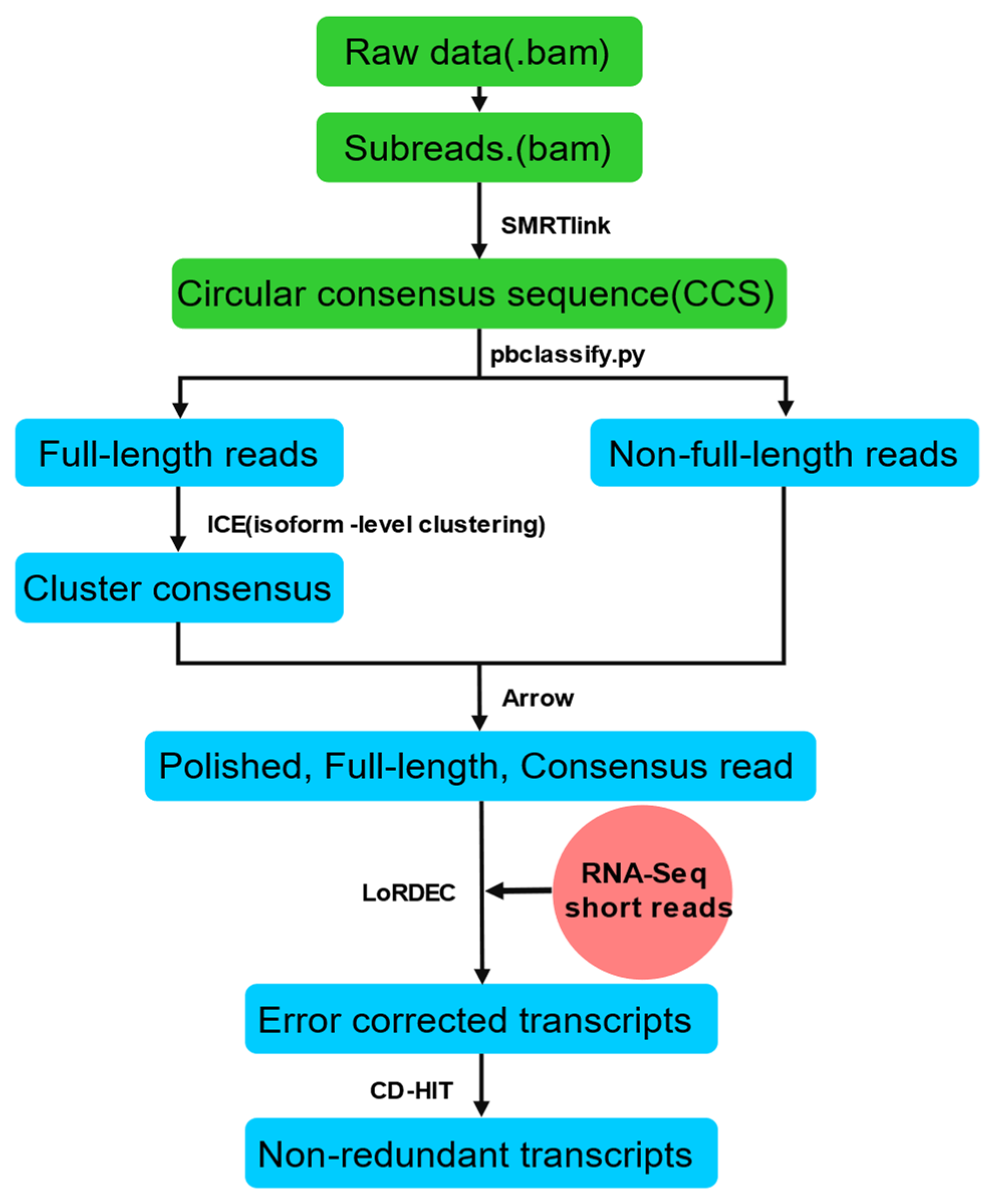
4.3. PacBio cDNA Library Construction and Single-Molecule Real-Time (SMRT) Sequencing
4.4. Functional Annotation of PacBio Isoforms
4.5. Identification of Alternative Splicing Events and lncRNA Prediction
4.6. Quantification of Gene Expression Levels and Differential Expression Analysis
4.7. Transcription Factor Analysis and Identification of R2R3-MYB Gene Family Members
4.8. Phylogenetic Analysis
4.9. Expression Pattern Analysis of the R2R3-MYB Gene Family
4.10. Subcellular Localization and Heterologous Expression
4.11. Heterologous Expression of LmMYB1, LmMYB8 and LmMYB9 in Yeast
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Vicente-Serrano, S.M. Foreword: Drought complexity and assessment under climate change conditions. Cuad. Investig. Geográfica 2016, 42, 7–11. [Google Scholar] [CrossRef]
- Folland, C.K.; Hannaford, J.; Bloomfield, J.P.; Kendon, M.; Wallace, E. Hydrology and Earth System Sciences. Hydrol. Earth Syst. Sci. 2015, 19, 2353–2375. [Google Scholar] [CrossRef] [Green Version]
- Lanen, H.V.; Wanders, N.; Tallaksen, L.M.; Loon, A.V. Hydrological drought across the world: Impact of climate and physical catchment structure. Hydrol. Earth Syst. Sci. 2013, 17, 1715–1732. [Google Scholar] [CrossRef] [Green Version]
- Kim, W.; Iizumi, T.; Nishimori, M. Global Patterns of Crop Production Losses Associated with Droughts from 1983 to 2009. J. Appl. Meteorol. Clim. 2019, 58, 1233–1244. [Google Scholar] [CrossRef]
- Pea-Gallardo, M.; Vicente-Serrano, S.M.; Hannaford, J.; Lorenzo-Lacruz, J.; Kenawy, A.E. Complex influences of meteorological drought time-scales on hydrological droughts in natural basins of the contiguous Unites States. J. Hydrol. 2018, 568, 611–625. [Google Scholar] [CrossRef] [Green Version]
- Webber, H.; Ewert, F.; Olesen, J.E.; Müller, C.; Fronzek, S.; Ruane, A.C.; Bourgault, M.; Martre, P.; Ababaei, B.; Bindi, M. Diverging importance of drought stress for maize and winter wheat in Europe. Nat. Commun. 2018, 9, 4249. [Google Scholar] [CrossRef] [Green Version]
- Vicente-Serrano, S.M.; Quiring, S.M.; Pea-Gallardo, M.; Yuan, S.; Domínguez-Castro, F. A review of environmental droughts: Increased risk under global warming? Earth Sci. Rev. 2019, 201, 102953. [Google Scholar] [CrossRef]
- Törnros, T.; Menzel, L. Addressing drought conditions under current and future climates in the Jordan River region. Hydrol. Earth Syst. Sci. 2013, 18, 305–318. [Google Scholar] [CrossRef] [Green Version]
- Giddens, A. The Politics of Climate Change, 2nd ed.; Polity: Cambridge, UK, 2011. [Google Scholar]
- Doede, A.L.; Deguzman, P.B. The Disappearing Lake: An Historical Analysis of Drought and the Salton Sea in the Context of the GeoHealth Framework. GeoHealth 2020, 4, e2020GH000271. [Google Scholar] [CrossRef]
- Knorst, V.; Yates, S.; Byrne, S.; Asp, T.; Widmer, F.; Studer, B.; Kölliker, R. Lliker First assembly of the gene-space of Lolium multiflorum and comparison to other Poaceae genomes. Grassl. Sci. 2019, 65, 125–134. [Google Scholar] [CrossRef] [Green Version]
- Xin-Yue, Z.; Yuan-Hua, L.I.; Wen-Long, G.; Rui-Zhen, Z. Research development of Italian ryegrass. Pratacultural Sci. 2009, 26, 55–60. [Google Scholar]
- Fang, Z.; Hu, Z.; Zhao, H.; Yang, L.; Ding, C.; Lou, L.; Cai, Q. Screening for cadmium tolerance of 21 cultivars from Italian ryegrass (Lolium multiflorum Lam) during germination. Grassl. Sci. 2017, 63, 36–45. [Google Scholar] [CrossRef]
- Mugica-Alvarez, V.; Cortés-Jiménez, V.; Vaca-Mier, M.; Domínguez-Soria, V. Phytoremediation of Mine Tailings Using Lolium Multiflorum. Int. J. Environ. Sci. Dev. 2015, 6, 246–251. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; He, X.; Chen, W.; Zhao, M. Ecotoxicological responses of three ornamental herb species to cadmium. Environ. Toxicol. Chem. 2013, 32, 1746–1751. [Google Scholar] [CrossRef] [PubMed]
- Yasuda, M.; Takenouchi, Y.; Nitta, Y.; Ishii, Y.; Ohta, K. Italian ryegrass (Lolium multiflorum Lam) as a High-Potential Bio-Ethanol Resource. Bioenerg. Res. 2015, 8, 1303–1309. [Google Scholar] [CrossRef]
- Choi, K.C.; Son, Y.O.; Hwang, J.M.; Kim, B.T.; Lee, J.C. Antioxidant, anti-inflammatory and anti-septic potential of phenolic acids and flavonoid fractions isolated from Lolium multiflorum. Pharm. Biol. 2017, 55, 611–619. [Google Scholar] [CrossRef] [Green Version]
- Cornish, M.A.; Hayward, M.D.; Lawrence, M.J. Self-incompatibility in ryegrass. Heredity 1979, 43, 95–106. [Google Scholar] [CrossRef]
- Morganti, S.; Tarantino, P.; Ferraro, E.; D’Amico, P.; Viale, G.; Trapani, D.; Duso, B.A.; Curigliano, G. Complexity of genome sequencing and reporting: Next generation sequencing (NGS) technologies and implementation of precision medicine in real life. Crit. Rev. Oncol. Hemetology 2019, 133, 171–182. [Google Scholar] [CrossRef]
- Au, K.F.; Sebastiano, V.; Afshar, P.T.; Durruthy, J.D.; Lee, L.; Williams, B.A.; Van Bakel, H.; Schadt, E.E.; Reijo-Pera, R.A.; Underwood, J.G. Characterization of the human ESC transcriptome by hybrid sequencing. Proc. Natl. Acad. Sci. USA 2013, 110, e4821–e4830. [Google Scholar] [CrossRef] [Green Version]
- Chaisson, M.J.P.; Huddleston, J.; Megan, Y.D.; Sudmant, P.H.; Malig, M. Resolving the complexity of the human genome using single-molecule sequencing. Nature 2015, 517, 608–611. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Bracht, J.; Goldman, A.; Dolzhenko, E.; Clay, D.; Swart, E.; Perlman, D.; Doak, T.; Stuart, A.; Amemiya, C. The Architecture of a Scrambled Genome Reveals Massive Levels of Genomic Rearrangement during Development. Cell 2014, 158, 1187–1198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ju, C.; Zhao, Z.; Wei, W. Efficient approach to correct read alignment for pseudogene abundance estimates. IEEE/ACM Trans. Comput. Biol. Bioinform. 2016, 14, 522–533. [Google Scholar] [CrossRef] [PubMed]
- Sharon, D.; Tilgner, H.; Grubert, F.; Snyder, M. A single-molecule long-read survey of the human transcriptome. Nat. Biotechnol. 2013, 31, 1009–1014. [Google Scholar] [CrossRef] [PubMed]
- Qla, B.; Yza, B.; Ywa, B.; Cga, B.; Sha, B.; Opd, C.; Hya, B. Combining single-molecule sequencing and next-generation sequencing to provide insight into the complex response of Iris halophila Pall. to Pb exposure. Ind. Crop. Prod. 2021, 168, 113623. [Google Scholar]
- Xu, D.; Lin, H.; Tang, Y.; Huang, L.; Xu, J.; Nian, S.; Zhao, Y. Integration of full-length transcriptomics and targeted metabolomics to identify benzylisoquinoline alkaloid biosynthetic genes in. Hortic. Res. 2021, 8, 16. [Google Scholar] [CrossRef]
- Au, K.F.; Underwood, J.G.; Lee, L.; Wong, W.H. Improving PacBio Long Read Accuracy by Short Read Alignment. PLoS ONE 2012, 7, e46679. [Google Scholar] [CrossRef] [Green Version]
- Cechin, J.; Piasecki, C.; Benemann, D.P.; Kremer, F.S.; Galli, V.; Maia, L.C.; Agostinetto, D.; Vargas, A.L. Lolium multiflorumTranscriptome Analysis Identifies Candidate Target Genes Involved in Glyphosate-Resistance Mechanism in. Plants 2020, 9, 685. [Google Scholar] [CrossRef]
- Pan, L.; Zhang, X.; Wang, J.; Ma, X.; Zhou, M.; Huang, L.; Nie, G.; Wang, P.; Yang, Z.; Li, J. Transcriptional Profiles of Drought-Related Genes in Modulating Metabolic Processes and Antioxidant Defenses in Lolium multiflorum. Front. Plant Sci. 2016, 7, 519. [Google Scholar] [CrossRef]
- Xue, H.W.; Chen, X.; Mei, Y. Function and regulation of phospholipid signalling in plants. Biochem. J. 2009, 421, 145–156. [Google Scholar] [CrossRef]
- Divecha, N.; Truong, O.; Hsuan, J.J.; Hinchliffe, K.A.; Irvine, R.F. The cloning and sequence of the C isoform of PtdIns4P 5-kinase. Biochem. J. 1995, 309, 715–719. [Google Scholar] [CrossRef] [Green Version]
- Mikami, K.; Katagiri, T.; Iuchi, S.; Yamaguchi-Shinozaki, K.; Shinozaki, K. A gene encoding phosphatidylinositol-4-phosphate 5-kinase is induced by water stress and abscisic acid in Arabidopsis thaliana. Plant J. Cell Mol. Biol. 1998, 15, 563–568. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Kim, Y.W.; Jeon, B.W.; Park, K.Y.; Suh, S.J.; Seo, J.; Kwak, J.M.; Martinoia, E.; Hwang, I.; Lee, Y. Phosphatidylinositol 4,5-bisphosphate is important for stomatal opening. Plant J. Cell Mol. Biol. 2007, 52, 803–816. [Google Scholar] [CrossRef] [PubMed]
- Kuroda, R.; Kato, M.; Tsuge, T.; Aoyama, T. Arabidopsis phosphatidylinositol 4-phosphate 5-kinase genes PIP5K7, PIP5K8, and PIP5K9 are redundantly involved in root growth adaptation to osmotic stress. Plant J. Cell Mol. Biol. 2021, 106, 913–927. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Zhai, S.; Zhao, Y.; Sun, B.; Liu, C.; Yang, A.; Zhang, J. Overexpression of the phosphatidylinositol synthase gene (ZmPIS) conferring drought stress tolerance by altering membrane lipid composition and increasing ABA synthesis in maize. Plant Cell Environ. 2013, 36, 1037–1055. [Google Scholar] [CrossRef]
- Lou, Y.; Gou, J.Y.; Xue, H.W. PIP5K9, an Arabidopsis phosphatidylinositol monophosphate kinase, interacts with a cytosolic invertase to negatively regulate sugar-mediated root growth. Plant Cell 2007, 19, 163–181. [Google Scholar] [CrossRef] [Green Version]
- Wen, B.Q.; Xing, M.Q.; Zhang, H.; Dai, C.; Xue, H.W. Rice homeobox transcription factor HOX1a positively regulates gibberellin responses by directly suppressing EL1. J. Integr. Plant Biol. 2011, 53, 869–878. [Google Scholar] [CrossRef]
- Corratgé-Faillie, C.; Jabnoune, M.; Zimmermann, S.; Véry, A.A.; Fizames, C.; Sentenac, H. Potassium and sodium transport in non-animal cells: The Trk/Ktr/HKT transporter family. Cell. Mol. Life Sci. CMLS 2010, 67, 2511–2532. [Google Scholar] [CrossRef]
- Han, M.; Wu, W.; Wu, W.H.; Wang, Y. Potassium Transporter KUP7 Is Involved in K(+) Acquisition and Translocation in Arabidopsis Root under K(+)-Limited Conditions. Mol. Plant 2016, 9, 437–446. [Google Scholar] [CrossRef] [Green Version]
- Véry, A.A.; Nieves-Cordones, M.; Daly, M.; Khan, I.; Fizames, C.; Sentenac, H. Molecular biology of K+ transport across the plant cell membrane: What do we learn from comparison between plant species? J. Plant Physiol. 2014, 171, 748–769. [Google Scholar] [CrossRef]
- Li, W.; Xu, G.; Alli, A.; Yu, L. Plant HAK/KUP/KT K transporters: Function and regulation. Semin. Cell Dev. Biol. 2018, 74, 133–141. [Google Scholar] [CrossRef]
- Qin, Y.J.; Wu, W.H.; Wang, Y. ZmHAK5 and ZmHAK1 function in K uptake and distribution in maize under low K conditions. J. Integr. Plant Biol. 2019, 61, 691–705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conesa, A.; Gotz, S.; Garcia-Gomez, J.M.; Terol, J.; Talon, M.; Robles, M. Blast2GO: A universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 2005, 21, 3674–3676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, J. WEGO: A web tool for plotting GO annotations. Nucleic Acids Res. 2006, 34, W293–W297. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Harata-Lee, Y.; Denton, M.D.; Feng, Q.; Rathjen, J.R.; Qu, Z.; Adelson, D.L. Long read reference genome-free reconstruction of a full-length transcriptome from Astragalus membranaceus reveals transcript variants involved in bioactive compound biosynthesis. Cell Discov. 2017, 3, 17031. [Google Scholar] [CrossRef] [PubMed]
- Alamancos, G.P.; Pagès, A.; Trincado, J.L.; Bellora, N.; Eyras, E. Leveraging transcript quantification for fast computation of alternative splicing profiles. RNA 2015, 21, 1521–1531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, L.; Luo, H.; Bu, D.; Zhao, G.; Yu, K.; Zhang, C.; Liu, Y.; Chen, R.; Zhao, Y. Utilizing sequence intrinsic composition to classify protein-coding and long non-coding transcripts. Nucleic Acids Res. 2013, 41, e166. [Google Scholar] [CrossRef]
- Kong, L.; Yong, Z.; Ye, Z.Q.; Liu, X.Q.; Ge, G. CPC: Assess the protein-coding potential of transcripts using sequence features and support vector machine. Nucleic Acids Res. 2007, 35, W345–W349. [Google Scholar] [CrossRef]
- Finn, R.D.; Penelope, C.; Eberhardt, R.Y.; Eddy, S.R.; Jaina, M.; Mitchell, A.L.; Potter, S.C.; Marco, P.; Matloob, Q.; Amaia, S.V. The Pfam protein families database: Towards a more sustainable future. Nucleic Acids Res. 2016, 44, D279–D285. [Google Scholar] [CrossRef]
- Li, A.; Zhang, J.; Zhou, Z. PLEK: A tool for predicting long non-coding RNAs and messenger RNAs based on an improved k-mer scheme. BMC Bioinform. 2014, 15, 311. [Google Scholar] [CrossRef] [Green Version]
- Dewey, C.N.; Li, B. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar]
- Kim, G.B.; Gao, Y.; Palsson, B.O.; Lee, S.Y. DeepTFactor: A deep learning-based tool for the prediction of transcription factors. Proc. Natl. Acad. Sci. USA 2021, 118, e2021171118. [Google Scholar] [CrossRef] [PubMed]
- Yi, Z.; Chen, J.; Sun, H.; Rosli, H.G.; Pombo, M.A.; Zhang, P.; Banf, M. iTAK: A program for genome-wide prediction and classification of plant transcription factors, transcriptional regulators, and protein kinases. Mol. Plant 2016, 9, 1667–1670. [Google Scholar]
- Eddy, S.R.; Pearson, W.R. Accelerated Profile HMM Searches. PLoS Comput. Biol. 2011, 7, e1002195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finn, R.D.; Alex, B.; Jody, C.; Penelope, C.; Eberhardt, R.Y.; Eddy, S.R.; Andreas, H.; Kirstie, H.; Liisa, H.; Jaina, M. Pfam: The protein families database. Nucleic Acids Res. 2014, 42, D222–D230. [Google Scholar] [CrossRef] [Green Version]
- Duvaud, S.; Gabella, C.; Lisacek, F.; Stockinger, H.; Durinx, C. Expasy, the Swiss Bioinformatics Resource Portal, as designed by its users. Nucleic Acids Res. 2021, 49, W216–W227. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [Green Version]
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.J.; Higgins, D.G. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 2011, 7, 539. [Google Scholar] [CrossRef]
- Sudhir, K.; Glen, S.; Koichiro, T. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar]
- Letunic, I.; Bork, P. Interactive Tree of Life (iTOL) v5: An online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 2021, 49, W293–W296. [Google Scholar] [CrossRef]
- Liu, Q.; Qi, X.; Yan, H.; Huang, L.; Nie, G.; Zhang, X. Reference Gene Selection for Quantitative Real-Time Reverse-Transcriptase PCR in Annual Ryegrass (Lolium multiflorum) Subjected to Various Abiotic Stresses. Molecules 2018, 23, 172. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CK | DR | Total | |||
|---|---|---|---|---|---|
| Subreads base (G) | 15.48 | 15.24 | 30.72 | ||
| number | 6,944,546 | 6,843,321 | 13,787,867 | ||
| Average length (bp) | 2229 | 2227 | 2228 | ||
| N50 (bp) | 2652 | 2694 | - | ||
| CCS | 488,868 | 538,115 | 1,026,983 | ||
| 5′-primer | 456,294 | 489,188 | 945,482 | ||
| 3′-primer | 462,375 | 501,744 | 964,119 | ||
| Poly-A | 458,539 | 497,870 | 956,409 | ||
| Full length | 424,911 | 454,129 | 879,040 | ||
| FLNC | 403,543 | 431,325 | 834,868 | ||
| Before Correction | After Correction | Before Correction | After Correction | After Correction | |
| Total nucleotides | 469,861,260 | 471,334,422 | 519,052,371 | 520,093,777 | 991,428,199 |
| Total number | 184,267 | 184,267 | 201,378 | 201,378 | 385,645 |
| Mean length (bp) | 2550 | 2558 | 2578 | 2583 | 2571 |
| Min length (bp) | 192 | 193 | 200 | 197 | 193 |
| Max length (bp) | 14,449 | 14,437 | 14,422 | 14,348 | 14,437 |
| N50 (bp) | 2755 | 2759 | 2795 | 2798 | - |
| N90 (bp) | 1774 | 1778 | 1782 | 1784 | - |
| Transcripts Length Interval | Number of Transcripts | Number of Unigenes | Number of Transcripts | Number of Unigenes | |
| <500 bp | 1973 | 1086 | 418 | 211 | |
| 500–1k bp | 4402 | 3289 | 3408 | 2299 | |
| 1–2k bp | 46,268 | 17,602 | 50,843 | 25,263 | |
| 2–3k bp | 80,106 | 40,389 | 87,946 | 45,276 | |
| >3k bp | 51,518 | 32,554 | 58,763 | 39,986 | |
| Total | 184,267 | 94,920 | 201,378 | 113,035 | |
| ORF Length (bp) | No. of AA | pI | Mw (kDa) | |
|---|---|---|---|---|
| LmMYB1 | 747 | 248 | 9.12 | 27.75 |
| LmMYB2 | 984 | 327 | 5.1 | 35.71 |
| LmMYB3 | 1155 | 384 | 6.68 | 42.41 |
| LmMYB4 | 1056 | 351 | 9.35 | 37.45 |
| LmMYB5 | 849 | 282 | 5.08 | 31.61 |
| LmMYB6 | 927 | 308 | 5.75 | 31.45 |
| LmMYB7 | 906 | 301 | 5.26 | 33.52 |
| LmMYB8 | 783 | 260 | 6.37 | 29.02 |
| LmMYB9 | 1059 | 352 | 5.21 | 38.58 |
| LmMYB10 | 720 | 239 | 7.13 | 27.23 |
| LmMYB11 | 978 | 325 | 5.46 | 34.48 |
| LmMYB12 | 1371 | 456 | 5.06 | 50.54 |
| LmMYB13 | 1095 | 364 | 5.19 | 40.74 |
| LmMYB14 | 1860 | 619 | 6.97 | 67.38 |
| LmMYB15 | 1287 | 428 | 6.19 | 47.15 |
| LmMYB16 | 873 | 290 | 6.39 | 32.21 |
| LmMYB17 | 756 | 251 | 6.09 | 28.21 |
| LmMYB18 | 915 | 304 | 5.27 | 33.87 |
| LmMYB19 | 2094 | 697 | 4.83 | 75.73 |
| LmMYB20 | 1872 | 623 | 6.97 | 67.75 |
| LmMYB21 | 1089 | 362 | 5.18 | 40.47 |
| LmMYB22 | 2889 | 962 | 5.11 | 105.65 |
| LmMYB23 | 2538 | 845 | 5.4 | 93.78 |
| LmMYB24 | 1002 | 333 | 5.21 | 37.37 |
| LmMYB25 | 1650 | 549 | 5.15 | 59.42 |
| LmMYB26 | 846 | 281 | 5.34 | 31.49 |
| LmMYB27 | 1065 | 354 | 5.16 | 39.49 |
| LmMYB28 | 2556 | 851 | 5.48 | 94.47 |
| LmMYB29 | 933 | 310 | 5.11 | 34.54 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, Q.; Wang, F.; Shuai, Y.; Huang, L.; Zhang, X. Integrated Analysis of Single-Molecule Real-Time Sequencing and Next-Generation Sequencing Eveals Insights into Drought Tolerance Mechanism of Lolium multiflorum. Int. J. Mol. Sci. 2022, 23, 7921. https://doi.org/10.3390/ijms23147921
Liu Q, Wang F, Shuai Y, Huang L, Zhang X. Integrated Analysis of Single-Molecule Real-Time Sequencing and Next-Generation Sequencing Eveals Insights into Drought Tolerance Mechanism of Lolium multiflorum. International Journal of Molecular Sciences. 2022; 23(14):7921. https://doi.org/10.3390/ijms23147921
Chicago/Turabian StyleLiu, Qiuxu, Fangyan Wang, Yang Shuai, Linkai Huang, and Xinquan Zhang. 2022. "Integrated Analysis of Single-Molecule Real-Time Sequencing and Next-Generation Sequencing Eveals Insights into Drought Tolerance Mechanism of Lolium multiflorum" International Journal of Molecular Sciences 23, no. 14: 7921. https://doi.org/10.3390/ijms23147921
APA StyleLiu, Q., Wang, F., Shuai, Y., Huang, L., & Zhang, X. (2022). Integrated Analysis of Single-Molecule Real-Time Sequencing and Next-Generation Sequencing Eveals Insights into Drought Tolerance Mechanism of Lolium multiflorum. International Journal of Molecular Sciences, 23(14), 7921. https://doi.org/10.3390/ijms23147921