
Statins Neuromuscular Adverse Effects


Abstract
:1. Introduction
2. Statins and Skeletal Muscle Adverse Effects
3. Pathogenesis of SAMS
4. Genetic Origin of SAMS
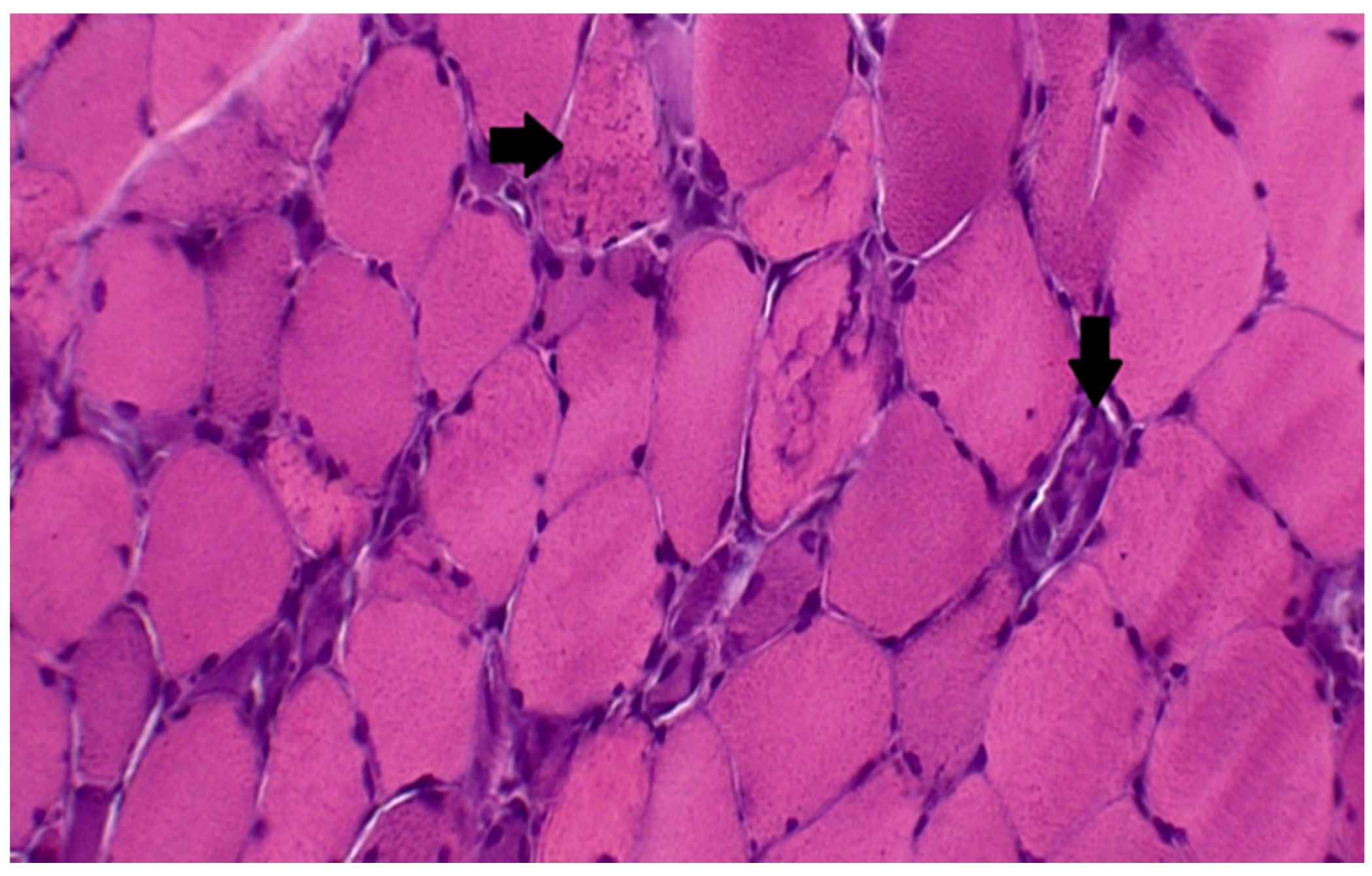
5. Classification of Statin-Related Myopathic Phenotypes
6. Statins and Peripheral Neuropathy
7. Statins and Autoimmune Illnesses
7.1. Statins and Myasthenia Gravis
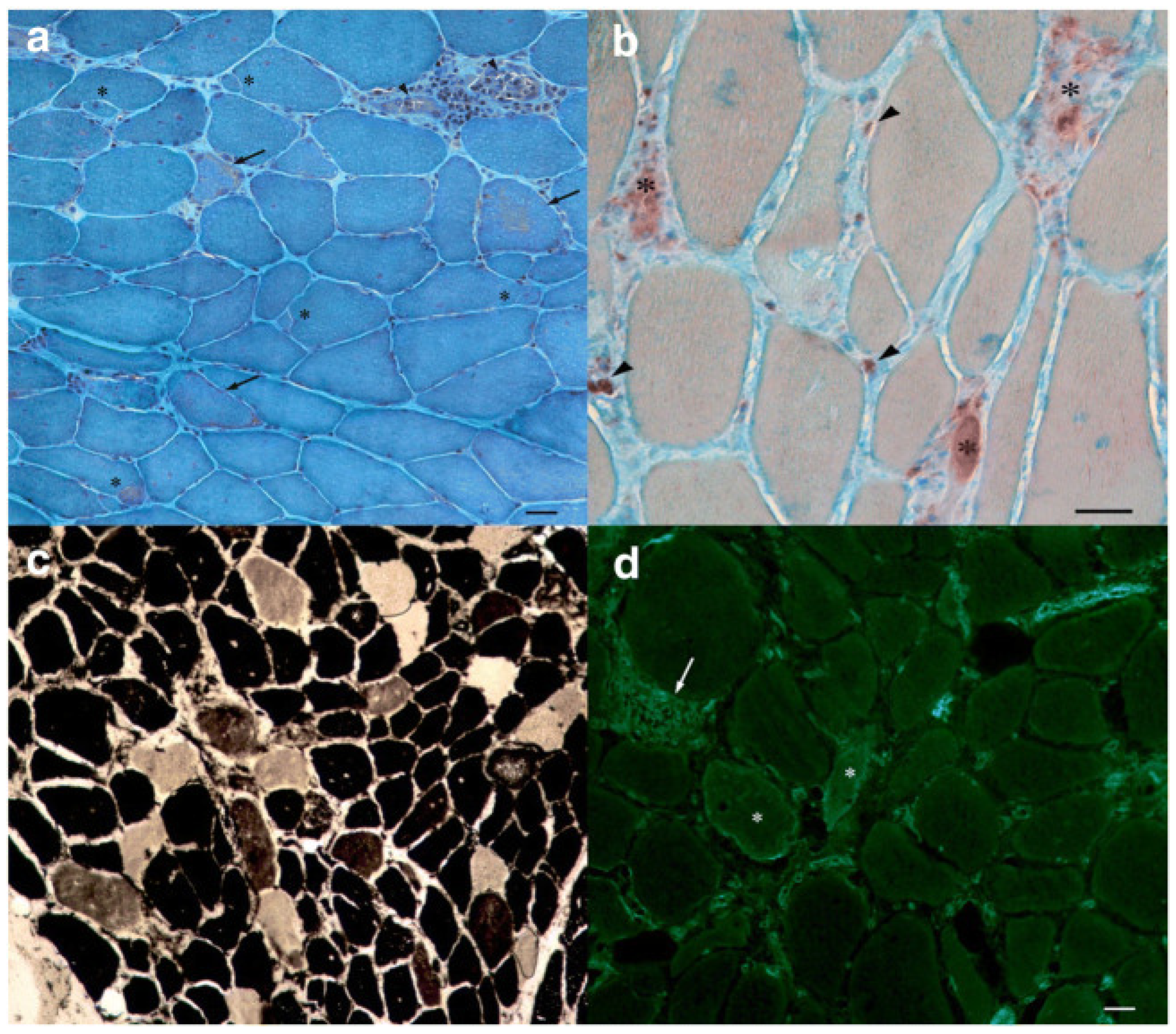
7.2. Immune-Mediated Necrotizing Myopathy Statin-Related
7.3. Treatment of IMNM
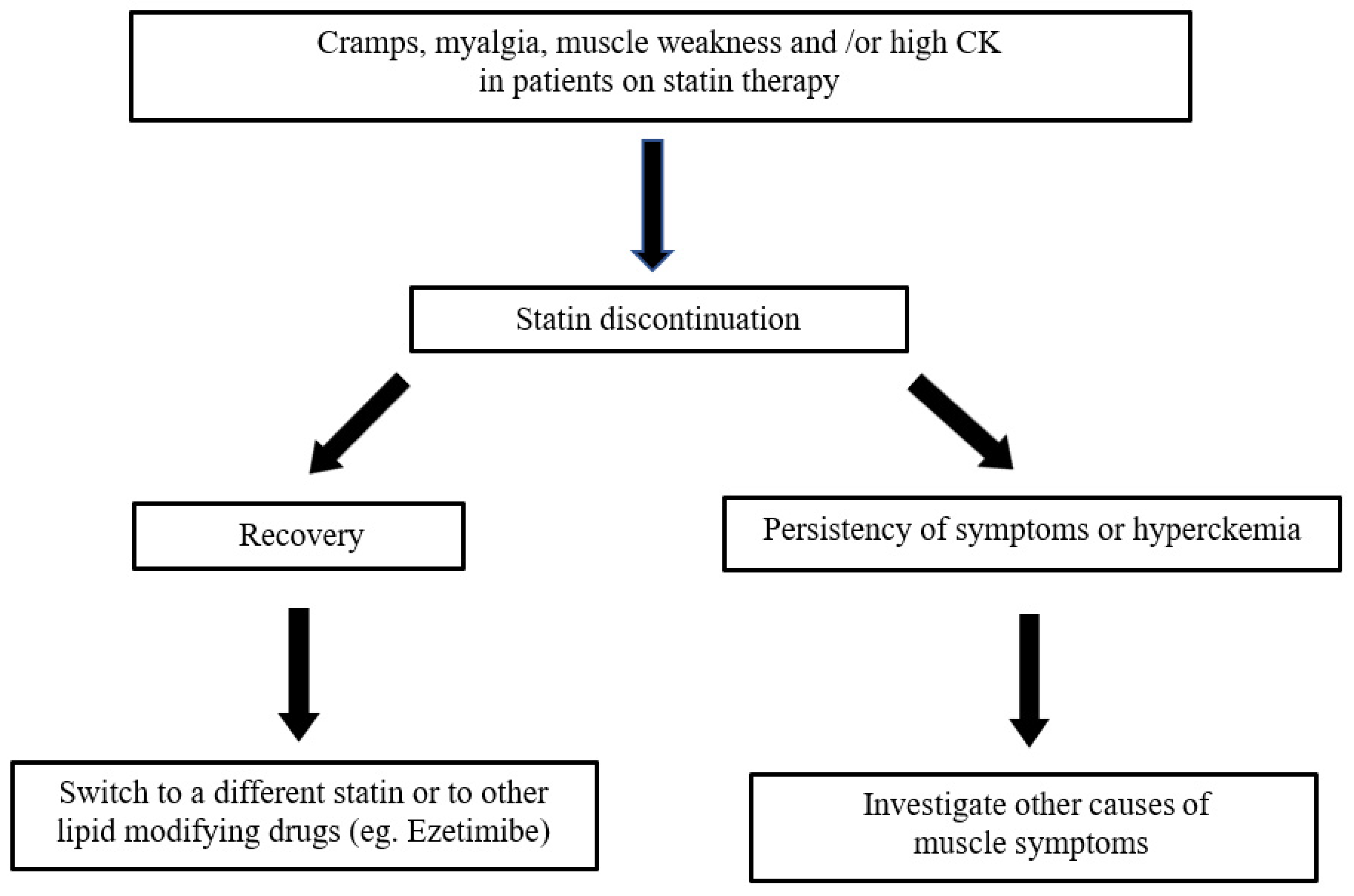
8. Management of Statin Adverse Effects
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mohammadkhani, N.; Gharbi, S.; Rajani, H.F.; Farzaneh, A.; Mahjoob, G.; Hoseinsalari, A.; Korsching, E. Statins: Complex outcomes but increasingly helpful treatment options for patients. Eur. J. Pharm. 2019, 863, 172704. [Google Scholar] [CrossRef] [PubMed]
- Sirtori, C.R. The pharmacology of statins. Pharmacol. Res. 2014, 88, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Oesterle, A.; Liao, J.K. The Pleiotropic Effects of Statins—From Coronary Artery Disease and Stroke to Atrial Fibrillation and Ventricular Tachyarrhythmia. Curr. Vasc. Pharmacol. 2019, 17, 222–232. [Google Scholar] [CrossRef] [PubMed]
- Perez-Matos, M.C.; Morales-Alvarez, M.C.; Mendivil, C.O. Lipids: A Suitable Therapeutic Target in Diabetic Neuropathy? J. Diabetes Res. 2017, 2017, 6943851. [Google Scholar] [CrossRef] [PubMed]
- Schachter, M. Chemical, pharmacokinetic and pharmacodynamic properties of statins: An update. Fundam. Clin. Pharmacol. 2005, 19, 117–125. [Google Scholar] [CrossRef]
- Thompson, P.D.; Clarkson, P.; Karas, R.H. Statin-associated myopathy. JAMA 2003, 289, 1681–1690. [Google Scholar] [CrossRef]
- Ahn, S.C. Neuromuscular complications of statins. Phys. Med. Rehabil. Clin. N. Am. 2008, 19, 47–59. [Google Scholar] [CrossRef]
- Ramkumar, S.; Raghunath, A.; Raghunath, S. Statin Therapy: Review of Safety and Potential Side Effects. Acta Cardiol. Sin. 2016, 32, 631–639. [Google Scholar]
- Alonso, R.; Cuevas, A.; Cafferata, A. Diagnosis and Management of Statin Intolerance. J. Atheroscler. Thromb. 2019, 26, 207–215. [Google Scholar] [CrossRef] [Green Version]
- Pohjola-Sintonen, S.; Julkunen, H. Muscle-related adverse effects of statins. Duodecim 2014, 130, 1622–1627. [Google Scholar]
- Abd, T.T.; Jacobson, T.A. Statin-induced myopathy: A review and update. Expert Opin. Drug Saf. 2011, 10, 373–387. [Google Scholar] [CrossRef]
- Abed, W.; Abujbara, M.; Batieha, A.; Ajlouni, K. Statin Induced Myopathy Among Patients Attending the National Center for Diabetes, endocrinology, & genetics. Ann. Med. Surg. 2022, 74, 103304. [Google Scholar]
- Horodinschi, R.N.; Stanescu, A.; Bratu, O.G.; Pantea Stoian, A.; Radavoi, D.G.; Diaconu, C.C. Treatment with Statins in Elderly Patients. Medicina 2019, 55, 721. [Google Scholar] [CrossRef] [Green Version]
- Taylor, B.A.; Thompson, P.D. Statin-Associated Muscle Disease: Advances in Diagnosis and Management. Neurother. J. Am. Soc. Exp. Neurother. 2018, 15, 1006–1017. [Google Scholar] [CrossRef] [Green Version]
- Camerino, G.M.; Musumeci, O.; Conte, E.; Musaraj, K.; Fonzino, A.; Barca, E.; Marino, M.; Rodolico, C.; Tricarico, D.; Camerino, C.; et al. Risk of Myopathy in Patients in Therapy with Statins: Identification of Biological Markers in a Pilot Study. Front. Pharmacol. 2017, 8, 500. [Google Scholar] [CrossRef]
- Ward, N.C.; Watts, G.F.; Eckel, R.H. Statin Toxicity. Circ. Res. 2019, 124, 328–350. [Google Scholar] [CrossRef]
- Catapano, A.L. Statin-induced myotoxicity: Pharmacokinetic differences among statins and the risk of rhabdomyolysis, with particular reference to pitavastatin. Curr. Vasc. Pharmacol. 2012, 10, 257–267. [Google Scholar] [CrossRef]
- Weng, T.C.; Yang, Y.H.; Lin, S.J.; Tai, S.H. A systematic review and meta-analysis on the therapeutic equivalence of statins. J. Clin. Pharm. Ther. 2010, 35, 139–151. [Google Scholar] [CrossRef]
- Golomb, B.A.; Evans, M.A. Statin adverse effects: A review of the literature and evidence for a mitochondrial mechanism. Am. J. Cardiovasc. Drugs 2008, 8, 373–418. [Google Scholar] [CrossRef]
- Vrablik, M.; Zlatohlavek, L.; Stulc, T.; Adamkova, V.; Prusikova, M.; Schwarzova, L.; Hubacek, J.A.; Ceska, R. Statin-associated myopathy: From genetic predisposition to clinical management. Physiol. Res. 2014, 63, S327–S334. [Google Scholar] [CrossRef]
- Rosenson, R.S.; Miller, K.; Bayliss, M.; Sanchez, R.J.; Baccara-Dinet, M.T.; Chibedi-De-Roche, D.; Taylor, B.; Khan, I.; Manvelian, G.; White, M.; et al. The Statin-Associated Muscle Symptom Clinical Index (SAMS-CI): Revision for Clinical Use, Content Validation, and Inter-rater Reliability. Cardiovasc. Drugs Ther. 2017, 31, 179–186. [Google Scholar] [CrossRef] [PubMed]
- Vinci, P.; Panizon, E.; Tosoni, L.M.; Cerrato, C.; Pellicori, F.; Mearelli, F.; Biasinutto, C.; Fiotti, N.; Di Girolamo, F.G.; Biolo, G. Statin-Associated Myopathy: Emphasis on Mechanisms and Targeted Therapy. Int. J. Mol. Sci. 2021, 22, 11687. [Google Scholar] [CrossRef] [PubMed]
- Stroes, E.S.; Thompson, P.D.; Corsini, A.; Vladutiu, G.D.; Raal, F.J.; Ray, K.K.; Roden, M.; Stein, E.; Tokgözoğlu, L.; Nordestgaard, B.G.; et al. Statin-associated muscle symptoms: Impact on statin therapy-European Atherosclerosis Society Consensus Panel Statement on Assessment, Aetiology and Management. Eur. Heart J. 2015, 36, 1012–1022. [Google Scholar] [CrossRef] [PubMed]
- Omar, M.A.; Wilson, J.P.; Cox, T.S. Rhabdomyolysis and HMG-CoA reductase inhibitors. Ann. Pharmacother. 2001, 35, 1096–1107. [Google Scholar] [CrossRef]
- Tomaszewski, M.; Stępień, K.M.; Tomaszewska, J.; Czuczwar, S.J. Statin-induced myopathies. Pharmacol. Rep. 2011, 63, 859–866. [Google Scholar] [CrossRef]
- Ramachandran, R.; Wierzbicki, A.S. Statins, Muscle Disease and Mitochondria. J. Clin. Med. 2017, 6, 75. [Google Scholar] [CrossRef] [Green Version]
- Apostolopoulou, M.; Corsini, A.; Roden, M. The role of mitochondria in statin-induced myopathy. Eur. J. Clin. Investig. 2015, 45, 745–754. [Google Scholar] [CrossRef] [Green Version]
- Zaleski, A.L.; Taylor, B.A.; Thompson, P.D. Coenzyme Q10 as Treatment for Statin-Associated Muscle Symptoms-A Good Idea, but…. Adv. Nutr. 2018, 9, 519S–523S. [Google Scholar] [CrossRef]
- Di Stasi, S.L.; MacLeod, T.D.; Winters, J.D.; Binder-Macleod, S.A. Effects of statins on skeletal muscle: A perspective for physical therapists. Phys. Ther. 2010, 90, 1530–1542. [Google Scholar] [CrossRef] [Green Version]
- Dirks, A.J.; Jones, K.M. Statin-induced apoptosis and skeletal myopathy. Am. J. Physiol. Cell Physiol. 2006, 291, C1208–C1212. [Google Scholar] [CrossRef]
- Brunham, L.R.; Baker, S.; Mammen, A.; Mancini, G.; Rosenson, R.S. Role of genetics in the prediction of statin-associated muscle symptoms and optimization of statin use and adherence. Cardiovasc. Res. 2018, 114, 1073–1081. [Google Scholar] [CrossRef]
- Turongkaravee, S.; Jittikoon, J.; Lukkunaprasit, T.; Sangroongruangsri, S.; Chaikledkaew, U.; Thakkinstian, A. A systematic review and meta-analysis of genotype-based and individualized data analysis of SLCO1B1 gene and statin-induced myopathy. Pharm. J. 2021, 21, 296–307. [Google Scholar] [CrossRef] [PubMed]
- Isackson, P.J.; Wang, J.; Zia, M.; Spurgeon, P.; Levesque, A.; Bard, J.; James, S.; Nowak, N.; Lee, T.K.; Vladutiu, G.D. RYR1 and CACNA1S genetic variants identified with statin-associated muscle symptoms. Pharmacogenomics 2018, 19, 1235–1249. [Google Scholar] [CrossRef]
- Turner, R.M.; Pirmohamed, M. Statin-Related Myotoxicity: A Comprehensive Review of Pharmacokinetic, Pharmacogenomic and Muscle Components. J. Clin. Med. 2019, 9, 22. [Google Scholar] [CrossRef] [Green Version]
- Muntean, D.M.; Thompson, P.D.; Catapano, A.L.; Stasiolek, M.; Fabis, J.; Muntner, P.; Serban, M.-C.; Banach, M. Statin-associated myopathy and the quest for biomarkers: Can we effectively predict statin-associated muscle symptoms? Drug Discov. Today 2017, 22, 85–96. [Google Scholar] [CrossRef]
- Moghadam-Kia, S.; Oddis, C.V.; Aggarwal, R. Approach to asymptomatic creatine kinase elevation. Clevel. Clin. J. Med. 2016, 83, 37–42. [Google Scholar] [CrossRef]
- Sikka, P.; Kapoor, S.; Bindra, V.K.; Sharma, M.; Vishwakarma, P.; Saxena, K.K. Statin intolerance: Now a solved problem. J. Postgrad. Med. 2011, 57, 321–328. [Google Scholar] [CrossRef]
- Sasson, M.; Pesach, S. Simvastatin-induced meralgia paresthetica. J. Am. Board Fam. Med. JABFM 2011, 24, 469–473. [Google Scholar] [PubMed]
- Özdemir, I.H.; Copkiran, Ö.; Tıkız, H.; Tıkız, C. Peripheral polyneuropathy in patients receiving long-term statin therapy. Turk Kardiyol. Dern. Ars. 2019, 47, 554–563. [Google Scholar]
- Al-Kuraishy, H.M.; Al-Gareeb, A.I.; Hussien, N.R.; Al-Naimi, M.S.; Rasheed, H.A. Statins an oft-prescribed drug is implicated in peripheral neuropathy: The time to know more. J. Pak. Med. Assoc. 2019, 69, S108–S112. [Google Scholar]
- De Langen, J.J.; van Puijenbroek, E.P. HMG-CoA-reductase inhibitors and neuropathy: Reports to the Netherlands Pharmacovigilance Centre. Neth. J. Med. 2006, 64, 334–338. [Google Scholar] [PubMed]
- Jones, M.R.; Urits, I.; Wolf, J.; Corrigan, D.; Colburn, L.; Peterson, E.; Williamson, A.; Viswanath, O. Drug-Induced Peripheral Neuropathy: A Narrative Review. Curr. Clin. Pharmacol. 2020, 15, 38–48. [Google Scholar] [CrossRef]
- Emad, M.; Arjmand, H.; Farpour, H.R.; Kardeh, B. Lipid-lowering drugs (statins) and peripheral neuropathy. Electron. Physician 2018, 10, 6527–6533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Otruba, P.; Kanovsky, P.; Hlustik, P. Treatment with statins and involvement of the peripheral nervous system: Results of a prospective clinical and neurophysiological follow-up. Biomed. Pap. Med. Fac. Univ. Palacky Olomouc Czechoslov. 2007, 151, 307–310. [Google Scholar] [CrossRef] [Green Version]
- Gurha, N.; Rehan, H.S.; Yadav, M.; Gupta, L.K. Association of statin induced reduction in serum coenzyme Q10 level and conduction deficits in motor and sensory nerves: An observational cross-sectional study. Clin. Neurol. Neurosurg. 2020, 196, 106046. [Google Scholar] [CrossRef]
- Svendsen, T.K.; Nørregaard Hansen, P.; García Rodríguez, L.A.; Andersen, L.; Hallas, J.; Sindrup, S.H.; Gaist, D. Statins and polyneuropathy revisited: Case-control study in Denmark, 1999–2013. Br. J. Clin. Pharmacol. 2017, 83, 2087–2095. [Google Scholar] [CrossRef] [Green Version]
- Lehrer, S.; Rheinstein, P.H. Statins combined with niacin reduce the risk of peripheral neuropathy. Int. J. Funct. Nutr. 2020, 1, 3. [Google Scholar] [CrossRef]
- Newman, C.B.; Preiss, D.; Tobert, J.A.; Jacobson, T.A.; Page, R.L.; Goldstein, L.B.; Chin, C.; Tannock, L.R.; Miller, M.; Raghuveer, G.; et al. Statin Safety and Associated Adverse Events: A Scientific Statement From the American Heart Association. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 38–81. [Google Scholar] [CrossRef] [Green Version]
- Keogh, M.J.; Findlay, J.M.; Leach, S.; Bowen, J. Statin-associated weakness in myasthenia gravis: A case report. J. Med. Case Rep. 2010, 4, 61. [Google Scholar] [CrossRef] [Green Version]
- Elsais, A.; Popperud, T.H.; Melien, Ø.; Kerty, E. Drugs that may trigger or exacerbate myasthenia gravis. Tidsskr. Den Nor. Laegeforen. 2013, 133, 296–299. [Google Scholar] [CrossRef] [Green Version]
- Oh, S.J.; Dhall, R.; Young, A.; Morgan, M.B.; Lu, L.; Claussen, G.C. Statins may aggravate myasthenia gravis. Muscle Nerve 2008, 38, 1101–1107. [Google Scholar] [CrossRef] [Green Version]
- Cartwright, M.S.; Jeffery, D.R.; Nuss, G.R.; Donofrio, P.D. Statin-associated exacerbation of myasthenia gravis. Neurology 2004, 63, 2188. [Google Scholar] [CrossRef]
- Purvin, V.; Kawasaki, A.; Smith, K.H.; Kesler, A. Statin-associated myasthenia gravis: Report of 4 cases and review of the literature. Medicine 2006, 85, 82–85. [Google Scholar] [CrossRef]
- De Sousa, E.; Howard, J. More evidence for the association between statins and myasthenia gravis. Muscle Nerve 2008, 38, 1085–1086. [Google Scholar] [CrossRef]
- Gale, J.; Danesh-Meyer, H.V. Statins can induce myasthenia gravis. J. Clin. Neurosci. 2014, 21, 195–197. [Google Scholar] [CrossRef]
- Winiarska, M.; Bil, J.; Wilczek, E.; Wilczynski, M.; Lekka, M.; Engelberts, J.P.; Mackus, J.M.; Gorska, E.; Bojarski, L.; Stoklosa, T.; et al. Statins impair antitumor effects of rituximab by inducing conformational changes of CD20. PLoS Med. 2008, 5, e64. [Google Scholar] [CrossRef] [Green Version]
- Dalakas, M.C. Role of complement, anti-complement therapeutics, and other targeted immunotherapies in myasthenia gravis. Expert Rev. Clin. Immunol. 2022, 18, 691–701. [Google Scholar] [CrossRef]
- Gilhus, N.E. Is it safe to use statins in patients with myasthenia gravis? Nat. Clin. Pract. Neurol. 2009, 5, 8–9. [Google Scholar] [CrossRef] [PubMed]
- Thomas, R.; Yeoh, S.A.; Berkeley, R.; Woods, A.; Stevens, M.; Marino, S.; Radunovic, A. Initial seronegative immune-mediated necrotising myopathy with subsequent anti-HMGCR antibody development and response to rituximab: Case report. BMC Rheumatol. 2020, 4, 29. [Google Scholar] [CrossRef]
- Mohassel, P.; Mammen, A.L. Anti-HMGCR Myopathy. J. Neuromuscul. Dis. 2018, 5, 11–20. [Google Scholar] [CrossRef] [Green Version]
- Scripko, P.D.; Amato, A.A.; Puig, A. Mystery case: A 63-year-old man with progressive proximal pain and weakness. Neurology 2014, 82, 26–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loganathan, P.; Oddis, C.V.; Aggarwal, R. Immune-mediated statin myopathy. Expert Rev. Clin. Immunol. 2016, 12, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Villa, L.; Lerario, A.; Calloni, S.; Peverelli, L.; Matinato, C.; DE Liso, F.; Ceriotti, F.; Tironi, R.; Sciacco, M.; Moggio, M.; et al. Immune-mediated necrotizing myopathy due to statins exposure. Acta Myol. 2018, 37, 257–262. [Google Scholar] [PubMed]
- Pinal-Fernandez, I.; Casal-Dominguez, M.; Mammen, A.L. Immune-Mediated Necrotizing Myopathy. Curr. Rheumatol. Rep. 2018, 20, 21. [Google Scholar] [CrossRef]
- Sharma, P.; Timilsina, B.; Adhikari, J.; Parajuli, P.; Dhital, R.; Tachamo, N. Statin-induced necrotizing autoimmune myopathy: An extremely rare adverse effect from statin use. J. Community Hosp. Intern. Med. Perspect. 2019, 9, 503–506. [Google Scholar] [CrossRef] [Green Version]
- Madgula, A.S.; Gadela, N.V.; Singh, M.; Chen, K. A Rare Case of Statin-induced Immune-mediated Necrotizing Myopathy. Cureus 2020, 12, e7500. [Google Scholar] [CrossRef] [Green Version]
- Barp, A.; Velardo, D.; Ciscato, P.; Sansone, V.A.; Lunetta, C. Anti-HMGCR myopathy misdiagnosed as motor neuron disease and complicated with COVID-19 infection. Neurol. Sci. 2021, 42, 1679–1682. [Google Scholar] [CrossRef]
- Selva-O’Callaghan, A.; Alvarado-Cardenas, M.; Pinal-Fernández, I.; Trallero-Araguás, E.; Milisenda, J.C.; Martínez, M.Á.; Marín, A.; Labrador-Horrillo, M.; Juárez, C.; Grau-Junyent, J.M. Statin-induced myalgia and myositis: An update on pathogenesis and clinical recommendations. Expert Rev. Clin. Immunol. 2018, 14, 215–224. [Google Scholar] [CrossRef]
- De Cock, E.; Hannon, H.; Moerman, V.; Schurgers, M. Statin-induced myopathy: A case report. Eur. Heart J. 2018, 2, 130. [Google Scholar] [CrossRef]
- Zaki, M.M.; Virk, Z.M.; Lopez, D.; Klubnick, J.; Ahrendsen, J.T.; Varma, H.; Kyttaris, V.; Abeles, I. A case of statin-associated immune-mediated necrotizing myopathy with atypical biopsy features. Eur. J. Rheumatol. 2021, 8, 36–39. [Google Scholar] [CrossRef]
- Liang, W.C.; Wang, C.H.; Chen, W.Z.; Kuo, Y.T.; Lin, H.F.; Suzuki, S.; Nishino, I.; Jong, Y.J. Treatment experience of Taiwanese patients with anti-3-hydroxy-3-methylglutaryl-coenzyme A reductase myopathy. Kaohsiung J. Med. Sci. 2020, 36, 649–655. [Google Scholar] [CrossRef]
- Cha, D.; Wang, F.; Mukerji, B.; Mukerji, V. Statin-Induced Necrotizing Autoimmune Myositis: Diagnosis and Management. Cureus 2021, 13, 13787. [Google Scholar] [CrossRef]
- Zhang, W.; Prince, H.M.; Reardon, K. Statin-induced anti-HMGCR antibody-related immune-mediated necrotising myositis achieving complete remission with rituximab. BMJ Case Rep. 2019, 12, 232406. [Google Scholar] [CrossRef]
- Irvine, N.J. Anti-HMGCR Myopathy: A Rare and Serious Side Effect of Statins. J. Am. Board Fam. Med. 2020, 33, 785–788. [Google Scholar] [CrossRef]
- Yeo, C.H.; Yaakub, A.; Wang, M.; Shim, S.A.; Chong, P.L.; Khalil, M.; Telisinghe, P.U.; Lim, K.C.; Tan, J.; Chong, V.H. Refractory Statin-Induced Immune-Mediated Necrotizing Myositis: Challenges and Perils in Its Management. Cureus 2022, 14, 24778. [Google Scholar] [CrossRef]
- Echaniz-Laguna, A.; Mohr, M.; Tranchant, C. Neuromuscular symptoms and elevated creatine kinase after statin withdrawal. N. Engl. J. Med. 2010, 362, 564–565. [Google Scholar] [CrossRef]
- Arca, M.; Pigna, G.; Favoccia, C. Management of statin-intolerant patient. Panminerva Med. 2012, 54, 105–118. [Google Scholar]
- Mach, F.; Baigent, C.; Catapano, A.L.; Koskinas, K.C.; Casula, M.; Badimon, L.; Chapman, M.J.; De Backer, G.G.; Delgado, V.; Ference, B.A.; et al. 2019 ESC/EAS Guidelines for the management of dyslipidaemias: Lipid modification to reduce cardiovascular risk. Eur. Heart J. 2020, 41, 111–188. [Google Scholar] [CrossRef]
- Grundy, S.M.; Stone, N.J.; Bailey, A.L.; Beam, C.; Birtcher, K.K.; Blumenthal, R.S.; Braun, L.T.; de Ferranti, S.; Faiella-Tommasino, J.; Forman, D.E.; et al. AHA/ACC/AACVPR/AAPA/ABC/ACPM/ADA/AGS/APhA/ASPC/NLA/PCNA Guideline on the Management of Blood Cholesterol: Executive Summary: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. J. Am. Coll. Cardiol. 2019, 73, 3168–3209. [Google Scholar] [CrossRef]
- Toth, P.P.; Patti, A.M.; Giglio, R.V.; Nikolic, D.; Castellino, G.; Rizzo, M.; Banach, M. Management of Statin Intolerance in 2018: Still More Questions Than Answers. Am. J. Cardiovasc. Drugs 2018, 18, 157–173. [Google Scholar] [CrossRef] [Green Version]
- Oddis, C.V. Myopathy for the general internist: Statins and much more. Clevel. Clin. J. Med. 2019, 86, 656–664. [Google Scholar] [CrossRef] [PubMed]
- Taylor, B.A. Does Coenzyme Q10 Supplementation Mitigate Statin-Associated Muscle Symptoms? Pharmacological and Methodological Considerations. Am. J. Cardiovasc. Drugs 2018, 18, 75–82. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Ochs-Balcom, H.M.; Ma, C.; Isackson, P.J.; Vladutiu, G.D.; Luzum, J.A. Coenzyme Q10 supplementation for the treatment of statin-associated muscle symptoms. Future Cardiol. 2022, 18, 461–470. [Google Scholar] [CrossRef] [PubMed]
- Argov, Z. Statins and the neuromuscular system: A neurologist’s perspective. Eur. J. Neurol. 2015, 22, 31–36. [Google Scholar] [CrossRef]
- Riche, K.D.; Arnall, J.; Rieser, K.; East, H.E.; Riche, D.M. Impact of vitamin D status on statin-induced myopathy. J. Clin. Transl. Endocrinol. 2016, 6, 56–59. [Google Scholar] [CrossRef]
- Hou, Q.; Pang, C.; Chen, Y. Association Between Vitamin D and Statin-Related Myopathy: A Meta-analysis. Am. J. Cardiovasc. Drugs 2022, 22, 183–193. [Google Scholar] [CrossRef]
- Balestrino, M.; Adriano, E. Creatine as a Candidate to Prevent Statin Myopathy. Biomolecules 2019, 9, 496. [Google Scholar] [CrossRef] [Green Version]
- Baker, S.K.; Samjoo, I.A. A neuromuscular approach to statin-related myotoxicity. Can. J. Neurol. Sci. 2008, 35, 8–21. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Lypophilic Statins | Hydrophilic Statins |
|---|---|
| Atorvastatin | Rosuvastatin |
| Fluvastatin | Pravastatin |
| Simvastatin | |
| Pitavastatin | |
| Lovastatin |
| Inhibitors of CYP3A4 (atorvastatin, simvastatin, lovastatin) | azole antifungals (ketoconazole, fluconazole), macrolide antibiotics (erythromycin, clarithromycin, troleandomycin, telithromycin), antidepressants (fluvoxamine, fluoxetine, sertraline, nefazodone) calcium channel antagonists (amlodipine, verapamil, diltiazem, mibefradil), protease inhibitors (indinavir, ritonavir), immunosuppresants (cyclosporine, erlotinib), clopidogrel, grapefruit juice |
| Inhibitors CYP2C9 (fluvastatin, rosuvastatin) | azole antifungals (ketoconazole, fluconazole, itraconazole), fluoxetine, amiodarone, warfarin |
| Inhibitors of organic anion transporting peptide 1B1 | gemfibrozil |
| Family or Personal History of Intolerance to Statins | References |
|---|---|
| Advanced age | [9,10,13,16] |
| Female gender | [9,16] |
| Asian ethnicity | [9,16] |
| Drugs altering statin plasma levels | [13,16] |
| Excessive physical activity | [9,16] |
| Muscle diseases | [9,16] |
| Liver diseases | [9,16] |
| Chronic kidney diseases | [9,16] |
| Uncontrolled hypothyroidism | [9,16] |
| Abdominal obesity and metabolic syndrome | [9,16] |
| Vitamin D deficiency | [9,16] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Attardo, S.; Musumeci, O.; Velardo, D.; Toscano, A. Statins Neuromuscular Adverse Effects. Int. J. Mol. Sci. 2022, 23, 8364. https://doi.org/10.3390/ijms23158364
Attardo S, Musumeci O, Velardo D, Toscano A. Statins Neuromuscular Adverse Effects. International Journal of Molecular Sciences. 2022; 23(15):8364. https://doi.org/10.3390/ijms23158364
Chicago/Turabian StyleAttardo, Silvia, Olimpia Musumeci, Daniele Velardo, and Antonio Toscano. 2022. "Statins Neuromuscular Adverse Effects" International Journal of Molecular Sciences 23, no. 15: 8364. https://doi.org/10.3390/ijms23158364
APA StyleAttardo, S., Musumeci, O., Velardo, D., & Toscano, A. (2022). Statins Neuromuscular Adverse Effects. International Journal of Molecular Sciences, 23(15), 8364. https://doi.org/10.3390/ijms23158364