
Exploring the Mechanism of Adjuvant Treatment of Glioblastoma Using Temozolomide and Metformin


 , and
, and
Abstract
:1. Introduction
2. Results
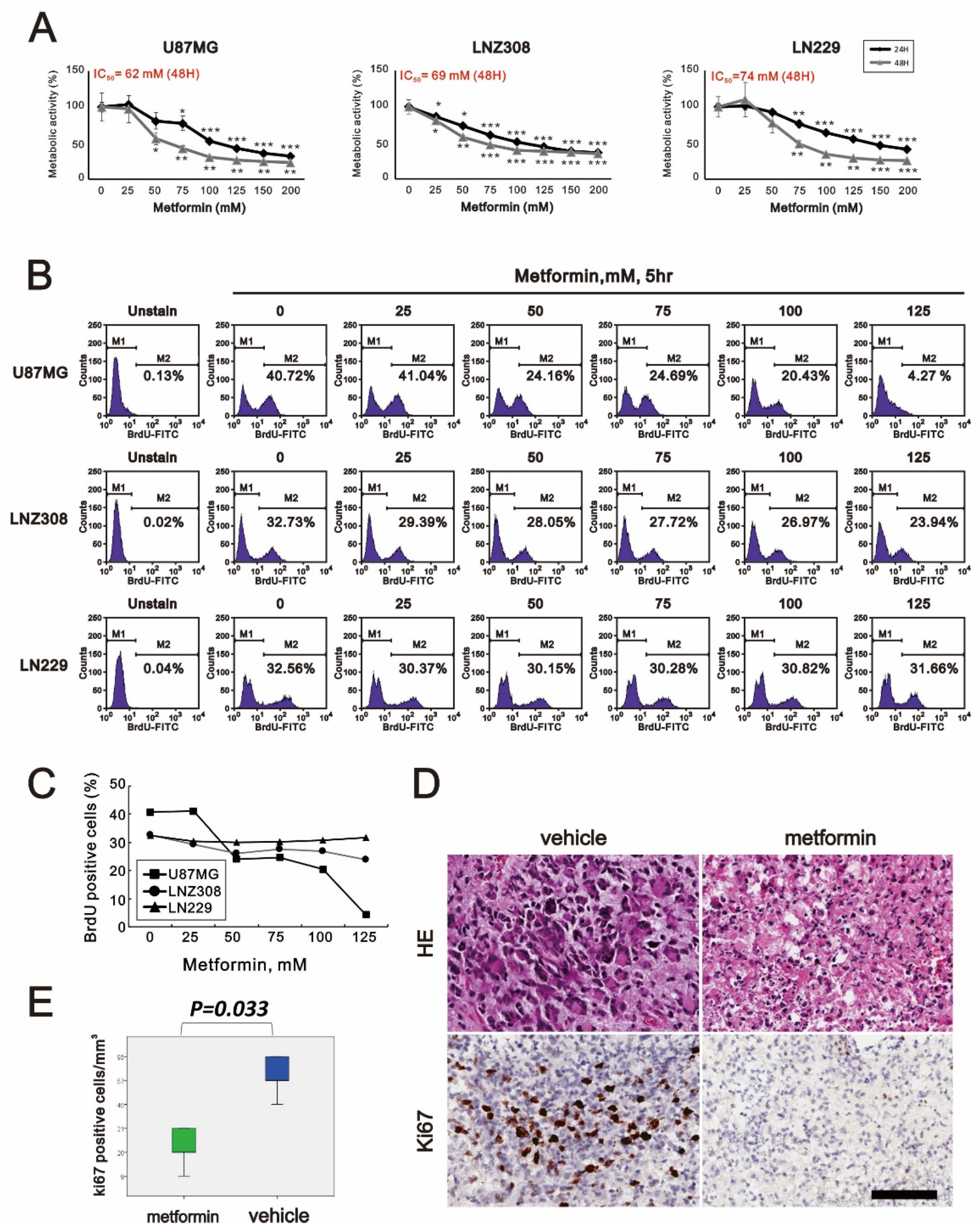
2.1. Metformin-Decreased Cell Viability and Proliferation in a Dose-Dependent Manner in Three GBM Cell Lines
2.2. Metformin Induced Apoptosis in All Three GBM Cell Lines
2.3. Metformin Inhibited Cell Migration and Disrupted Epithelial–Mesenchymal Transition in GBM Cells
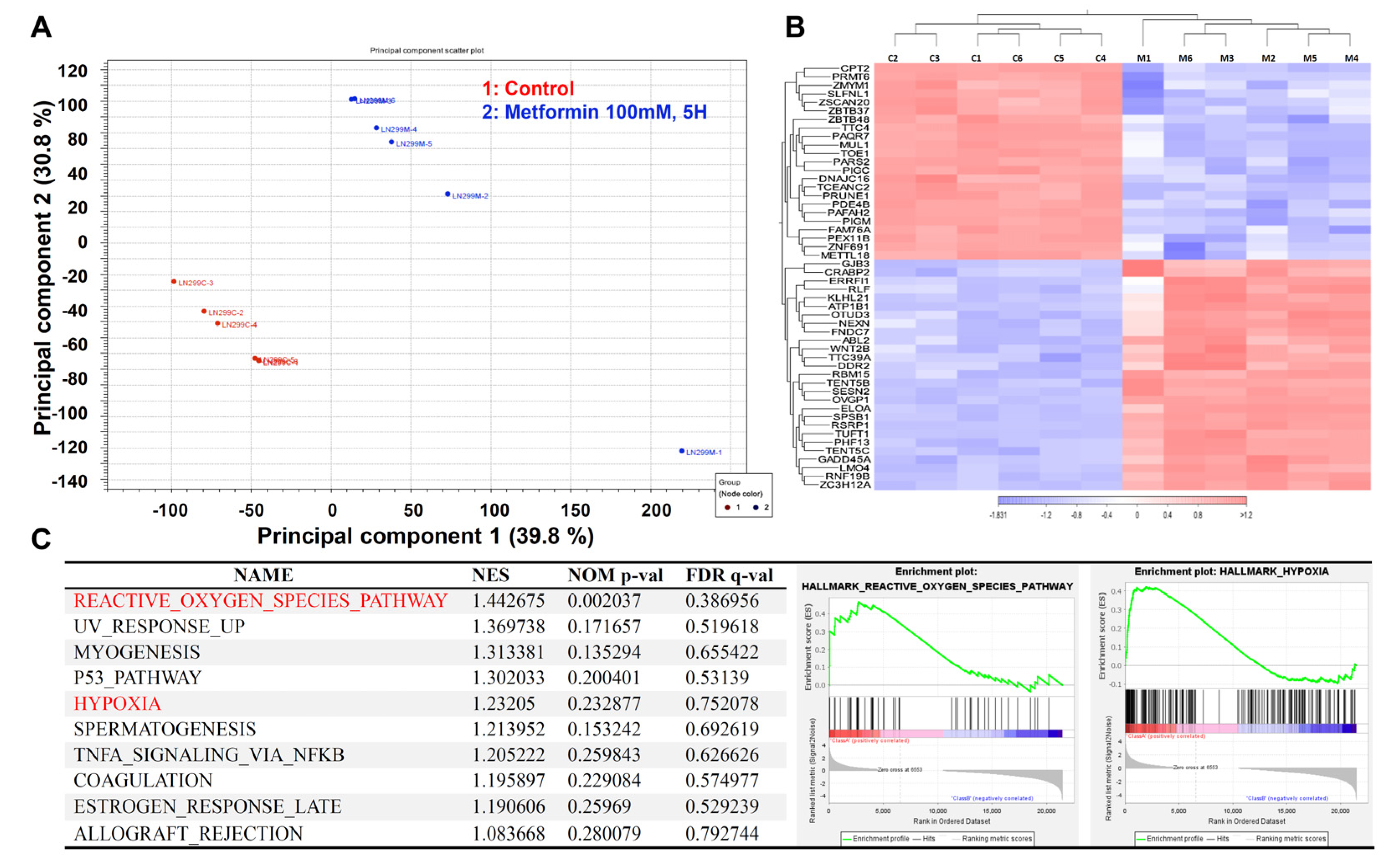
2.4. The Transcriptome of the Metformin-Treated Group Revealed Distinct Signatures Compared to the Control with an Associated Increase in Reactive Oxygen Species and Hypoxia
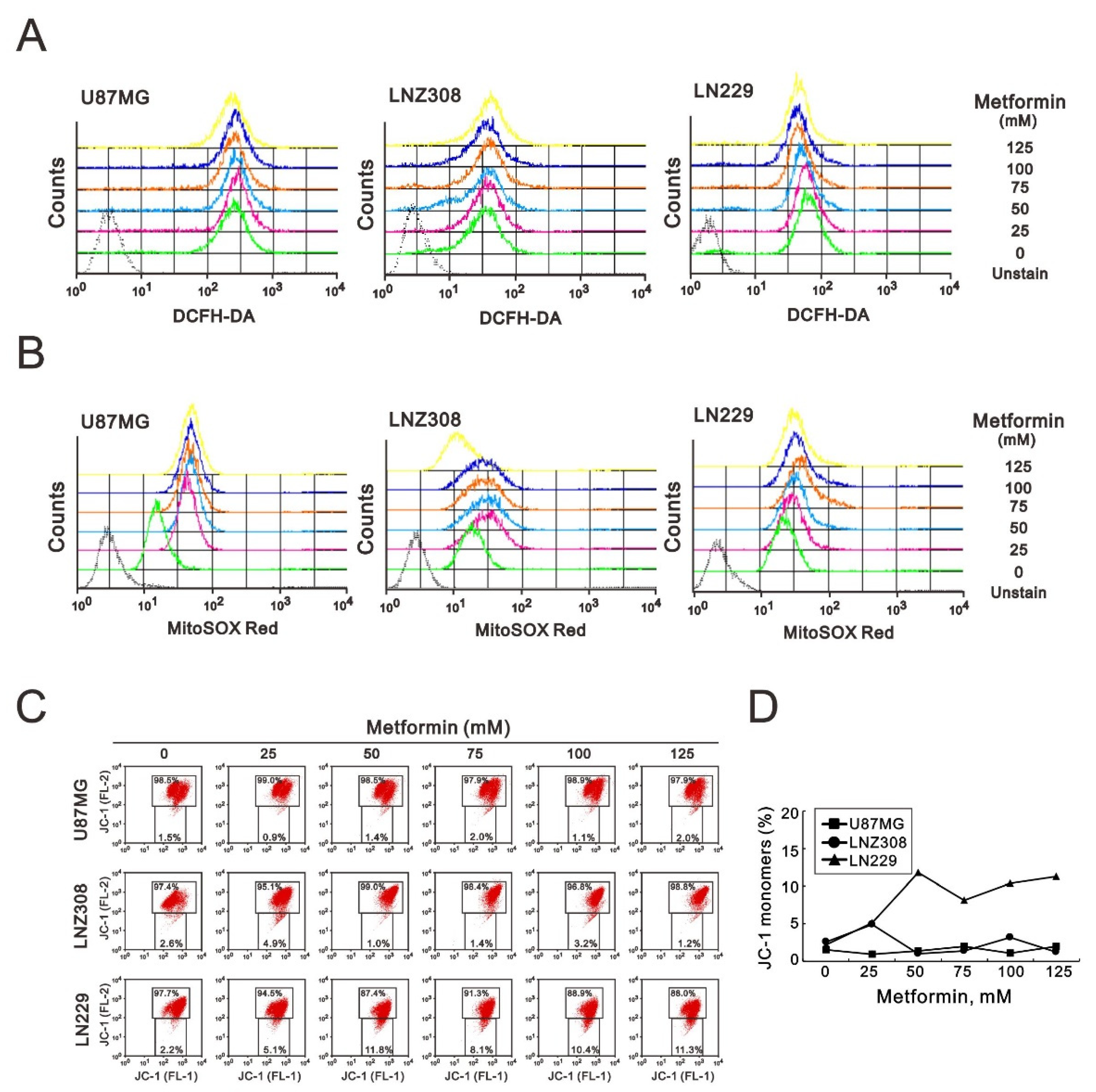
2.5. Metformin Increased ROS Production, Impaired Mitochondrial Membrane Potential (MMP), and Reduced Mitochondrial Biogenesis in GBM Cells
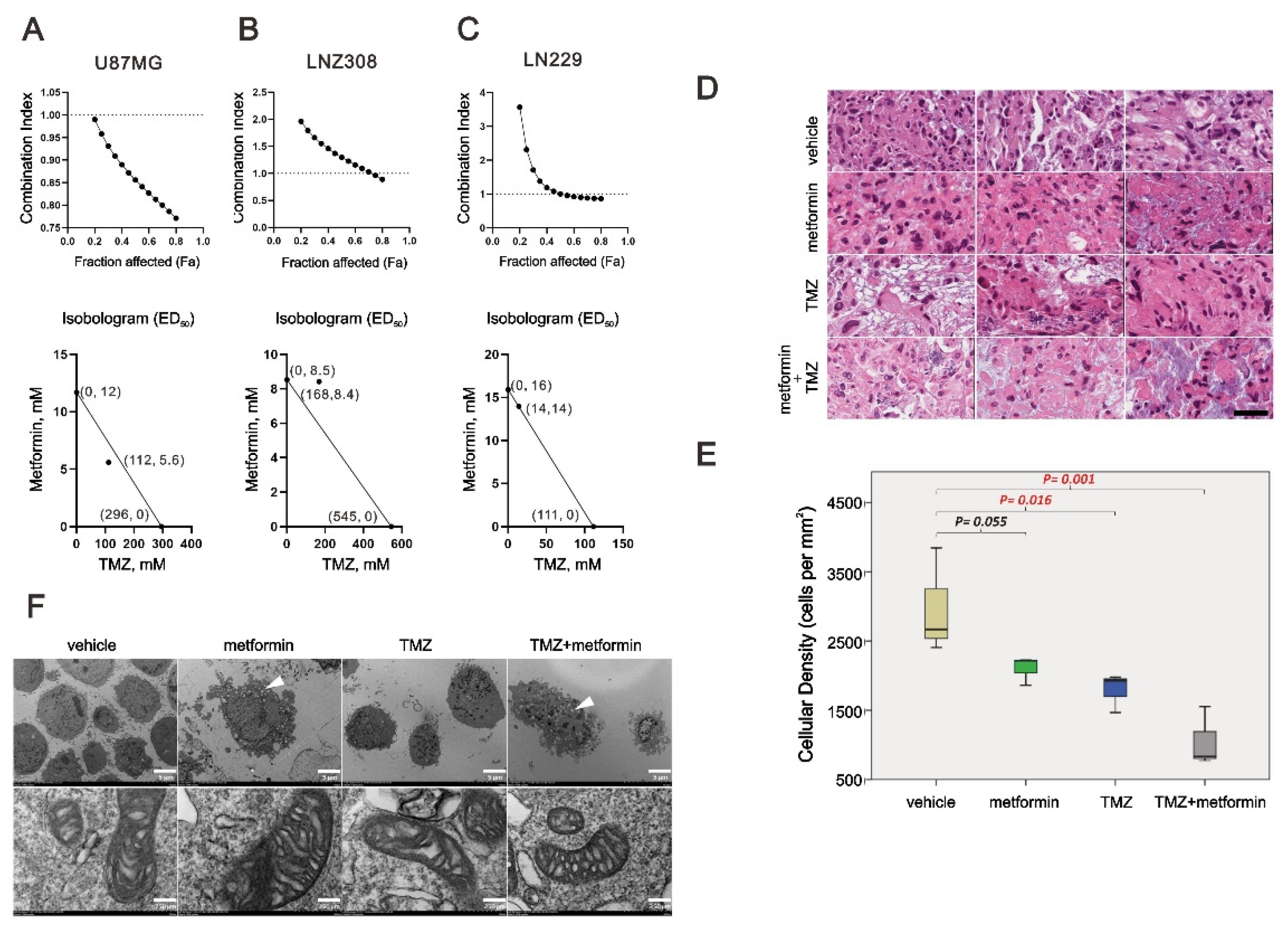
2.6. The Combinatory Effect of TMZ with Metformin on GBM Cells
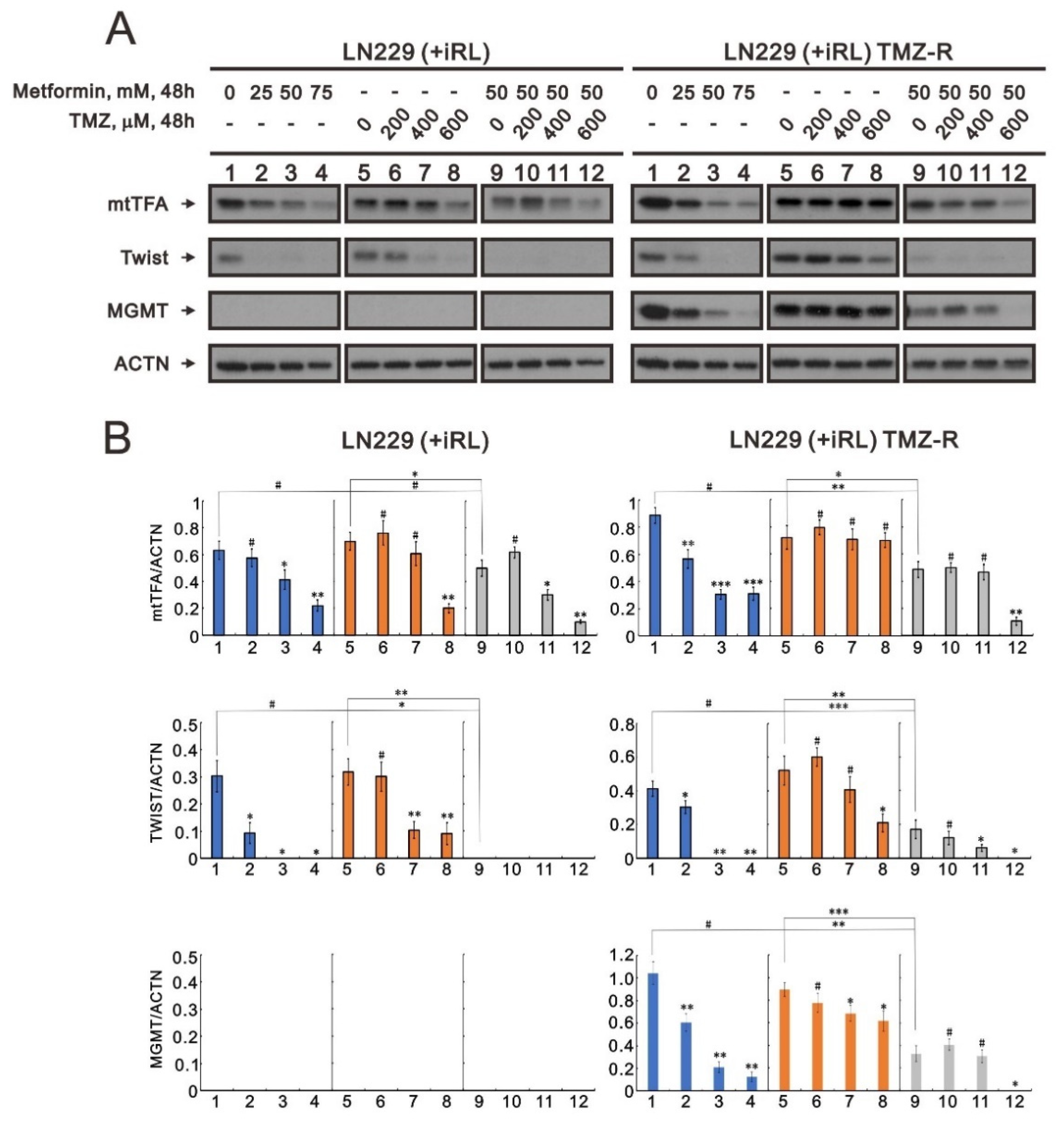
2.7. The Effect of Metformin on Mitochondrial Biogenesis and EMT in TMZ-Sensitive and TMZ-Resistant (TMZ-R) Glioma Cells
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Chemicals
4.2. Metabolic Activity Analysis and the Combination Index
4.3. Fluorescence-Activated Cell Sorting (FACS), Cell-Cycle Profiling, Cellular Proliferation, and Analyses
4.4. Cytosolic and Mitochondrial Reactive Oxygen Species (ROS) Assays
4.5. Mitochondrial Membrane Potential (MMP) Analysis
4.6. Western Blotting Analysis
4.7. Organotypic Slice Culture of Glioblastoma and Immunohistochemistry
4.8. RNA Sequencing Analysis
4.9. Gene Set Enrichment Analysis
4.10. Transmission Electron Microscopy Analysis
4.11. Preparation of Patient-Derived Organoid Culture
4.12. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Louis, D.N.; Perry, A.; Wesseling, P.; Brat, D.J.; Cree, I.A.; Figarella-Branger, D.; Hawkins, C.; Ng, H.K.; Pfister, S.M.; Reifenberger, G.; et al. The 2021 WHO Classification of Tumors of the Central Nervous System: A summary. Neuro Oncol. 2021, 23, 1231–1251. [Google Scholar] [CrossRef]
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parsons, D.W.; Jones, S.; Zhang, X.; Lin, J.C.; Leary, R.J.; Angenendt, P.; Mankoo, P.; Carter, H.; Siu, I.M.; Gallia, G.L.; et al. An integrated genomic analysis of human glioblastoma multiforme. Science 2008, 321, 1807–1812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, H.; Parsons, D.W.; Jin, G.; McLendon, R.; Rasheed, B.A.; Yuan, W.; Kos, I.; Batinic-Haberle, I.; Jones, S.; Riggins, G.J.; et al. IDH1 and IDH2 mutations in gliomas. N. Engl. J. Med. 2009, 360, 765–773. [Google Scholar] [CrossRef]
- Brat, D.J.; Aldape, K.; Colman, H.; Holland, E.C.; Louis, D.N.; Jenkins, R.B.; Kleinschmidt-DeMasters, B.K.; Perry, A.; Reifenberger, G.; Stupp, R.; et al. cIMPACT-NOW update 3: Recommended diagnostic criteria for "Diffuse astrocytic glioma, IDH-wildtype, with molecular features of glioblastoma, WHO grade IV". Acta Neuropathol. 2018, 136, 805–810. [Google Scholar] [CrossRef] [Green Version]
- Brat, D.J.; Aldape, K.; Colman, H.; Figrarella-Branger, D.; Fuller, G.N.; Giannini, C.; Holland, E.C.; Jenkins, R.B.; Kleinschmidt-DeMasters, B.; Komori, T.; et al. cIMPACT-NOW update 5: Recommended grading criteria and terminologies for IDH-mutant astrocytomas. Acta Neuropathol. 2020, 139, 603–608. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef] [PubMed]
- Nanegrungsunk, D.; Onchan, W.; Chattipakorn, N.; Chattipakorn, S.C. Current evidence of temozolomide and bevacizumab in treatment of gliomas. Neurol. Res. 2015, 37, 167–183. [Google Scholar] [CrossRef]
- Jiapaer, S.; Furuta, T.; Tanaka, S.; Kitabayashi, T.; Nakada, M. Potential Strategies Overcoming the Temozolomide Resistance for Glioblastoma. Neurol. Med. Chir. 2018, 58, 405–421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Mir, M.Y.; Nogueira, V.; Fontaine, E.; Avéret, N.; Rigoulet, M.; Leverve, X. Dimethylbiguanide inhibits cell respiration via an indirect effect targeted on the respiratory chain complex I. J. Biol. Chem. 2000, 275, 223–228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rena, G.; Hardie, D.G.; Pearson, E.R. The mechanisms of action of metformin. Diabetologia 2017, 60, 1577–1585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rojas, L.B.A.; Gomes, M.B. Metformin: An old but still the best treatment for type 2 diabetes. Diabetol. Metab. Syndr. 2013, 5, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wróbel, M.P.; Marek, B.; Kajdaniuk, D.; Rokicka, D.; Szymborska-Kajanek, A.; Strojek, K. Metformin—A new old drug. Endokrynologia Polska 2017, 68, 482–496. [Google Scholar] [CrossRef]
- Salpeter, S.; Greyber, E.; Pasternak, G.; Salpeter, E. Risk of fatal and nonfatal lactic acidosis with metformin use in type 2 diabetes mellitus. Cochrane Database Syst. Rev. 2006, 4, CD002967. [Google Scholar]
- Defronzo, R.; Fleming, G.A.; Chen, K.; Bicsak, T.A. Metformin-associated lactic acidosis: Current perspectives on causes and risk. Metab. Clin. Exp. 2016, 65, 20–29. [Google Scholar] [CrossRef] [Green Version]
- Maruthur, N.M.; Tseng, E.; Hutfless, S.; Wilson, L.M.; Suarez-Cuervo, C.; Berger, Z.; Chu, Y.; Iyoha, E.; Segal, J.B.; Bolen, S. Diabetes medications as monotherapy or metformin-based combination therapy for type 2 diabetes: A systematic review and meta-analysis. Ann. Intern. Med. 2016, 164, 740–751. [Google Scholar] [CrossRef] [PubMed]
- Menendez, J.A.; Quirantes-Pine, R.; Rodriguez-Gallego, E.; Cufi, S.; Corominas-Faja, B.; Cuyas, E.; Bosch-Barrera, J.; Martin-Castillo, B.; Segura-Carretero, A.; Joven, J. Oncobiguanides: Paracelsus’ law and nonconventional routes for administering diabetobiguanides for cancer treatment. Oncotarget 2014, 5, 2344–2348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saito, T.; Itoh, M.; Tohda, S. Metformin suppresses the growth of leukemia cells partly through downregulation of AXL receptor tyrosine kinase. Leuk. Res. 2020, 94, 106383. [Google Scholar] [CrossRef] [PubMed]
- Erkinantti, S.; Marttila, M.; Sund, R.; Arffman, M.; Urpilainen, E.; Puistola, U.; Hautakoski, A.; Karihtala, P.; Läärä, E.; Jukkola, A. Association of Metformin, Other Antidiabetic Medications, and Statins With Incidence of Colon Cancer in Patients With Type 2 Diabetes. Clin. Colorectal. Cancer 2021, 20, e113–e119. [Google Scholar] [CrossRef] [PubMed]
- Urbonas, V.; Rutenberge, J.; Patasius, A.; Dulskas, A.; Burokiene, N.; Smailyte, G. The impact of metformin on survival in patients with melanoma-national cohort study. Ann. Epidemiol. 2020, 52, 23–25. [Google Scholar] [CrossRef]
- Tomic, T.; Botton, T.; Cerezo, M.; Robert, G.; Luciano, F.; Puissant, A.; Gounon, P.; Allegra, M.; Bertolotto, C.; Bereder, J.M.; et al. Metformin inhibits melanoma development through autophagy and apoptosis mechanisms. Cell Death Dis. 2011, 2, e199. [Google Scholar] [CrossRef] [Green Version]
- Janjetovic, K.; Harhaji-Trajkovic, L.; Misirkic-Marjanovic, M.; Vucicevic, L.; Stevanovic, D.; Zogovic, N.; Sumarac-Dumanovic, M.; Micic, D.; Trajkovic, V. In vitro and in vivo anti-melanoma action of metformin. Eur. J. Pharmacol. 2011, 668, 373–382. [Google Scholar] [CrossRef] [PubMed]
- Zakikhani, M.; Dowling, R.; Fantus, I.G.; Sonenberg, N.; Pollak, M. Metformin is an AMP kinase-dependent growth inhibitor for breast cancer cells. Cancer Res. 2006, 66, 10269–10273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahn, H.K.; Lee, Y.H.; Koo, K.C. Current Status and Application of Metformin for Prostate Cancer: A Comprehensive Review. Int. J. Mol. Sci. 2020, 21, 8540. [Google Scholar] [CrossRef]
- Zhou, P.T.; Li, B.; Liu, F.R.; Zhang, M.C.; Wang, Q.; Li, Y.Y.; Xu, C.; Liu, Y.H.; Yao, Y.; Li, D. Metformin is associated with survival benefit in pancreatic cancer patients with diabetes: A systematic review and metaanalysis. Oncotarget 2017, 8, 25242–25250. [Google Scholar] [CrossRef] [Green Version]
- Vincent, A.; Herman, J.; Schulick, R.; Hruban, R.H.; Goggins, M. Pancreatic cancer. Lancet 2011, 378, 607–620. [Google Scholar] [CrossRef]
- Gong, H.; Chen, Y.; Zhou, D. Prognostic significance of metformin treatment in endometrial cancer: A meta-analysis. Pharmazie 2020, 75, 401–406. [Google Scholar]
- Yousef, M.; Tsiani, E. Metformin in lung cancer: Review of in vitro and in vivo animal studies. Cancers 2017, 9, 45. [Google Scholar] [CrossRef] [PubMed]
- Seliger, C.; Luber, C.; Gerken, M.; Schaertl, J.; Proescholdt, M.; Riemenschneider, M.J.; Meier, C.R.; Bogdahn, U.; Leitzmann, M.F.; Klinkhammer-Schalke, M.; et al. Use of metformin and survival of patients with high-grade glioma. Int. J. Cancer 2019, 144, 273–280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jornayvaz, F.R.; Shulman, G.I. Regulation of mitochondrial biogenesis. Essays Biochem. 2010, 47, 69–84. [Google Scholar]
- Scarpulla, R.C. Metabolic control of mitochondrial biogenesis through the PGC-1 family regulatory network. Biochim. Biophys. Acta 2011, 1813, 1269–1278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, W.J.; He, D.S.; Tang, R.X.; Ren, F.H.; Chen, G. Ki-67 is a valuable prognostic factor in gliomas: Evidence from a systematic review and meta-analysis. Asian Pac. J. Cancer Prev. 2015, 16, 411–420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, B.; Zhou, P.; Xu, K.; Chen, T.; Jiao, J.; Wei, H.; Yang, X.; Xu, W.; Wan, W.; Xiao, J. Metformin induces cell cycle arrest, apoptosis and autophagy through ROS/JNK signaling pathway in human osteosarcoma. Int. J. Biol. Sci. 2020, 16, 74–84. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Xu, W.; Yan, Z.; Zhao, W.; Mi, J.; Li, J.; Yan, H. Metformin induces autophagy and G0/G1 phase cell cycle arrest in myeloma by targeting the AMPK/mTORC1 and mTORC2 pathways. J. Exp. Clin. Cancer Res. 2018, 37, 63. [Google Scholar] [CrossRef]
- Mogavero, A.; Maiorana, M.V.; Zanutto, S.; Varinelli, L.; Bozzi, F.; Belfiore, A.; Volpi, C.C.; Gloghini, A.; Pierotti, M.A.; Gariboldi, M. Metformin transiently inhibits colorectal cancer cell proliferation as a result of either AMPK activation or increased ROS production. Sci. Rep. 2017, 7, 15992. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiong, Z.S.; Gong, S.F.; Si, W.; Jiang, T.; Li, Q.L.; Wang, T.J.; Wang, W.J.; Wu, R.Y.; Jiang, K. Effect of metformin on cell proliferation, apoptosis, migration and invasion in A172 glioma cells and its mechanisms. Mol. Med. Rep. 2019, 20, 887–894. [Google Scholar] [CrossRef]
- Burger, P.C.; Heinz, E.R.; Shibata, T.; Kleihues, P. Topographic anatomy and CT correlations in the untreated glioblastoma multiforme. J. Neurosurg. 1988, 68, 698–704. [Google Scholar] [CrossRef] [PubMed]
- Bao, B.; Wang, Z.; Ali, S.; Ahmad, A.; Azmi, A.S.; Sarkar, S.H.; Banerjee, S.; Kong, D.; Li, Y.; Thakur, S.; et al. Metformin inhibits cell proliferation, migration and invasion by attenuating CSC function mediated by deregulating miRNAs in pancreatic cancer cells. Cancer Prev. Res. 2012, 5, 355–364. [Google Scholar] [CrossRef] [Green Version]
- Xiao, Z.; Wu, W.; Poltoratsky, V. Metformin Suppressed CXCL8 Expression and Cell Migration in HEK293/TLR4 Cell Line. Mediators Inflamm. 2017, 2017, 6589423. [Google Scholar] [CrossRef] [Green Version]
- Ferretti, A.C.; Hidalgo, F.; Tonucci, F.M.; Almada, E.; Pariani, A.; Larocca, M.C.; Favre, C. Metformin and glucose starvation decrease the migratory ability of hepatocellular carcinoma cells: Targeting AMPK activation to control migration. Sci. Rep. 2019, 9, 2815. [Google Scholar] [CrossRef] [PubMed]
- Kast, R.E.; Skuli, N.; Karpel-Massler, G.; Frosina, G.; Ryken, T.; Halatsch, M.E. Blocking epithelial-to-mesenchymal transition in glioblastoma with a sextet of repurposed drugs: The EIS regimen. Oncotarget 2017, 8, 60727–60749. [Google Scholar] [CrossRef]
- Zhang, P.; Zhao, S.; Lu, X.; Shi, Z.; Liu, H.; Zhu, B. Metformin enhances the sensitivity of colorectal cancer cells to cisplatin through ROS-mediated PI3K/Akt signaling pathway. Gene 2020, 745, 144623. [Google Scholar] [CrossRef]
- Sharma, P.; Kumar, S. Metformin inhibits human breast cancer cell growth by promoting apoptosis via a ROS-independent pathway involving mitochondrial dysfunction: Pivotal role of superoxide dismutase (SOD). Cell Oncol. 2018, 41, 637–650. [Google Scholar] [CrossRef]
- Momcilovic, M.; Jones, A.; Bailey, S.T.; Waldmann, C.M.; Li, R.; Lee, J.T.; Abdelhady, G.; Gomez, A.; Holloway, T.; Schmid, E.; et al. In vivo imaging of mitochondrial membrane potential in non-small-cell lung cancer. Nature 2019, 575, 380–384. [Google Scholar] [CrossRef]
- Gottlieb, E.; Armour, S.M.; Harris, M.H.; Thompson, C.B. Mitochondrial membrane potential regulates matrix configuration and cytochrome c release during apoptosis. Cell Death Differ. 2003, 10, 709–717. [Google Scholar] [CrossRef]
- Vyas, S.; Zaganjor, E.; Haigis, M.C. Mitochondria and Cancer. Cell 2016, 166, 555–566. [Google Scholar] [CrossRef] [Green Version]
- Jäger, S.; Handschin, C.; St-Pierre, J.; Spiegelman, B.M. AMP-activated protein kinase (AMPK) action in skeletal muscle via direct phosphorylation of PGC-1alpha. Proc. Natl. Acad. Sci. USA 2007, 104, 12017–12022. [Google Scholar] [CrossRef] [Green Version]
- Bost, F.; Kaminski, L. The metabolic modulator PGC-1α in cancer. Am. J. Cancer Res. 2019, 9, 198–211. [Google Scholar]
- Gilbert, M.R.; Wang, M.; Aldape, K.D.; Stupp, R.; Hegi, M.E.; Jaeckle, K.A.; Armstrong, T.S.; Wefel, J.S.; Won, M.; Blumenthal, D.T.; et al. Dose-dense temozolomide for newly diagnosed glioblastoma: A randomized phase III clinical trial. J. Clin. Oncol. 2013, 31, 4085–4091. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.H.; Guo, X.L. Combinational strategies of metformin and chemotherapy in cancers. Cancer Chemother. Pharmacol. 2016, 78, 13–26. [Google Scholar] [CrossRef]
- Ying, M.A.; Maruschak, N.; Mansur, R.; Carvalho, A.F.; Cha, D.S.; McIntyre, R.S. Metformin: Repurposing opportunities for cognitive and mood dysfunction. CNS Neurol. Disord. Drug. Targets 2014, 13, 1836–1845. [Google Scholar] [CrossRef]
- Wang, Y.; Meng, Y.; Zhang, S.; Wu, H.; Yang, D.; Nie, C.; Hu, Q. Phenformin and metformin inhibit growth and migration of LN229 glioma cells in vitro and in vivo. OncoTargets Ther. 2018, 11, 6039–6048. [Google Scholar] [CrossRef] [Green Version]
- Cuyàs, E.; Fernández-Arroyo, S.; Corominas-Faja, B.; Rodríguez-Gallego, E.; Bosch-Barrera, J.; Martin-Castillo, B.; De Llorens, R.; Joven, J.; Menendez, J.A. Oncometabolic mutation IDH1 R132H confers a metformin-hypersensitive phenotype. Oncotarget 2015, 6, 12279–12296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mazurek, M.; Litak, J.; Kamieniak, P.; Kulesza, B.; Jonak, K.; Baj, J.; Grochowski, C. Metformin as Potential Therapy for High-Grade Glioma. Cancers 2020, 12, 210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chou, T.C. Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacol. Rev. 2006, 58, 621–681. [Google Scholar] [CrossRef]
- Chang, Y.L.; Hsu, Y.J.; Chen, Y.; Wang, Y.W.; Huang, S.M. Theophylline exhibits anti-cancer activity via suppressing SRSF3 in cervical and breast cancer cell lines. Oncotarget 2017, 8, 101461–101474. [Google Scholar] [CrossRef] [Green Version]
- Kuo, C.L.; Hsieh Li, S.M.; Liang, S.Y.; Liu, S.T.; Huang, L.C.; Wang, W.M.; Yen, L.C.; Huang, S.M. The antitumor properties of metformin and phenformin reflect their ability to inhibit the actions of differentiated embryo chondrocyte 1. Cancer Manag. Res. 2019, 11, 6567–6579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stoppini, L.; Buchs, P.A.; Muller, D. A simple method for organotypic cultures of nervous tissue. J. Neurosci. Methods 1991, 37, 173–182. [Google Scholar] [CrossRef]
- Li, Y.F.; Scerif, F.; Picker, S.R.; Stone, T.J.; Pickles, J.C.; Moulding, D.A.; Avery, A.; Virasami, A.; Fairchild, A.R.; Tisdall, M.; et al. Identifying cellular signalling molecules in developmental disorders of the brain: Evidence from focal cortical dysplasia and tuberous sclerosis. Neuropathol. Appl. Neurobiol. 2021, 47, 781–795. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacob, F.; Ming, G.L.; Song, H. Generation and biobanking of patient-derived glioblastoma organoids and their application in CAR T cell testing. Nat. Protoc. 2020, 15, 4000–4033. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Cell line | ||
|---|---|---|---|
| U87MG | LNZ308 | LN229 | |
| IDH1 | Mutant (R132H) | WT | WT |
| p53 | WT/WT | intragenic deletion loss by translocation | Codon 98 CCT(Pro)→CTT(Lys) |
| ATRX | Mutant | Mutant | Mutant |
| PTEN | splice (del. exon 3) in frame deletion | splice (del. exon 6) frameshift | WT |
| p16 | del | WT | del |
| p14ARF | del | WT | del |
| Integrated Dx | IDH-mutant astrocytoma, Gr 4 | IDH-wildtype glioblastoma, Gr 4 | IDH-wildtype glioblastoma, Gr 4 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Feng, S.-W.; Chang, P.-C.; Chen, H.-Y.; Hueng, D.-Y.; Li, Y.-F.; Huang, S.-M. Exploring the Mechanism of Adjuvant Treatment of Glioblastoma Using Temozolomide and Metformin. Int. J. Mol. Sci. 2022, 23, 8171. https://doi.org/10.3390/ijms23158171
Feng S-W, Chang P-C, Chen H-Y, Hueng D-Y, Li Y-F, Huang S-M. Exploring the Mechanism of Adjuvant Treatment of Glioblastoma Using Temozolomide and Metformin. International Journal of Molecular Sciences. 2022; 23(15):8171. https://doi.org/10.3390/ijms23158171
Chicago/Turabian StyleFeng, Shao-Wei, Pei-Chi Chang, Hsuan-Yu Chen, Dueng-Yuan Hueng, Yao-Feng Li, and Shih-Ming Huang. 2022. "Exploring the Mechanism of Adjuvant Treatment of Glioblastoma Using Temozolomide and Metformin" International Journal of Molecular Sciences 23, no. 15: 8171. https://doi.org/10.3390/ijms23158171
APA StyleFeng, S. -W., Chang, P. -C., Chen, H. -Y., Hueng, D. -Y., Li, Y. -F., & Huang, S. -M. (2022). Exploring the Mechanism of Adjuvant Treatment of Glioblastoma Using Temozolomide and Metformin. International Journal of Molecular Sciences, 23(15), 8171. https://doi.org/10.3390/ijms23158171