
Sanguinarine Inhibition of TNF-α-Induced CCL2, IKBKE/NF-κB/ERK1/2 Signaling Pathway, and Cell Migration in Human Triple-Negative Breast Cancer Cells


Abstract
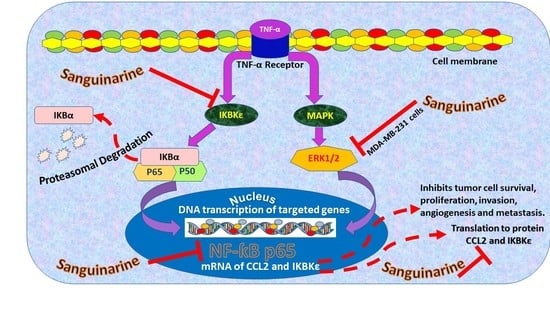
:
1. Introduction
2. Results
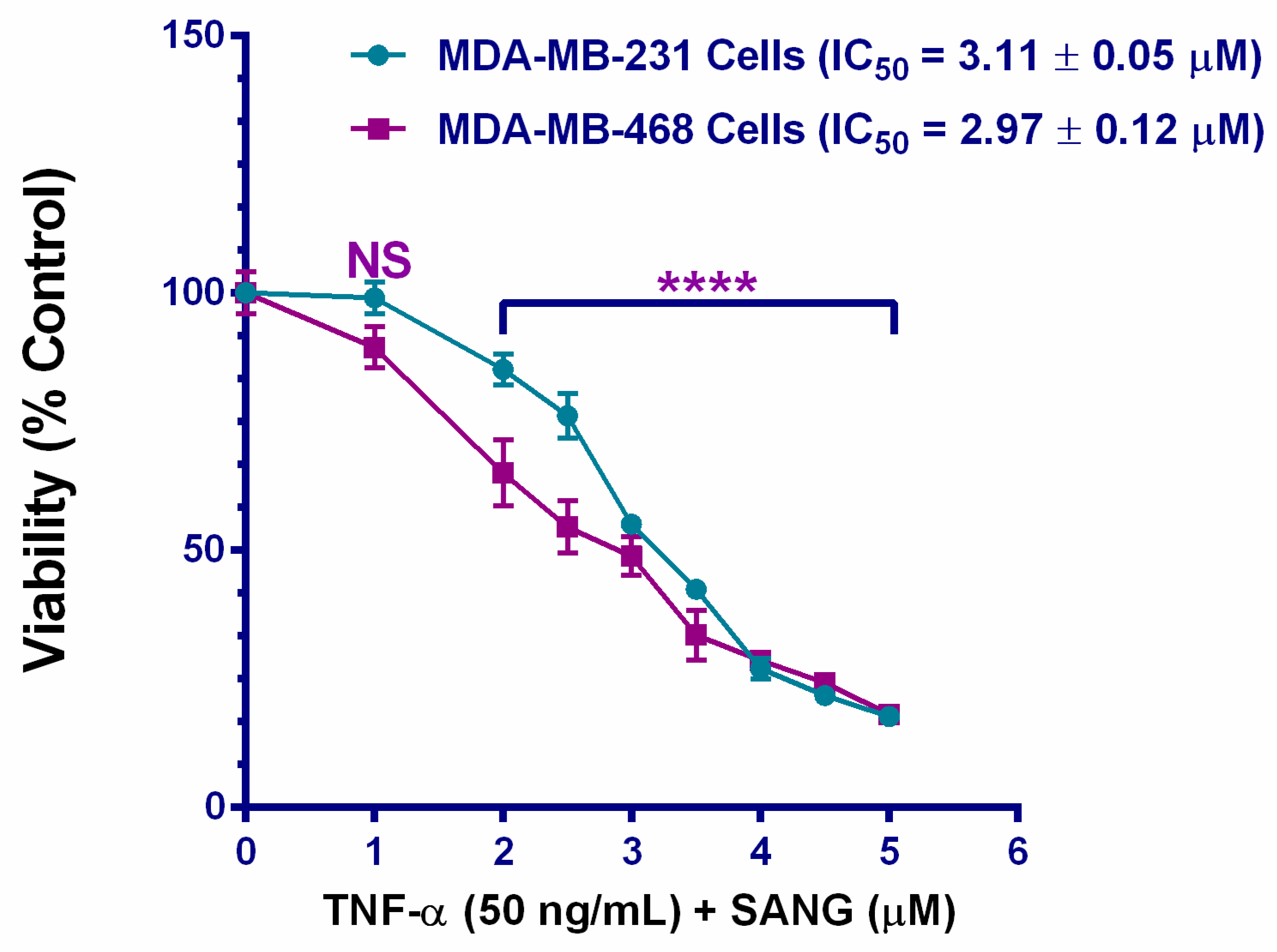
2.1. SANG Decreases the Viability of TNF-α-Treated TNBC Cells
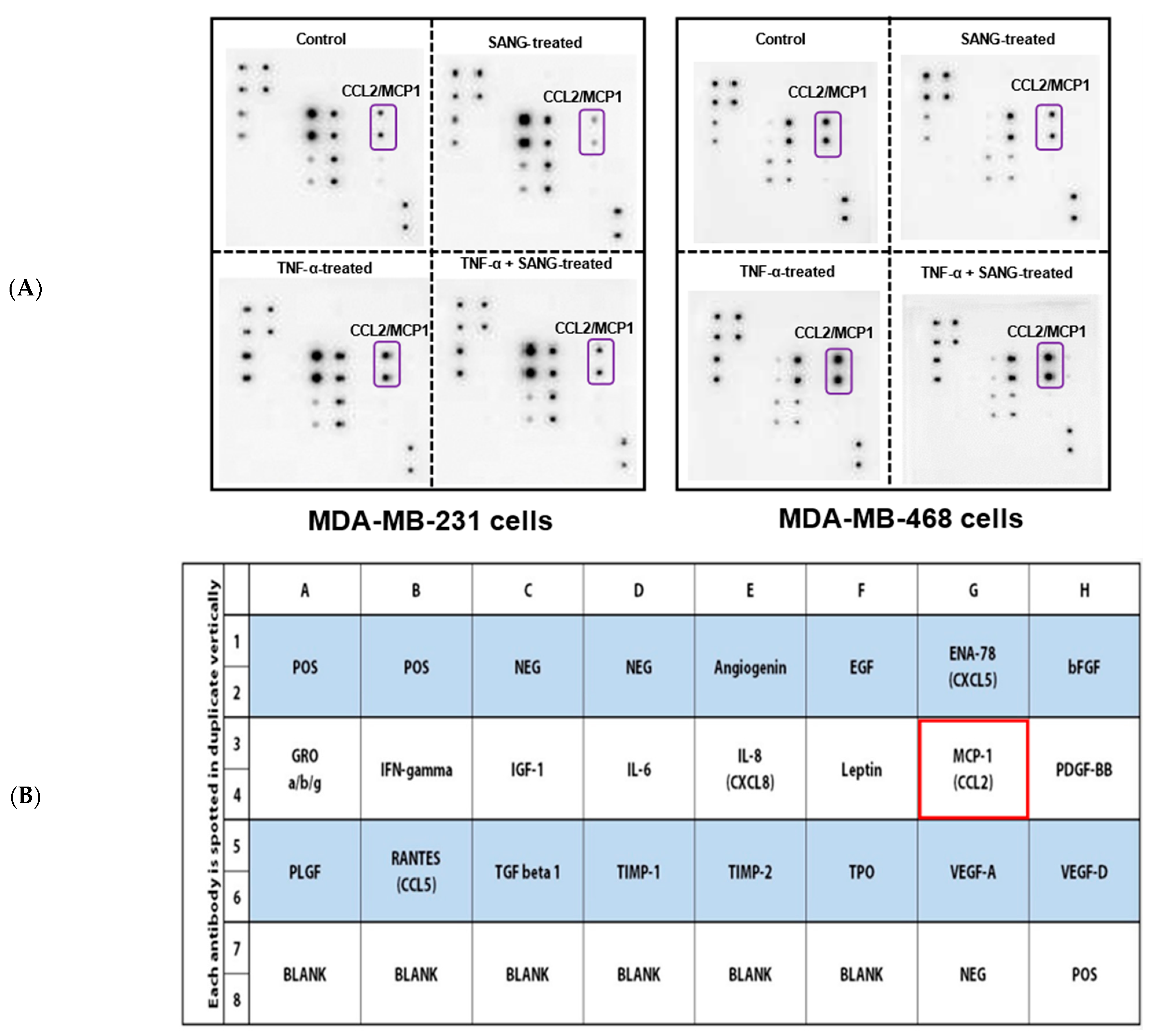
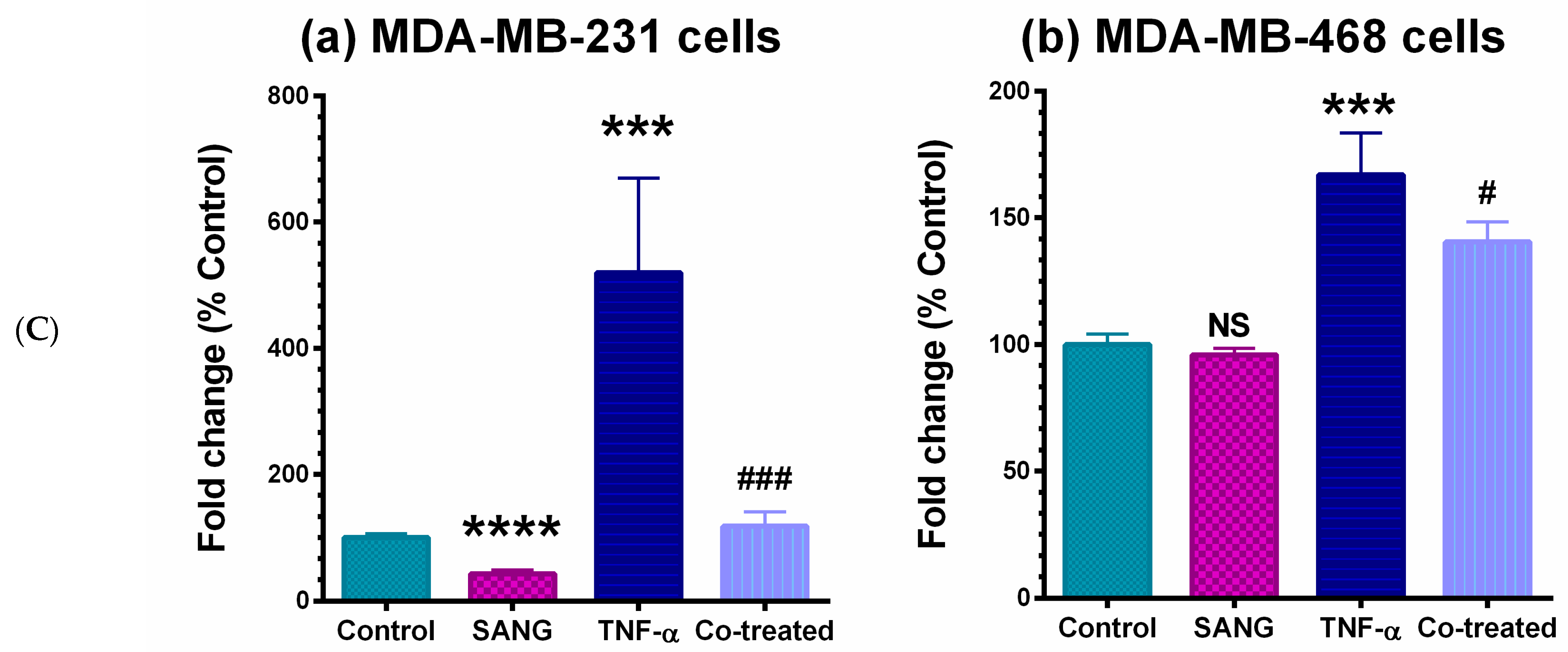
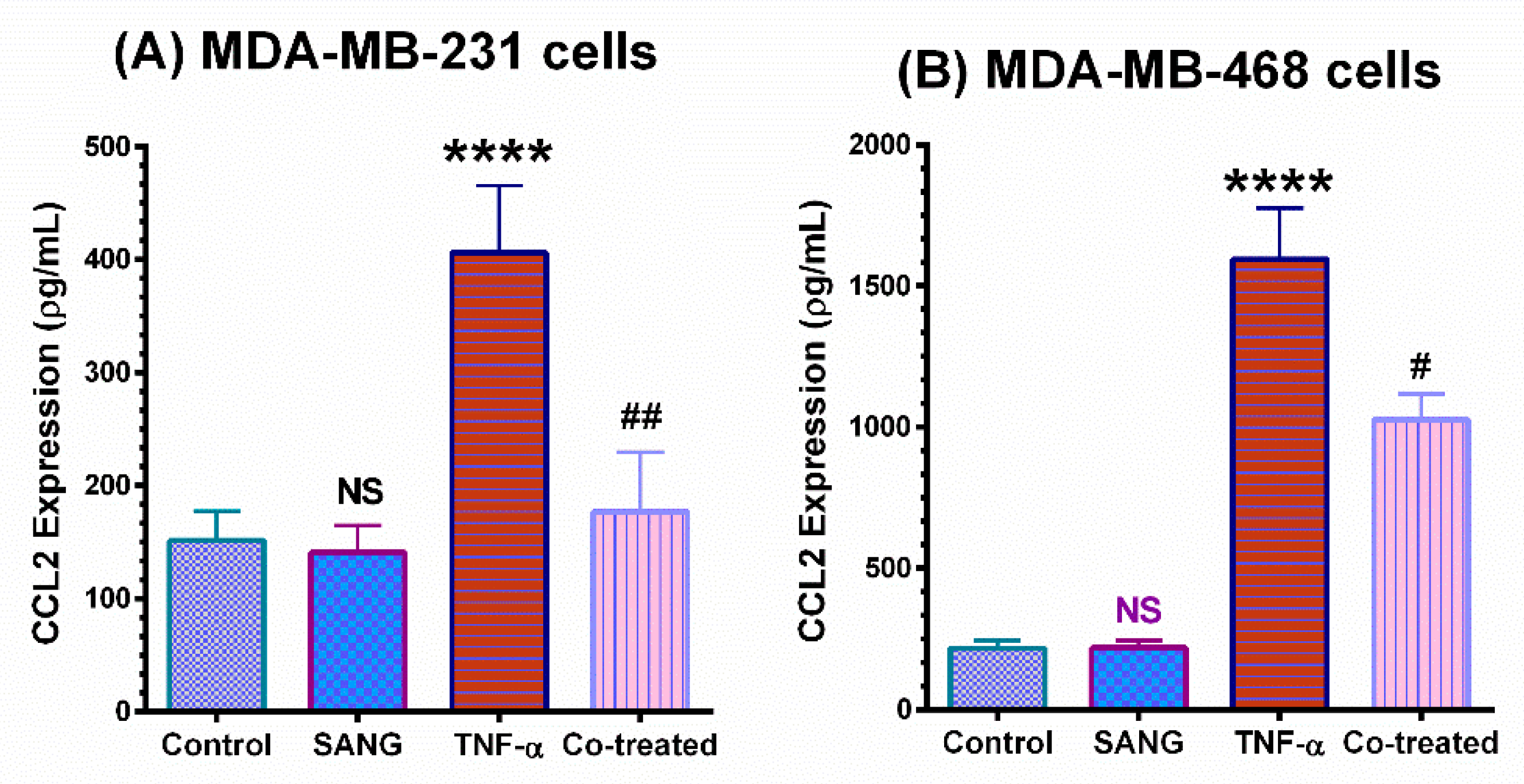
2.2. SANG Inhibits the Release of CCL2 Cytokine in TNBC Cells
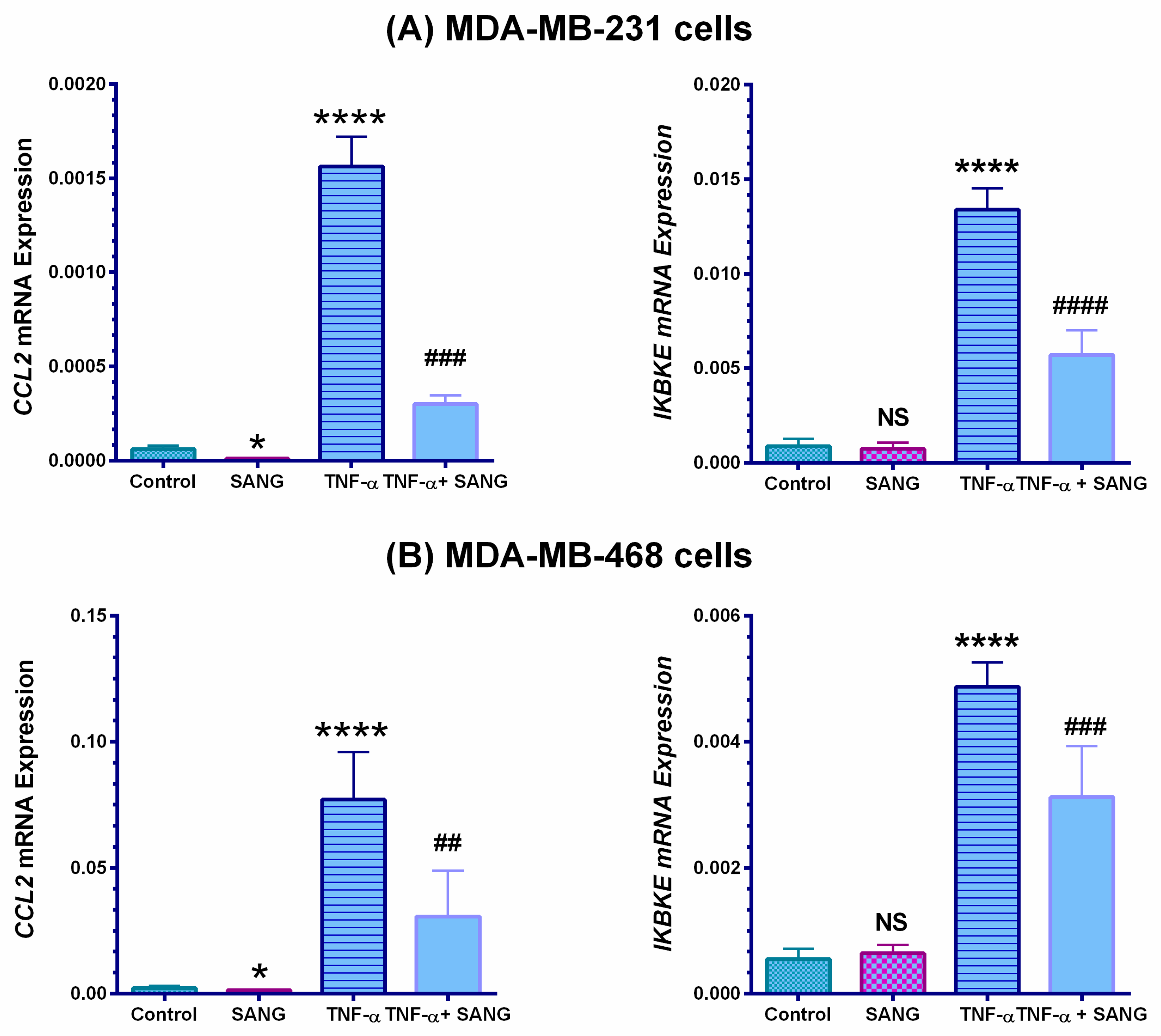
2.3. SANG Represses the Expression of the IKBKE Gene Promoting CCL2 Release in Stimulated TNBC Cells
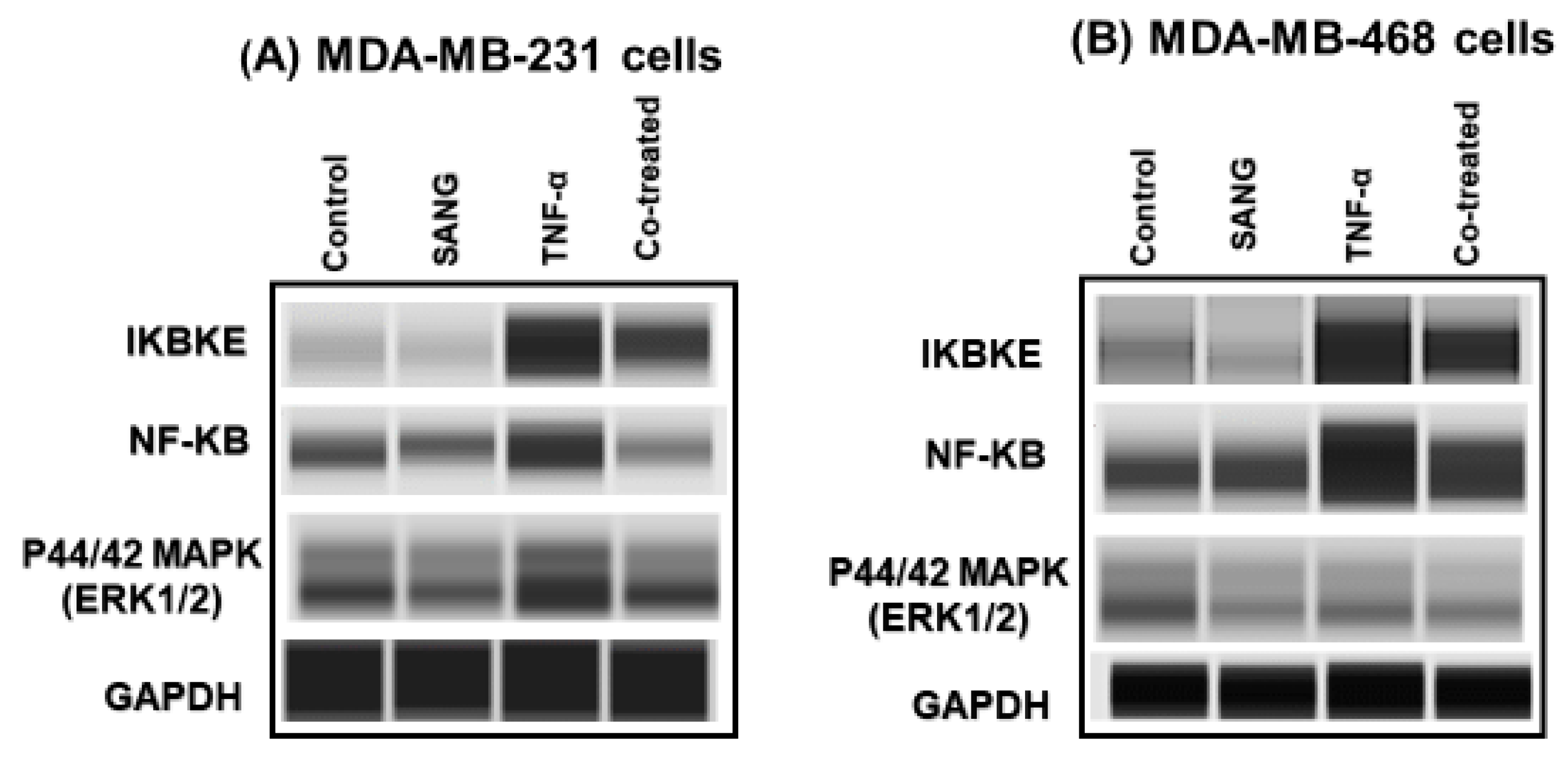
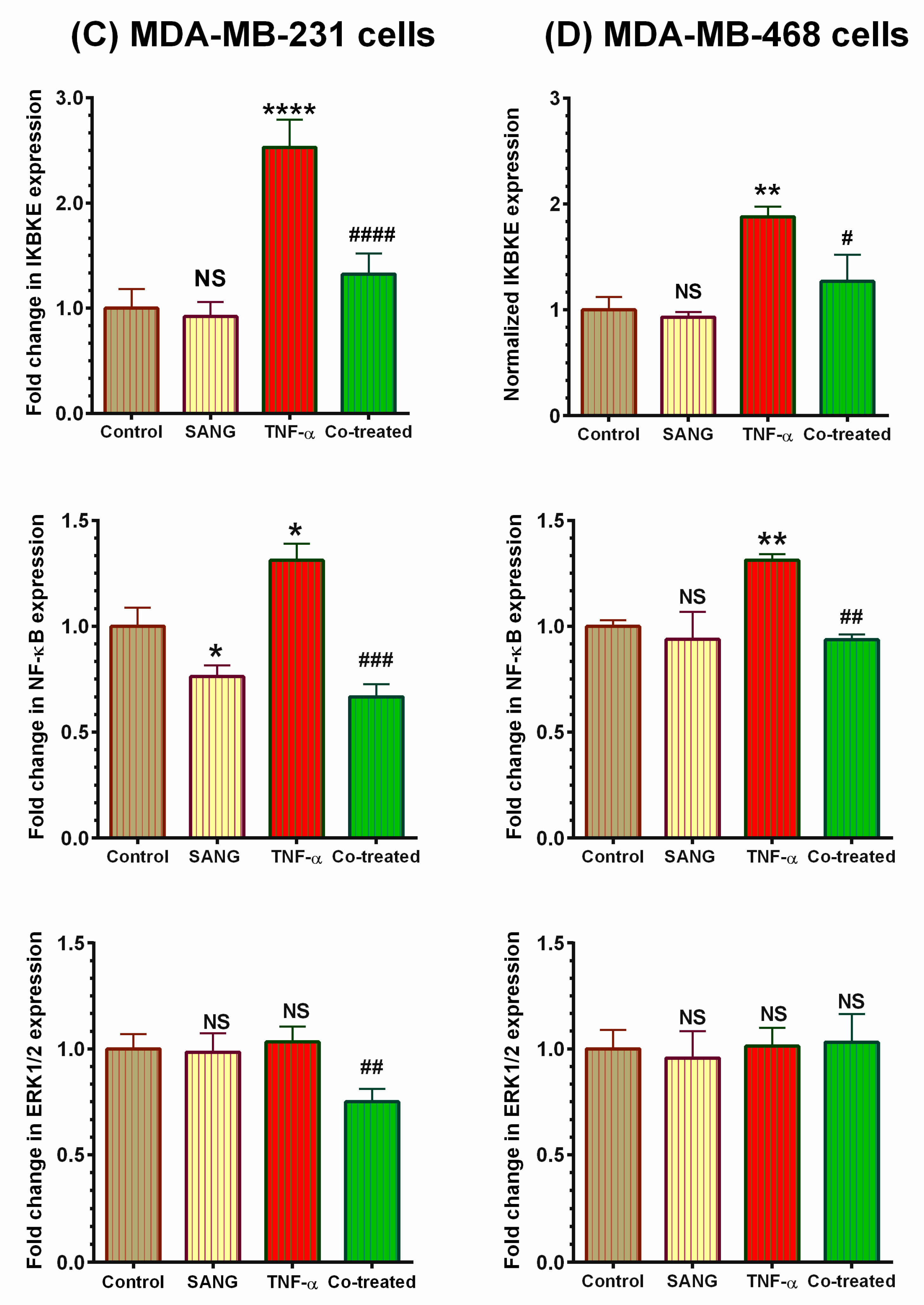
2.4. SANG Modulates Protein Expression of Signaling Pathways Mediating CCL2 Release in TNF-α-Stimulated TNBC Cells
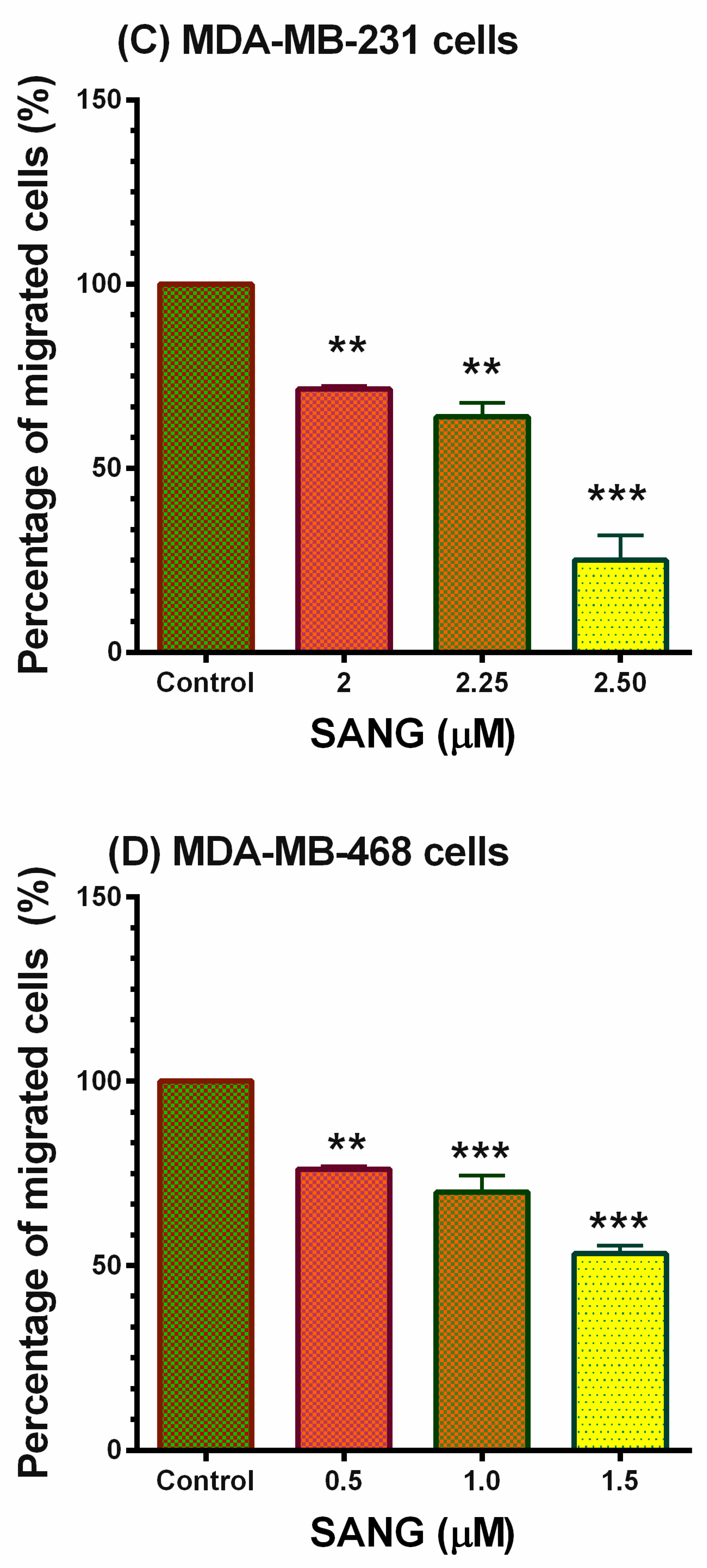
2.5. SANG Inhibits Migration in TNBC Cells
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Media
4.2. Cell Viability Assay
4.3. Human Angiogenesis Array
4.4. Enzyme-Linked Immunosorbent Assay (ELISA)
4.5. Gene Expression Study
4.5.1. RNA Extraction
4.5.2. Complementary DNA (cDNA) Synthesis
4.5.3. Quantitative Reverse Transcription-Polymerase Chain Reaction (qRT-PCR)
4.6. Capillary Electrophoresis Western Analysis
4.7. Migration Assay
4.8. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Folkman, J. Tumor angiogenesis: Therapeutic implications. N. Engl. J. Med. 1971, 285, 1182–1186. [Google Scholar] [CrossRef] [PubMed]
- Lugano, R.; Ramachandran, M.; Dimberg, A. Tumor angiogenesis: Causes, consequences, challenges, and opportunities. Cell. Mol. Life Sci. 2020, 77, 1745–1770. [Google Scholar] [CrossRef] [Green Version]
- Borri, F.; Granaglia, A. Pathology of triple-negative breast cancer. Semin. Cancer Biol. 2021, 72, 136–145. [Google Scholar] [CrossRef]
- Garrido-Castro, A.C.; Lin, N.U.; Polyak, K. Insights into Molecular Classifications of Triple-Negative Breast Cancer: Improving Patient Selection for Treatment. Cancer Discov. 2019, 9, 176–198. [Google Scholar] [CrossRef] [Green Version]
- Howard, F.M.; Olopade, O.I. Epidemiology of Triple-Negative Breast Cancer: A Review. Cancer J. 2021, 27, 8–16. [Google Scholar] [CrossRef]
- Yin, L.; Duan, J.J.; Bian, X.W.; Yu, S.C. Triple-negative breast cancer molecular subtyping and treatment progress. Breast Cancer Res. 2020, 22, 61. [Google Scholar] [CrossRef]
- Bergin, A.R.T.; Loi, S. Triple-negative breast cancer: Recent treatment advances. F1000Research 2019, 8, 1342. [Google Scholar] [CrossRef]
- Lyons, T.G. Targeted Therapies for Triple-Negative Breast Cancer. Curr. Treat. Options Oncol. 2019, 20, 82. [Google Scholar] [CrossRef]
- Keenan, T.E.; Tolaney, S.M. Role of Immunotherapy in Triple-Negative Breast Cancer. J. Natl. Compr. Cancer Netw. 2020, 18, 479–489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papetti, M.; Herman, I.M. Mechanisms of normal and tumor-derived angiogenesis. Am. J. Physiol. Cell Physiol. 2002, 282, C947–C970. [Google Scholar] [CrossRef] [Green Version]
- Ahn, J.B.; Rha, S.Y.; Shin, S.J.; Jeung, H.C.; Kim, T.S.; Zhang, X.; Park, K.H.; Noh, S.H.; Roh, J.K.; Chung, H.C. Circulating endothelial progenitor cells (EPC) for tumor vasculogenesis in gastric cancer patients. Cancer Lett. 2010, 288, 124–132. [Google Scholar] [CrossRef]
- Greenfield, J.P.; Cobb, W.S.; Lyden, D. Resisting arrest: A switch from angiogenesis to vasculogenesis in recurrent malignant gliomas. J. Clin. Investig. 2010, 120, 663–667. [Google Scholar] [CrossRef]
- Badodekar, N.; Sharma, A.; Patil, V.; Telang, G.; Sharma, R.; Patil, S.; Vyas, N.; Somasundaram, I. Angiogenesis induction in breast cancer: A paracrine paradigm. Cell Biochem. Funct. 2021, 39, 860–873. [Google Scholar] [CrossRef]
- Tímár, J.; Döme, B.; Fazekas, K.; Janovics, A.; Paku, S. Angiogenesis-dependent diseases and angiogenesis therapy. Pathol. Oncol. Res. 2001, 7, 85–94. [Google Scholar] [CrossRef] [PubMed]
- Vartanian, R.K.; Weidner, N. Correlation of intratumoral endothelial cell proliferation with microvessel density (tumor angiogenesis) and tumor cell proliferation in breast carcinoma. Am. J. Pathol. 1994, 144, 1188–1194. [Google Scholar] [PubMed]
- Weidner, N.; Semple, J.P.; Welch, W.R.; Folkman, J. Tumor angiogenesis and metastasis—Correlation in invasive breast carcinoma. N. Engl. J. Med. 1991, 324, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Carmeliet, P.; Jain, R.K. Molecular mechanisms and clinical applications of angiogenesis. Nature 2011, 473, 298–307. [Google Scholar] [CrossRef] [Green Version]
- Bodolay, E.; Koch, A.E.; Kim, J.; Szegedi, G.; Szekanecz, Z. Angiogenesis and chemokines in rheumatoid arthritis and other systemic inflammatory rheumatic diseases. J. Cell. Mol. Med. 2002, 6, 357–376. [Google Scholar] [CrossRef]
- Koch, A.E. Review: Angiogenesis: Implications for rheumatoid arthritis. Arthritis Rheum. 1998, 41, 951–962. [Google Scholar] [CrossRef]
- Szekanecz, Z.; Gáspár, L.; Koch, A.E. Angiogenesis in rheumatoid arthritis. Front. Biosci. 2005, 10, 1739–1753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, X.; Wang, J.; Deng, X.; Xiong, F.; Zhang, S.; Gong, Z.; Li, X.; Cao, K.; Deng, H.; He, Y.; et al. The role of microenvironment in tumor angiogenesis. J. Exp. Clin. Cancer Res. 2020, 39, 204. [Google Scholar] [CrossRef] [PubMed]
- Comunanza, V.; Bussolino, F. Therapy for Cancer: Strategy of Combining Anti-Angiogenic and Target Therapies. Front. Cell Dev. Biol. 2017, 5, 101. [Google Scholar] [CrossRef] [Green Version]
- Teleanu, R.I.; Chircov, C.; Grumezescu, A.M.; Teleanu, D.M. Tumor Angiogenesis and Anti-Angiogenic Strategies for Cancer Treatment. J. Clin. Med. 2019, 9, 84. [Google Scholar] [CrossRef] [Green Version]
- Rust, R.; Gantner, C.; Schwab, M.E. Pro- and antiangiogenic therapies: Current status and clinical implications. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2019, 33, 34–48. [Google Scholar] [CrossRef]
- Voss, V.; Senft, C.; Lang, V.; Ronellenfitsch, M.W.; Steinbach, J.P.; Seifert, V.; Kögel, D. The pan-Bcl-2 inhibitor (−)-gossypol triggers autophagic cell death in malignant glioma. Mol. Cancer Res. 2010, 8, 1002–1016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanahan, D.; Folkman, J. Patterns and emerging mechanisms of the angiogenic switch during tumorigenesis. Cell 1996, 86, 353–364. [Google Scholar] [CrossRef] [Green Version]
- Linderholm, B.; Tavelin, B.; Grankvist, K.; Henriksson, R. Does vascular endothelial growth factor (VEGF) predict local relapse and survival in radiotherapy-treated node-negative breast cancer? Br. J. Cancer 1999, 81, 727–732. [Google Scholar] [CrossRef] [Green Version]
- George, M.L.; Tutton, M.G.; Janssen, F.; Arnaout, A.; Abulafi, A.M.; Eccles, S.A.; Swift, R.I. VEGF-A, VEGF-C, and VEGF-D in colorectal cancer progression. Neoplasia 2001, 3, 420–427. [Google Scholar] [CrossRef] [Green Version]
- Ridiandries, A.; Tan, J.T.; Bursill, C.A. The Role of CC-Chemokines in the Regulation of Angiogenesis. Int. J. Mol. Sci. 2016, 17, 1856. [Google Scholar] [CrossRef] [Green Version]
- Szekanecz, Z.; Koch, A.E. Macrophages and their products in rheumatoid arthritis. Curr. Opin. Rheumatol. 2007, 19, 289–295. [Google Scholar] [CrossRef] [PubMed]
- Relf, M.; LeJeune, S.; Scott, P.A.; Fox, S.; Smith, K.; Leek, R.; Moghaddam, A.; Whitehouse, R.; Bicknell, R.; Harris, A.L. Expression of the angiogenic factors vascular endothelial cell growth factor, acidic and basic fibroblast growth factor, tumor growth factor beta-1, platelet-derived endothelial cell growth factor, placenta growth factor, and pleiotrophin in human primary breast cancer and its relation to angiogenesis. Cancer Res. 1997, 57, 963–969. [Google Scholar]
- Folkman, J. Angiogenesis in cancer, vascular, rheumatoid and other disease. Nat. Med. 1995, 1, 27–31. [Google Scholar] [CrossRef]
- Szekanecz, Z.; Koch, A.E. Chemokines and angiogenesis. Curr. Opin. Rheumatol. 2001, 13, 202–208. [Google Scholar] [CrossRef] [PubMed]
- Szekanecz, Z.; Koch, A.E. Mechanisms of Disease: Angiogenesis in inflammatory diseases. Nat. Clin. Pract. Rheumatol. 2007, 3, 635–643. [Google Scholar] [CrossRef]
- Walsh, D.A. Angiogenesis and arthritis. Rheumatology 1999, 38, 103–112. [Google Scholar] [CrossRef] [Green Version]
- Øynebråten, I.; Bakke, O.; Brandtzaeg, P.; Johansen, F.E.; Haraldsen, G. Rapid chemokine secretion from endothelial cells originates from 2 distinct compartments. Blood 2004, 104, 314–320. [Google Scholar] [CrossRef] [Green Version]
- Chan, A. Antiangiogenic therapy for metastatic breast cancer: Current status and future directions. Drugs 2009, 69, 167–181. [Google Scholar] [CrossRef]
- Fernandez, E.J.; Lolis, E. Structure, function, and inhibition of chemokines. Annu. Rev. Pharmacol. Toxicol. 2002, 42, 469–499. [Google Scholar] [CrossRef] [PubMed]
- Rossi, D.; Zlotnik, A. The biology of chemokines and their receptors. Annu. Rev. Immunol. 2000, 18, 217–242. [Google Scholar] [CrossRef]
- Förster, R.; Davalos-Misslitz, A.C.; Rot, A. CCR7 and its ligands: Balancing immunity and tolerance. Nat. Rev. Immunol. 2008, 8, 362–371. [Google Scholar] [CrossRef]
- Laurence, A.D. Location, movement and survival: The role of chemokines in haematopoiesis and malignancy. Br. J. Haematol. 2006, 132, 255–267. [Google Scholar] [CrossRef] [PubMed]
- Stamatovic, S.M.; Keep, R.F.; Mostarica-Stojkovic, M.; Andjelkovic, A.V. CCL2 regulates angiogenesis via activation of Ets-1 transcription factor. J. Immunol. 2006, 177, 2651–2661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hao, Q.; Vadgama, J.V.; Wang, P. CCL2/CCR2 signaling in cancer pathogenesis. Cell Commun. Signal. 2020, 18, 82. [Google Scholar] [CrossRef]
- Kadomoto, S.; Izumi, K.; Mizokami, A. Roles of CCL2-CCR2 Axis in the Tumor Microenvironment. Int. J. Mol. Sci. 2021, 22, 8530. [Google Scholar] [CrossRef] [PubMed]
- Brummer, G.; Fang, W.; Smart, C.; Zinda, B.; Alissa, N.; Berkland, C.; Miller, D.; Cheng, N. CCR2 signaling in breast carcinoma cells promotes tumor growth and invasion by promoting CCL2 and suppressing CD154 effects on the angiogenic and immune microenvironments. Oncogene 2020, 39, 2275–2289. [Google Scholar] [CrossRef]
- Gálvez, B.G.; Genís, L.; Matías-Román, S.; Oblander, S.A.; Tryggvason, K.; Apte, S.S.; Arroyo, A.G. Membrane type 1-matrix metalloproteinase is regulated by chemokines monocyte-chemoattractant protein-1/ccl2 and interleukin-8/CXCL8 in endothelial cells during angiogenesis. J. Biol. Chem. 2005, 280, 1292–1298. [Google Scholar] [CrossRef] [Green Version]
- Aguilar-Cazares, D.; Chavez-Dominguez, R.; Carlos-Reyes, A.; Lopez-Camarillo, C.; Hernadez de la Cruz, O.N.; Lopez-Gonzalez, J.S. Contribution of Angiogenesis to Inflammation and Cancer. Front. Oncol. 2019, 9, 1399. [Google Scholar] [CrossRef] [Green Version]
- Pausch, T.M.; Aue, E.; Wirsik, N.M.; Freire Valls, A.; Shen, Y.; Radhakrishnan, P.; Hackert, T.; Schneider, M.; Schmidt, T. Metastasis-associated fibroblasts promote angiogenesis in metastasized pancreatic cancer via the CXCL8 and the CCL2 axes. Sci. Rep. 2020, 10, 5420. [Google Scholar] [CrossRef]
- Guo, D.; Zhou, H.; Wu, Y.; Zhou, F.; Xu, G.; Wen, H.; Zhang, X. Involvement of ERK1/2/NF-κB signal transduction pathway in TF/FVIIa/PAR2-induced proliferation and migration of colon cancer cell SW620. Tumour Biol. 2011, 32, 921–930. [Google Scholar] [CrossRef]
- Charo, I.F.; Ransohoff, R.M. The many roles of chemokines and chemokine receptors in inflammation. N. Engl. J. Med. 2006, 354, 610–621. [Google Scholar] [CrossRef] [PubMed]
- Weyrich, A.S.; McIntyre, T.M.; McEver, R.P.; Prescott, S.M.; Zimmerman, G.A. Monocyte tethering by P-selectin regulates monocyte chemotactic protein-1 and tumor necrosis factor-alpha secretion. Signal integration and NF-kappa B translocation. J. Clin. Investig. 1995, 95, 2297–2303. [Google Scholar] [CrossRef] [PubMed]
- Werts, C.; le Bourhis, L.; Liu, J.; Magalhaes, J.G.; Carneiro, L.A.; Fritz, J.H.; Stockinger, S.; Balloy, V.; Chignard, M.; Decker, T.; et al. Nod1 and Nod2 induce CCL5/RANTES through the NF-kappaB pathway. Eur. J. Immunol. 2007, 37, 2499–2508. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, S.; Matsuda, M.; Hou, Z.; Bailey, J.P.; Kitazawa, R.; Herbst, M.P.; Horseman, N.D. Receptor activator of NF-kappaB ligand induction via Jak2 and Stat5a in mammary epithelial cells. J. Biol. Chem. 2003, 278, 46171–46178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cabebe, E.; Wakelee, H. Role of anti-angiogenesis agents in treating NSCLC: Focus on bevacizumab and VEGFR tyrosine kinase inhibitors. Curr. Treat. Options Oncol. 2007, 8, 15–27. [Google Scholar] [CrossRef]
- Mourad, J.J.; des Guetz, G.; Debbabi, H.; Levy, B.I. Blood pressure rise following angiogenesis inhibition by bevacizumab. A crucial role for microcirculation. Ann. Oncol. 2008, 19, 927–934. [Google Scholar] [CrossRef] [PubMed]
- Thiery, J.P.; Sastre-Garau, X.; Vincent-Salomon, B.; Sigal-Zafrani, X.; Pierga, J.Y.; Decraene, C.; Meyniel, J.P.; Gravier, E.; Asselain, B.; De Rycke, Y.; et al. Challenges in the stratification of breast tumors for tailored therapies. Bull. Cancer 2006, 93, E81–E89. [Google Scholar]
- Albini, A.; Noonan, D.M.; Ferrari, N. Molecular pathways for cancer angioprevention. Clin. Cancer Res. 2007, 13, 4320–4325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, C.; Guan, G.; Wang, H. The Anticancer Effect of Sanguinarine: A Review. Curr. Pharm. Des. 2018, 24, 2760–2764. [Google Scholar] [CrossRef]
- Mackraj, I.; Govender, T.; Gathiram, P. Sanguinarine. Cardiovasc. Ther. 2008, 26, 75–83. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Mao, K.; Gu, Q.; Wu, X. The Antiangiogenic Effect of Sanguinarine Chloride on Experimental Choroidal Neovacularization in Mice via Inhibiting Vascular Endothelial Growth Factor. Front. Pharmacol. 2021, 12, 638215. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.; Liu, C.; Nadiminty, N.; Lou, W.; Zhu, Y.; Yang, J.; Evans, C.P.; Zhou, Q.; Gao, A.C. Inhibition of Stat3 activation by sanguinarine suppresses prostate cancer cell growth and invasion. Prostate 2012, 72, 82–89. [Google Scholar] [CrossRef] [Green Version]
- De Stefano, I.; Raspaglio, G.; Zannoni, G.F.; Travaglia, D.; Prisco, M.G.; Mosca, M.; Ferlini, C.; Scambia, G.; Gallo, D. Antiproliferative and antiangiogenic effects of the benzophenanthridine alkaloid sanguinarine in melanoma. Biochem. Pharmacol. 2009, 78, 1374–1381. [Google Scholar] [CrossRef] [Green Version]
- Jang, B.C.; Park, J.G.; Song, D.K.; Baek, W.K.; Yoo, S.K.; Jung, K.H.; Park, G.Y.; Lee, T.Y.; Suh, S.I. Sanguinarine induces apoptosis in A549 human lung cancer cells primarily via cellular glutathione depletion. Toxicol. Vitro 2009, 23, 281–287. [Google Scholar] [CrossRef] [PubMed]
- Tsukamoto, H.; Kondo, S.; Mukudai, Y.; Nagumo, T.; Yasuda, A.; Kurihara, Y.; Kamatani, T.; Shintani, S. Evaluation of anticancer activities of benzo[c]phenanthridine alkaloid sanguinarine in oral squamous cell carcinoma cell line. Anticancer Res. 2011, 31, 2841–2846. [Google Scholar] [PubMed]
- Xu, J.Y.; Meng, Q.H.; Chong, Y.; Jiao, Y.; Zhao, L.; Rosen, E.M.; Fan, S. Sanguinarine is a novel VEGF inhibitor involved in the suppression of angiogenesis and cell migration. Mol. Clin. Oncol. 2013, 1, 331–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaziano, R.; Moroni, G.; Buè, C.; Miele, M.T.; Sinibaldi-Vallebona, P.; Pica, F. Antitumor effects of the benzophenanthridine alkaloid sanguinarine: Evidence and perspectives. World J. Gastrointest. Oncol. 2016, 8, 30–39. [Google Scholar] [CrossRef]
- Achkar, I.W.; Mraiche, F.; Mohammad, R.M.; Uddin, S. Anticancer potential of sanguinarine for various human malignancies. Future Med. Chem. 2017, 9, 933–950. [Google Scholar] [CrossRef]
- Pica, F.; Balestrieri, E.; Serafino, A.; Sorrentino, R.; Gaziano, R.; Moroni, G.; Moroni, N.; Palmieri, G.; Mattei, M.; Garaci, E.; et al. Antitumor effects of the benzophenanthridine alkaloid sanguinarine in a rat syngeneic model of colorectal cancer. Anticancer Drugs 2012, 23, 32–42. [Google Scholar] [CrossRef]
- Galadari, S.; Rahman, A.; Pallichankandy, S.; Thayyullathil, F. Molecular targets and anticancer potential of sanguinarine-a benzophenanthridine alkaloid. Phytomedicine 2017, 34, 143–153. [Google Scholar] [CrossRef]
- Eun, J.P.; Koh, G.Y. Suppression of angiogenesis by the plant alkaloid, sanguinarine. Biochem. Biophys. Res. Commun. 2004, 317, 618–624. [Google Scholar] [CrossRef] [PubMed]
- Basini, G.; Bussolati, S.; Santini, S.E.; Grasselli, F. Sanguinarine inhibits VEGF-induced angiogenesis in a fibrin gel matrix. Biofactors 2007, 29, 11–18. [Google Scholar] [CrossRef]
- Basini, G.; Santini, S.E.; Bussolati, S.; Grasselli, F. The plant alkaloid sanguinarine is a potential inhibitor of follicular angiogenesis. J. Reprod. Dev. 2007, 53, 573–579. [Google Scholar] [CrossRef] [Green Version]
- Carmeliet, P. Angiogenesis in health and disease. Nat. Med. 2003, 9, 653–660. [Google Scholar] [CrossRef]
- Li, Y.; Wu, Z.; Yuan, J.; Sun, L.; Lin, L.; Huang, N.; Bin, J.; Liao, Y.; Liao, W. Long non-coding RNA MALAT1 promotes gastric cancer tumorigenicity and metastasis by regulating vasculogenic mimicry and angiogenesis. Cancer Lett. 2017, 395, 31–44. [Google Scholar] [CrossRef]
- Shen, J.; Zhou, S.; Shi, L.; Liu, X.; Lin, H.; Yu, H.; Xiaoliang; Tang, J.; Yu, T.; Cai, X. DUSP1 inhibits cell proliferation, metastasis and invasion and angiogenesis in gallbladder cancer. Oncotarget 2017, 8, 12133–12144. [Google Scholar] [CrossRef] [Green Version]
- Xu, H.; Zhang, Y.; Peña, M.M.; Pirisi, L.; Creek, K.E. Six1 promotes colorectal cancer growth and metastasis by stimulating angiogenesis and recruiting tumor-associated macrophages. Carcinogenesis 2017, 38, 281–292. [Google Scholar] [CrossRef] [Green Version]
- Fan, T.P.; Jaggar, R.; Bicknell, R. Controlling the vasculature: Angiogenesis, anti-angiogenesis and vascular targeting of gene therapy. Trends Pharmacol. Sci. 1995, 16, 57–66. [Google Scholar] [CrossRef]
- Jayson, G.C.; Kerbel, R.; Ellis, L.M.; Harris, A.L. Antiangiogenic therapy in oncology: Current status and future directions. Lancet 2016, 388, 518–529. [Google Scholar] [CrossRef]
- Munir, S.; Shah, A.A.; Shahid, M.; Ahmed, M.S.; Shahid, A.; Riaz Rajoka, M.S.; Hamid Akash, M.S.; Akram, M.; Khurshid, M. Anti-angiogenesis potential of phytochemicals for the therapeutic management of tumors. Curr. Pharm. Des. 2019, 26, 265–278. [Google Scholar] [CrossRef]
- Bauer, D.; Redmon, N.; Mazzio, E.; Soliman, K.F. Apigenin inhibits TNFα/IL-1α-induced CCL2 release through IKBK-epsilon signaling in MDA-MB-231 human breast cancer cells. PLoS ONE 2017, 12, e0175558. [Google Scholar] [CrossRef]
- Messeha, S.S.; Zarmouh, N.O.; Mendonca, P.; Alwagdani, H.; Kolta, M.G.; Soliman, K.F.A. The inhibitory effects of plumbagin on the NF-қB pathway and CCL2 release in racially different triple-negative breast cancer cells. PLoS ONE 2018, 13, e0201116. [Google Scholar] [CrossRef] [Green Version]
- Mendonca, P.; Horton, A.; Bauer, D.; Messeha, S.; Soliman, K.F.A. The inhibitory effects of butein on cell proliferation and TNF-α-induced CCL2 release in racially different triple negative breast cancer cells. PLoS ONE 2019, 14, e0215269. [Google Scholar] [CrossRef] [Green Version]
- Huth, H.W.; Santos, D.M.; Gravina, H.D.; Resende, J.M.; Goes, A.M.; de Lima, M.E.; Ropert, C. Upregulation of p38 pathway accelerates proliferation and migration of MDA-MB-231 breast cancer cells. Oncol. Rep. 2017, 37, 2497–2505. [Google Scholar] [CrossRef] [Green Version]
- Boehm, J.S.; Zhao, J.J.; Yao, J.; Kim, S.Y.; Firestein, R.; Dunn, I.F.; Sjostrom, S.K.; Garraway, L.A.; Weremowicz, S.; Richardson, A.L.; et al. Integrative genomic approaches identify IKBKE as a breast cancer oncogene. Cell 2007, 129, 1065–1079. [Google Scholar] [CrossRef] [Green Version]
- Hoeben, A.; Landuyt, B.; Highley, M.S.; Wildiers, H.; Van Oosterom, A.T.; De Bruijn, E.A. Vascular endothelial growth factor and angiogenesis. Pharmacol. Rev. 2004, 56, 549–580. [Google Scholar] [CrossRef]
- Giraudo, E.; Primo, L.; Audero, E.; Gerber, H.P.; Koolwijk, P.; Soker, S.; Klagsbrun, M.; Ferrara, N.; Bussolino, F. Tumor necrosis factor-alpha regulates expression of vascular endothelial growth factor receptor-2 and of its co-receptor neuropilin-1 in human vascular endothelial cells. J. Biol. Chem. 1998, 273, 22128–22135. [Google Scholar] [CrossRef] [Green Version]
- Shin, M.R.; Kang, S.K.; Kim, Y.S.; Lee, S.Y.; Hong, S.C.; Kim, E.C. TNF-α and LPS activate angiogenesis via VEGF and SIRT1 signalling in human dental pulp cells. Int. Endod. J. 2015, 48, 705–716. [Google Scholar] [CrossRef]
- Mu, H.Q.; He, Y.H.; Wang, S.B.; Yang, S.; Wang, Y.J.; Nan, C.J.; Bao, Y.F.; Xie, Q.P.; Chen, Y.H. MiR-130b/TNF-α/NF-κB/VEGFA loop inhibits prostate cancer angiogenesis. Clin. Transl. Oncol. 2020, 22, 111–121. [Google Scholar] [CrossRef]
- Leek, R.D.; Landers, R.; Fox, S.B.; Ng, F.; Harris, A.L.; Lewis, C.E. Association of tumour necrosis factor alpha and its receptors with thymidine phosphorylase expression in invasive breast carcinoma. Br. J. Cancer 1998, 77, 2246–2251. [Google Scholar] [CrossRef] [Green Version]
- Baumgarten, S.C.; Frasor, J. Minireview: Inflammation: An instigator of more aggressive estrogen receptor (ER) positive breast cancers. Mol. Endocrinol. 2012, 26, 360–371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, Y.; Chen, X.; Shu, Y. Gene expression of the invasive phenotype of TNF-alpha-treated MCF-7 cells. Biomed. Pharmacother. 2009, 63, 421–428. [Google Scholar] [CrossRef]
- Ueno, T.; Toi, M.; Saji, H.; Muta, M.; Bando, H.; Kuroi, K.; Koike, M.; Inadera, H.; Matsushima, K. Significance of macrophage chemoattractant protein-1 in macrophage recruitment, angiogenesis, and survival in human breast cancer. Clin. Cancer Res. 2000, 6, 3282–3289. [Google Scholar] [PubMed]
- Messeha, S.S.; Zarmouh, N.O.; Mendonca, P.; Cotton, C.; Soliman, K.F.A. Molecular mechanism of gossypol mediating CCL2 and IL-8 attenuation in triple-negative breast cancer cells. Mol. Med. Rep. 2020, 22, 1213–1226. [Google Scholar] [CrossRef]
- Saji, H.; Koike, M.; Yamori, T.; Saji, S.; Seiki, M.; Matsushima, K.; Toi, M. Significant correlation of monocyte chemoattractant protein-1 expression with neovascularization and progression of breast carcinoma. Cancer 2001, 92, 1085–1091. [Google Scholar] [CrossRef]
- Balkwill, F. TNF-alpha in promotion and progression of cancer. Cancer Metastasis Rev. 2006, 25, 409–416. [Google Scholar] [CrossRef]
- Matsushima, K.; Larsen, C.G.; DuBois, G.C.; Oppenheim, J.J. Purification and characterization of a novel monocyte chemotactic and activating factor produced by a human myelomonocytic cell line. J. Exp. Med. 1989, 169, 1485–1490. [Google Scholar] [CrossRef] [Green Version]
- Goede, V.; Brogelli, L.; Ziche, M.; Augustin, H.G. Induction of inflammatory angiogenesis by monocyte chemoattractant protein-1. Int. J. Cancer 1999, 82, 765–770. [Google Scholar] [CrossRef]
- Fang, W.B.; Yao, M.; Brummer, G.; Acevedo, D.; Alhakamy, N.; Berkland, C.; Cheng, N. Targeted gene silencing of CCL2 inhibits triple negative breast cancer progression by blocking cancer stem cell renewal and M2 macrophage recruitment. Oncotarget 2016, 7, 49349–49367. [Google Scholar] [CrossRef]
- Espinoza-Sánchez, N.A.; Chimal-Ramírez, G.K.; Mantilla, A.; Fuentes-Pananá, E.M. IL-1β, IL-8, and Matrix Metalloproteinases-1, -2, and -10 Are Enriched upon Monocyte-Breast Cancer Cell Cocultivation in a Matrigel-Based Three-Dimensional System. Front. Immunol. 2017, 8, 205. [Google Scholar] [CrossRef] [Green Version]
- Barcelos, L.S.; Talvani, A.; Teixeira, A.S.; Cassali, G.D.; Andrade, S.P.; Teixeira, M.M. Production and in vivo effects of chemokines CXCL1-3/KC and CCL2/JE in a model of inflammatory angiogenesis in mice. Inflamm. Res. 2004, 53, 576–584. [Google Scholar] [CrossRef]
- Nam, J.S.; Kang, M.J.; Suchar, A.M.; Shimamura, T.; Kohn, E.A.; Michalowska, A.M.; Jordan, V.C.; Hirohashi, S.; Wakefield, L.M. Chemokine (C-C motif) ligand 2 mediates the prometastatic effect of dysadherin in human breast cancer cells. Cancer Res. 2006, 66, 7176–7184. [Google Scholar] [CrossRef] [Green Version]
- Qian, B.Z.; Li, J.; Zhang, H.; Kitamura, T.; Zhang, J.; Campion, L.R.; Kaiser, E.A.; Snyder, L.A.; Pollard, J.W. CCL2 recruits inflammatory monocytes to facilitate breast-tumour metastasis. Nature 2011, 475, 222–225. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, Y.; Gaynor, R.B. Therapeutic potential of inhibition of the NF-kappaB pathway in the treatment of inflammation and cancer. J. Clin. Investig. 2001, 107, 135–142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peters, R.T.; Liao, S.M.; Maniatis, T. IKKepsilon is part of a novel PMA-inducible IkappaB kinase complex. Mol. Cell 2000, 5, 513–522. [Google Scholar] [CrossRef]
- Harris, J.; Olière, S.; Sharma, S.; Sun, Q.; Lin, R.; Hiscott, J.; Grandvaux, N. Nuclear accumulation of cRel following C-terminal phosphorylation by TBK1/IKK epsilon. J. Immunol. 2006, 177, 2527–2535. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, S.; Yoshida, A.; Ishibashi, T. Induction of IL-8, MCP-1, and bFGF by TNF-alpha in retinal glial cells: Implications for retinal neovascularization during post-ischemic inflammation. Graefes Arch. Clin. Exp. Ophthalmol. 2004, 242, 409–413. [Google Scholar] [CrossRef]
- Yin, M.; Wang, X.; Lu, J. Advances in IKBKE as a potential target for cancer therapy. Cancer Med. 2020, 9, 247–258. [Google Scholar] [CrossRef]
- Williams, V.; Grosset, A.A.; Zamorano Cuervo, N.; St-Pierre, Y.; Sylvestre, M.P.; Gaboury, L.; Grandvaux, N. Detection of IKKε by immunohistochemistry in primary breast cancer: Association with EGFR expression and absence of lymph node metastasis. BMC Cancer 2017, 17, 356. [Google Scholar] [CrossRef] [Green Version]
- Tang, X.; Jin, L.; Cao, P.; Cao, K.; Huang, C.; Luo, Y.; Ma, J.; Shen, S.; Tan, M.; Li, X.; et al. MicroRNA-16 sensitizes breast cancer cells to paclitaxel through suppression of IKBKB expression. Oncotarget 2016, 7, 23668–23683. [Google Scholar] [CrossRef] [Green Version]
- Guo, J.P.; Shu, S.K.; Esposito, N.N.; Coppola, D.; Koomen, J.M.; Cheng, J.Q. IKKϵ phosphorylation of estrogen receptor α Ser-167 and contribution to tamoxifen resistance in breast cancer. J. Biol. Chem. 2016, 291, 22857. [Google Scholar] [CrossRef] [Green Version]
- Qin, B.; Cheng, K. Silencing of the IKKε gene by siRNA inhibits invasiveness and growth of breast cancer cells. Breast Cancer Res. 2010, 12, R74. [Google Scholar] [CrossRef] [Green Version]
- Barbie, T.U.; Alexe, G.; Aref, A.R.; Li, S.; Zhu, Z.; Zhang, X.; Imamura, Y.; Thai, T.C.; Huang, Y.; Bowden, M.; et al. Targeting an IKBKE cytokine network impairs triple-negative breast cancer growth. J. Clin. Investig. 2014, 124, 5411–5423. [Google Scholar] [CrossRef]
- Fang, W.B.; Jokar, I.; Zou, A.; Lambert, D.; Dendukuri, P.; Cheng, N. CCL2/CCR2 chemokine signaling coordinates survival and motility of breast cancer cells through Smad3 protein- and p42/44 mitogen-activated protein kinase (MAPK)-dependent mechanisms. J. Biol. Chem. 2012, 287, 36593–36608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carmeliet, P. Mechanisms of angiogenesis and arteriogenesis. Nat. Med. 2000, 6, 389–395. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Yuan, H.B.; Zhao, S.; Liu, F.F.; Jiang, Y.Q.; Guo, Y.X.; Wang, X.L. Activation of GABA(B) Receptor Suppresses Diabetic Neuropathic Pain through Toll-Like Receptor 4 Signaling Pathway in the Spinal Dorsal Horn. Mediat. Inflamm. 2018, 2018, 6016272. [Google Scholar] [CrossRef] [Green Version]
- Xia, Y.; Shen, S.; Verma, I.M. NF-κB, an active player in human cancers. Cancer Immunol. Res. 2014, 2, 823–830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giridharan, S.; Srinivasan, M. Mechanisms of NF-κB p65 and strategies for therapeutic manipulation. J. Inflamm. Res. 2018, 11, 407–419. [Google Scholar] [CrossRef] [Green Version]
- Adli, M.; Baldwin, A.S. IKK-i/IKKepsilon controls constitutive, cancer cell-associated NF-kappaB activity via regulation of Ser-536 p65/RelA phosphorylation. J. Biol. Chem. 2006, 281, 26976–26984. [Google Scholar] [CrossRef] [Green Version]
- Mattioli, I.; Geng, H.; Sebald, A.; Hodel, M.; Bucher, C.; Kracht, M.; Schmitz, M.L. Inducible phosphorylation of NF-kappa B p65 at serine 468 by T cell costimulation is mediated by IKK epsilon. J. Biol. Chem. 2006, 281, 6175–6183. [Google Scholar] [CrossRef] [Green Version]
- Dreesen, O.; Brivanlou, A.H. Signaling pathways in cancer and embryonic stem cells. Stem Cell Rev. 2007, 3, 7–17. [Google Scholar] [CrossRef]
- Huang, F.; Xiong, X.; Wang, H.; You, S.; Zeng, H. Leptin-induced vascular smooth muscle cell proliferation via regulating cell cycle, activating ERK1/2 and NF-kappaB. Acta Biochim. Biophys. Sin. 2010, 42, 325–331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moon, S.K.; Cha, B.Y.; Kim, C.H. ERK1/2 mediates TNF-alpha-induced matrix metalloproteinase-9 expression in human vascular smooth muscle cells via the regulation of NF-kappaB and AP-1: Involvement of the ras dependent pathway. J. Cell Physiol. 2004, 198, 417–427. [Google Scholar] [CrossRef] [PubMed]
- Bartholomeusz, C.; Gonzalez-Angulo, A.M.; Liu, P.; Hayashi, N.; Lluch, A.; Ferrer-Lozano, J.; Hortobágyi, G.N. High ERK protein expression levels correlate with shorter survival in triple-negative breast cancer patients. Oncologist 2012, 17, 766–774. [Google Scholar] [CrossRef] [Green Version]
- Seddighzadeh, M.; Zhou, J.N.; Kronenwett, U.; Shoshan, M.C.; Auer, G.; Sten-Linder, M.; Wiman, B.; Linder, S. ERK signalling in metastatic human MDA-MB-231 breast carcinoma cells is adapted to obtain high urokinase expression and rapid cell proliferation. Clin. Exp. Metastasis 1999, 17, 649–654. [Google Scholar] [CrossRef] [PubMed]
- Wen, S.; Hou, Y.; Fu, L.; Xi, L.; Yang, D.; Zhao, M.; Qin, Y.; Sun, K.; Teng, Y.; Liu, M. Cancer-associated fibroblast (CAF)-derived IL32 promotes breast cancer cell invasion and metastasis via integrin β3-p38 MAPK signalling. Cancer Lett. 2019, 442, 320–332. [Google Scholar] [CrossRef]
- Samatar, A.A.; Poulikakos, P.I. Targeting RAS-ERK signalling in cancer: Promises and challenges. Nat. Rev. Drug Discov. 2014, 13, 928–942. [Google Scholar] [CrossRef]
- Kyriakis, J.M.; Avruch, J. Mammalian MAPK signal transduction pathways activated by stress and inflammation: A 10-year update. Physiol. Rev. 2012, 92, 689–737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cargnello, M.; Roux, P.P. Activation and function of the MAPKs and their substrates, the MAPK-activated protein kinases. Microbiol. Mol. Biol. Rev. 2011, 75, 50–83. [Google Scholar] [CrossRef] [Green Version]
- Lim, S.Y.; Yuzhalin, A.E.; Gordon-Weeks, A.N.; Muschel, R.J. Targeting the CCL2-CCR2 signaling axis in cancer metastasis. Oncotarget 2016, 7, 28697–28710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiu, H.Y.; Sun, K.H.; Chen, S.Y.; Wang, H.H.; Lee, M.Y.; Tsou, Y.C.; Jwo, S.C.; Sun, G.H.; Tang, S.J. Autocrine CCL2 promotes cell migration and invasion via PKC activation and tyrosine phosphorylation of paxillin in bladder cancer cells. Cytokine 2012, 59, 423–432. [Google Scholar] [CrossRef] [PubMed]
- Monti, P.; Leone, B.E.; Marchesi, F.; Balzano, G.; Zerbi, A.; Scaltrini, F.; Pasquali, C.; Calori, G.; Pessi, F.; Sperti, C.; et al. The CC chemokine MCP-1/CCL2 in pancreatic cancer progression: Regulation of expression and potential mechanisms of antimalignant activity. Cancer Res. 2003, 63, 7451–7461. [Google Scholar] [PubMed]
- Kitamura, T.; Qian, B.Z.; Soong, D.; Cassetta, L.; Noy, R.; Sugano, G.; Kato, Y.; Li, J.; Pollard, J.W. CCL2-induced chemokine cascade promotes breast cancer metastasis by enhancing retention of metastasis-associated macrophages. J. Exp. Med. 2015, 212, 1043–1059. [Google Scholar] [CrossRef] [PubMed]
- Salcedo, R.; Ponce, M.L.; Young, H.A.; Wasserman, K.; Ward, J.M.; Kleinman, H.K.; Oppenheim, J.J.; Murphy, W.J. Human endothelial cells express CCR2 and respond to MCP-1: Direct role of MCP-1 in angiogenesis and tumor progression. Blood 2000, 96, 34–40. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.R.; Kumari, N.; Thi Vu, H.; Kim, H.; Park, C.K.; Choi, S.H. Increased Antiangiogenic Effect by Blocking CCL2-dependent Macrophages in a Rodent Glioblastoma Model: Correlation Study with Dynamic Susceptibility Contrast Perfusion MRI. Sci. Rep. 2019, 9, 11085. [Google Scholar] [CrossRef]
- Geiger, T.R.; Peeper, D.S. Metastasis mechanisms. Biochim. Biophys. Acta 2009, 1796, 293–308. [Google Scholar] [CrossRef] [PubMed]
- Aziz, M.N.M.; Rahim, N.F.C.; Hussin, Y.; Yeap, S.K.; Masarudin, M.J.; Mohamad, N.E.; Akhtar, M.N.; Osman, M.A.; Cheah, Y.K.; Alitheen, N.B. Anti-Metastatic and Anti-Angiogenic Effects of Curcumin Analog DK1 on Human Osteosarcoma Cells In Vitro. Pharmaceuticals 2021, 14, 532. [Google Scholar] [CrossRef]
- Kozłowski, J.; Kozłowska, A.; Kocki, J. Breast cancer metastasis—Insight into selected molecular mechanisms of the phenomenon. Postepy Hig. Med. Dosw. 2015, 69, 447–451. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Spezia, M.; Huang, S.; Yuan, C.; Zeng, Z.; Zhang, L.; Ji, X.; Liu, W.; Huang, B.; Luo, W.; et al. Breast cancer development and progression: Risk factors, cancer stem cells, signaling pathways, genomics, and molecular pathogenesis. Genes Dis. 2018, 5, 77–106. [Google Scholar] [CrossRef]
- Peart, O. Metastatic Breast Cancer. Radiol. Technol. 2017, 88, 519m–539m. [Google Scholar]
- Scully, O.J.; Bay, B.H.; Yip, G.; Yu, Y. Breast cancer metastasis. Cancer Genom. Proteom. 2012, 9, 311–320. [Google Scholar]
- Messeha, S.S.; Zarmouh, N.O.; Asiri, A.; Soliman, K.F.A. Gene Expression Alterations Associated with Oleuropein-Induced Antiproliferative Effects and S-Phase Cell Cycle Arrest in Triple-Negative Breast Cancer Cells. Nutrients 2020, 12, 3755. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer Name | GenBank Accession No. | Amplicon Context Sequence |
|---|---|---|
| MCP1/CCL2 | NM_002982.4 | ACTGAAGCTCGCACTCTCGCCTCCAGCATGAAAGTCTCTGCCGCCCTTCTGTGCCTGCTGCTCATAGCAGCCACCTTCATTCCCCAAGGGCTCGCTCAGCCAGATGCAATCAATGCCCCAGTCACCTGCTGTTATAACTTCACCAATAGGAAGATCTCAGTGCAGAGGCTCGCGAGCTAT |
| IKBKE | NM_014002.4 | GGCTTGGCTACAACGAGGAGCAGATTCACAAGCTGGATAAGGTGAATTTCAGTCATTTAGCCAAAAGACTCCTGCAGGTGTTCCAGGAGGAGTGCGTGCAGAAGTATCAAGCGTCCTTAGTCACACACGGCAAGAGGATGAGGGTGGTGCACGAG |
| GAPDH | NM_002046.7 | GTATGACAACGAATTTGGCTACAGCAACAGGGTGGTGGACCTCATGGCCCACATGGCCTCCAAGGAGTAAGACCCCTGGACCACCAGCCCCAGCAAGAGCACAAGAGGAAGAGAGAGACCCTCACTGCTGGGGAGTCCCTGCCACAC |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Messeha, S.S.; Zarmouh, N.O.; Antonie, L.; Soliman, K.F.A. Sanguinarine Inhibition of TNF-α-Induced CCL2, IKBKE/NF-κB/ERK1/2 Signaling Pathway, and Cell Migration in Human Triple-Negative Breast Cancer Cells. Int. J. Mol. Sci. 2022, 23, 8329. https://doi.org/10.3390/ijms23158329
Messeha SS, Zarmouh NO, Antonie L, Soliman KFA. Sanguinarine Inhibition of TNF-α-Induced CCL2, IKBKE/NF-κB/ERK1/2 Signaling Pathway, and Cell Migration in Human Triple-Negative Breast Cancer Cells. International Journal of Molecular Sciences. 2022; 23(15):8329. https://doi.org/10.3390/ijms23158329
Chicago/Turabian StyleMesseha, Samia S., Najla O. Zarmouh, Lovely Antonie, and Karam F. A. Soliman. 2022. "Sanguinarine Inhibition of TNF-α-Induced CCL2, IKBKE/NF-κB/ERK1/2 Signaling Pathway, and Cell Migration in Human Triple-Negative Breast Cancer Cells" International Journal of Molecular Sciences 23, no. 15: 8329. https://doi.org/10.3390/ijms23158329
APA StyleMesseha, S. S., Zarmouh, N. O., Antonie, L., & Soliman, K. F. A. (2022). Sanguinarine Inhibition of TNF-α-Induced CCL2, IKBKE/NF-κB/ERK1/2 Signaling Pathway, and Cell Migration in Human Triple-Negative Breast Cancer Cells. International Journal of Molecular Sciences, 23(15), 8329. https://doi.org/10.3390/ijms23158329