
Early Expression of Tet1 and Tet2 in Mouse Zygotes Altered DNA Methylation Status and Affected Embryonic Development


Abstract
: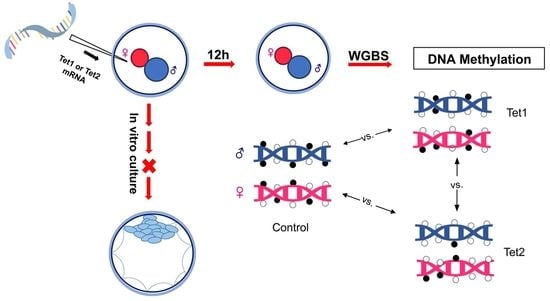
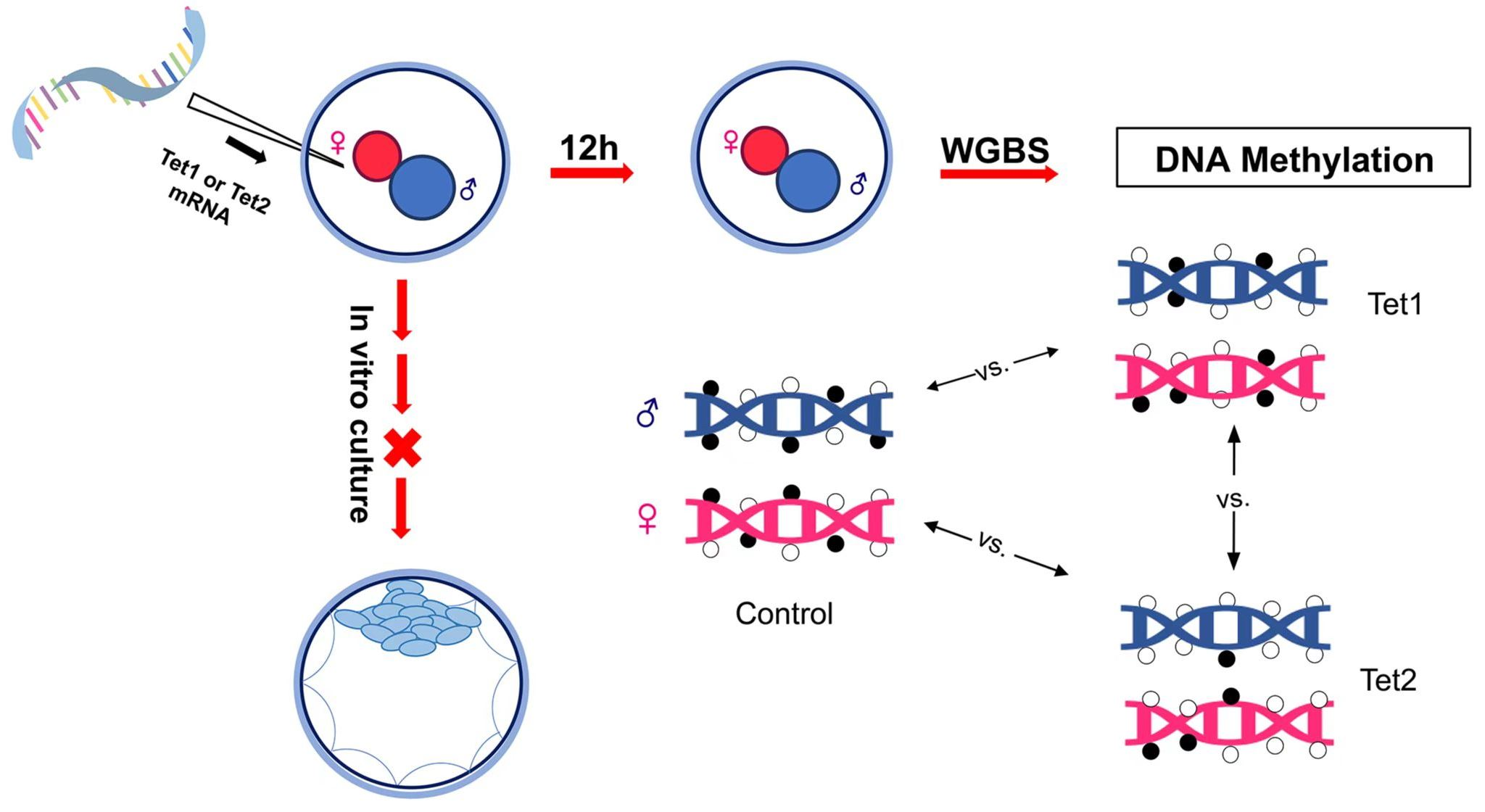{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. Overexpression of Tet1 and Tet2 in Zygotes Increased the DNA Oxidation of the Maternal Genome
2.2. Whole-Genome Bisulfite Sequencing (WGBS) Analysis of the Methylome
2.3. Effects of Tet1 and Tet2 on Various Genomic Regions in Zygotes
2.4. Methylation Sites Affected by Tet1 and Tet2
2.5. The Propensity of Tet1 and Tet2 to Base Sequences
2.6. Influences of Tet1 and Tet2 on Germline-Specific DMRs
2.7. Roles of Tet1 and Tet2 on Imprinting Control Regions (ICRs)
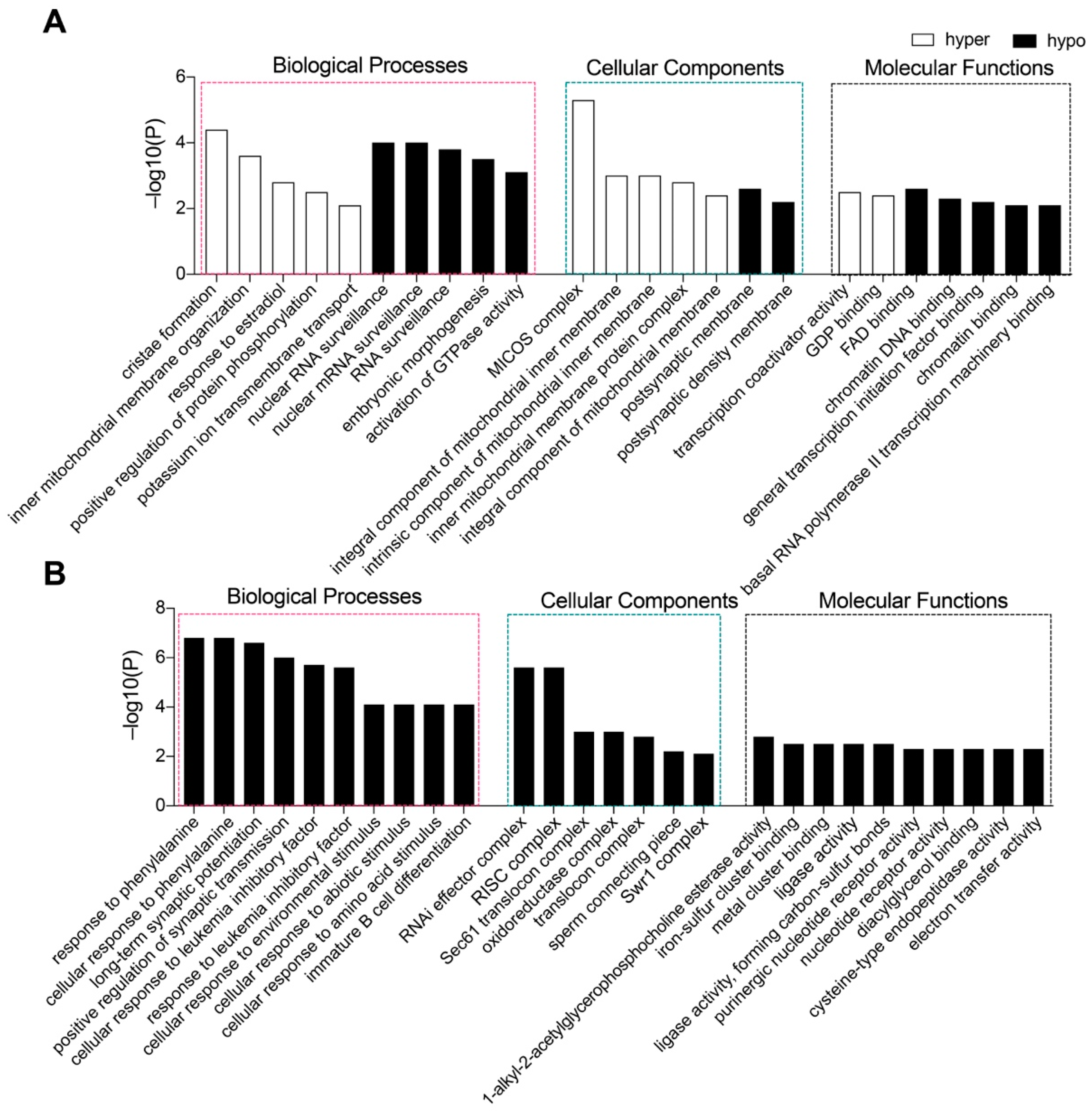
2.8. Genes with Altered Promoter Methylation by Tet1 and Tet2
2.9. Early Expression of Tet1 or Tet2 Damages Embryonic Development
3. Discussion
4. Materials and Methods
4.1. Mice
4.2. Embryo Collection and Culture
4.3. In Vitro Transcription and Microinjection
4.4. Immunofluorescence Staining
4.5. Single-Cell Library Preparation
4.6. Data Processing and Analysis
4.7. Single-Nucleotide Polymorphisms (SNPs) Calling
4.8. Statistical Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Reik, W.; Dean, W.; Walter, J. Epigenetic reprogramming in mammalian development. Science 2001, 293, 1089–1093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, Z.D.; Meissner, A. DNA methylation: Roles in mammalian development. Nat. Rev. Genet. 2013, 14, 204–220. [Google Scholar] [CrossRef] [PubMed]
- Saitou, M.; Kagiwada, S.; Kurimoto, K. Epigenetic reprogramming in mouse pre-implantation development and primordial germ cells. Development 2012, 139, 15–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mayer, W.; Niveleau, A.; Walter, J.; Fundele, R.; Haaf, T. Demethylation of the zygotic paternal genome. Nature 2000, 403, 501–502. [Google Scholar] [CrossRef]
- Santos, F.; Hendrich, B.; Reik, W.; Dean, W. Dynamic reprogramming of DNA methylation in the early mouse embryo. Dev. Biol. 2002, 241, 172–182. [Google Scholar] [CrossRef] [Green Version]
- Ito, S.; D’Alessio, A.C.; Taranova, O.V.; Hong, K.; Sowers, L.C.; Zhang, Y. Role of Tet proteins in 5mC to 5hmC conversion, ES-cell self-renewal and inner cell mass specification. Nature 2010, 466, 1129–1133. [Google Scholar] [CrossRef] [Green Version]
- Tahiliani, M.; Koh, K.P.; Shen, Y.; Pastor, W.A.; Bandukwala, H.; Brudno, Y.; Agarwal, S.; Iyer, L.M.; Liu, D.R.; Aravind, L.; et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science 2009, 324, 930–935. [Google Scholar] [CrossRef] [Green Version]
- Ito, S.; Shen, L.; Dai, Q.; Wu, S.C.; Collins, L.B.; Swenberg, J.A.; He, C.; Zhang, Y. Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science 2011, 333, 1300–1303. [Google Scholar] [CrossRef] [Green Version]
- He, Y.F.; Li, B.Z.; Li, Z.; Liu, P.; Wang, Y.; Tang, Q.; Ding, J.; Jia, Y.; Chen, Z.; Li, L.; et al. Tet-mediated formation of 5-carboxylcytosine and its excision by TDG in mammalian DNA. Science 2011, 333, 1303–1307. [Google Scholar] [CrossRef] [Green Version]
- Inoue, A.; Shen, L.; Dai, Q.; He, C.; Zhang, Y. Generation and replication-dependent dilution of 5fC and 5caC during mouse preimplantation development. Cell Res. 2011, 21, 1670–1676. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Lu, X.; Lu, J.; Liang, H.; Dai, Q.; Xu, G.L.; Luo, C.; Jiang, H.; He, C. Thymine DNA glycosylase specifically recognizes 5-carboxylcytosine-modified DNA. Nat. Chem. Biol. 2012, 8, 328–330. [Google Scholar] [CrossRef] [Green Version]
- Shen, L.; Inoue, A.; He, J.; Liu, Y.; Lu, F.; Zhang, Y. Tet3 and DNA replication mediate demethylation of both the maternal and paternal genomes in mouse zygotes. Cell Stem Cell 2014, 15, 459–471. [Google Scholar] [CrossRef] [Green Version]
- Wu, X.; Zhang, Y. TET-mediated active DNA demethylation: Mechanism, function and beyond. Nat. Rev. Genet. 2017, 18, 517–534. [Google Scholar] [CrossRef]
- Iqbal, K.; Jin, S.G.; Pfeifer, G.P.; Szabo, P.E. Reprogramming of the paternal genome upon fertilization involves genome-wide oxidation of 5-methylcytosine. Proc. Natl. Acad. Sci. USA 2011, 108, 3642–3647. [Google Scholar] [CrossRef] [Green Version]
- Wossidlo, M.; Nakamura, T.; Lepikhov, K.; Marques, C.J.; Zakhartchenko, V.; Boiani, M.; Arand, J.; Nakano, T.; Reik, W.; Walter, J. 5-Hydroxymethylcytosine in the mammalian zygote is linked with epigenetic reprogramming. Nat. Commun. 2011, 2, 241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, T.P.; Guo, F.; Yang, H.; Wu, H.P.; Xu, G.F.; Liu, W.; Xie, Z.G.; Shi, L.; He, X.; Jin, S.G.; et al. The role of Tet3 DNA dioxygenase in epigenetic reprogramming by oocytes. Nature 2011, 477, 606–610. [Google Scholar] [CrossRef] [PubMed]
- Guo, F.; Li, X.; Liang, D.; Li, T.; Zhu, P.; Guo, H.; Wu, X.; Wen, L.; Gu, T.P.; Hu, B.; et al. Active and passive demethylation of male and female pronuclear DNA in the mammalian zygote. Cell Stem Cell 2014, 15, 447–459. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, T.; Liu, Y.J.; Nakashima, H.; Umehara, H.; Inoue, K.; Matoba, S.; Tachibana, M.; Ogura, A.; Shinkai, Y.; Nakano, T. PGC7 binds histone H3K9me2 to protect against conversion of 5mC to 5hmC in early embryos. Nature 2012, 486, 415–419. [Google Scholar] [CrossRef]
- Kang, J.; Lienhard, M.; Pastor, W.A.; Chawla, A.; Novotny, M.; Tsagaratou, A.; Lasken, R.S.; Thompson, E.C.; Surani, M.A.; Koralov, S.B.; et al. Simultaneous deletion of the methylcytosine oxidases Tet1 and Tet3 increases transcriptome variability in early embryogenesis. Proc. Natl. Acad. Sci. USA 2015, 112, E4236–E4245. [Google Scholar] [CrossRef] [Green Version]
- Dawlaty, M.M.; Ganz, K.; Powell, B.E.; Hu, Y.C.; Markoulaki, S.; Cheng, A.W.; Gao, Q.; Kim, J.; Choi, S.W.; Page, D.C.; et al. Tet1 is dispensable for maintaining pluripotency and its loss is compatible with embryonic and postnatal development. Cell Stem Cell 2011, 9, 166–175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.; Cai, X.; Cai, C.L.; Wang, J.; Zhang, W.; Petersen, B.E.; Yang, F.C.; Xu, M. Deletion of Tet2 in mice leads to dysregulated hematopoietic stem cells and subsequent development of myeloid malignancies. Blood 2011, 118, 4509–4518. [Google Scholar] [CrossRef] [Green Version]
- Dawlaty, M.M.; Breiling, A.; Le, T.; Raddatz, G.; Barrasa, M.I.; Cheng, A.W.; Gao, Q.; Powell, B.E.; Li, Z.; Xu, M.; et al. Combined deficiency of Tet1 and Tet2 causes epigenetic abnormalities but is compatible with postnatal development. Dev. Cell 2013, 24, 310–323. [Google Scholar] [CrossRef] [Green Version]
- Vincent, J.J.; Huang, Y.; Chen, P.Y.; Feng, S.; Calvopina, J.H.; Nee, K.; Lee, S.A.; Le, T.; Yoon, A.J.; Faull, K.; et al. Stage-specific roles for tet1 and tet2 in DNA demethylation in primordial germ cells. Cell Stem Cell 2013, 12, 470–478. [Google Scholar] [CrossRef] [Green Version]
- Piccolo, F.M.; Bagci, H.; Brown, K.E.; Landeira, D.; Soza-Ried, J.; Feytout, A.; Mooijman, D.; Hajkova, P.; Leitch, H.G.; Tada, T.; et al. Different roles for Tet1 and Tet2 proteins in reprogramming-mediated erasure of imprints induced by EGC fusion. Mol. Cell 2013, 49, 1023–1033. [Google Scholar] [CrossRef] [Green Version]
- Yamaguchi, S.; Shen, L.; Liu, Y.; Sendler, D.; Zhang, Y. Role of Tet1 in erasure of genomic imprinting. Nature 2013, 504, 460–464. [Google Scholar] [CrossRef]
- Hackett, J.A.; Sengupta, R.; Zylicz, J.J.; Murakami, K.; Lee, C.; Down, T.A.; Surani, M.A. Germline DNA demethylation dynamics and imprint erasure through 5-hydroxymethylcytosine. Science 2013, 339, 448–452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Booth, M.J.; Branco, M.R.; Ficz, G.; Oxley, D.; Krueger, F.; Reik, W.; Balasubramanian, S. Quantitative sequencing of 5-methylcytosine and 5-hydroxymethylcytosine at single-base resolution. Science 2012, 336, 934–937. [Google Scholar] [CrossRef] [Green Version]
- Guo, F.; Li, L.; Li, J.; Wu, X.; Hu, B.; Zhu, P.; Wen, L.; Tang, F. Single-cell multi-omics sequencing of mouse early embryos and embryonic stem cells. Cell Res. 2017, 27, 967–988. [Google Scholar] [CrossRef]
- Smith, Z.D.; Chan, M.M.; Mikkelsen, T.S.; Gu, H.; Gnirke, A.; Regev, A.; Meissner, A. A unique regulatory phase of DNA methylation in the early mammalian embryo. Nature 2012, 484, 339–344. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, H.; Sakurai, T.; Imai, M.; Takahashi, N.; Fukuda, A.; Yayoi, O.; Sato, S.; Nakabayashi, K.; Hata, K.; Sotomaru, Y.; et al. Contribution of intragenic DNA methylation in mouse gametic DNA methylomes to establish oocyte-specific heritable marks. PLoS Genet. 2012, 8, e1002440. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Zhang, J.; Duan, J.; Gao, X.; Zhu, W.; Lu, X.; Yang, L.; Zhang, J.; Li, G.; Ci, W.; et al. Programming and inheritance of parental DNA methylomes in mammals. Cell 2014, 157, 979–991. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andersen, I.S.; Reiner, A.H.; Aanes, H.; Alestrom, P.; Collas, P. Developmental features of DNA methylation during activation of the embryonic zebrafish genome. Genome Biol. 2012, 13, R65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dahlet, T.; Argueso Lleida, A.; Al Adhami, H.; Dumas, M.; Bender, A.; Ngondo, R.P.; Tanguy, M.; Vallet, J.; Auclair, G.; Bardet, A.F.; et al. Genome-wide analysis in the mouse embryo reveals the importance of DNA methylation for transcription integrity. Nat. Commun. 2020, 11, 3153. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Xia, W.; Wang, Q.; Towers, A.J.; Chen, J.; Gao, R.; Zhang, Y.; Yen, C.A.; Lee, A.Y.; Li, Y.; et al. Isoform switch of TET1 regulates DNA demethylation and mouse development. Mol. Cell 2016, 64, 1062–1073. [Google Scholar] [CrossRef] [Green Version]
- Ko, M.; An, J.; Bandukwala, H.S.; Chavez, L.; Aijö, T.; Pastor, W.A.; Segal, M.F.; Li, H.; Koh, K.P.; Lähdesmäki, H.; et al. Modulation of TET2 expression and 5-methylcytosine oxidation by the CXXC domain protein IDAX. Nature 2013, 497, 122–126. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Wang, M.; Deng, W.; Schmidt, C.S.; Qin, W.; Leonhardt, H.; Spada, F. Intrinsic and extrinsic connections of Tet3 dioxygenase with CXXC zinc finger modules. PLoS ONE 2013, 8, e62755. [Google Scholar] [CrossRef] [Green Version]
- Jin, S.G.; Zhang, Z.M.; Dunwell, T.L.; Harter, M.R.; Wu, X.; Johnson, J.; Li, Z.; Liu, J.; Szabó, P.E.; Lu, Q.; et al. Tet3 reads 5-Carboxylcytosine through its CXXC domain and is a potential guardian against neurodegeneration. Cell Rep. 2016, 14, 493–505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joshi, K.; Liu, S.; Breslin, S.J.P.; Zhang, J. Mechanisms that regulate the activities of TET proteins. Cell Mol. Life Sci. 2022, 79, 363. [Google Scholar] [CrossRef]
- Nakamura, T.; Arai, Y.; Umehara, H.; Masuhara, M.; Kimura, T.; Taniguchi, H.; Sekimoto, T.; Ikawa, M.; Yoneda, Y.; Okabe, M.; et al. PGC7/Stella protects against DNA demethylation in early embryogenesis. Nat. Cell Biol. 2007, 9, 64–71. [Google Scholar] [CrossRef]
- Bian, C.; Yu, X. PGC7 suppresses TET3 for protecting DNA methylation. Nucleic Acids Res. 2014, 42, 2893–2905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Zhang, Z.; Chen, J.; Liu, W.; Lai, W.; Liu, B.; Li, X.; Liu, L.; Xu, S.; Dong, Q.; et al. Stella safeguards the oocyte methylome by preventing de novo methylation mediated by DNMT1. Nature 2018, 564, 136–140. [Google Scholar] [CrossRef] [PubMed]
- Au Yeung, W.K.; Brind’Amour, J.; Hatano, Y.; Yamagata, K.; Feil, R.; Lorincz, M.C.; Tachibana, M.; Shinkai, Y.; Sasaki, H. Histone H3K9 methyltransferase G9a in oocytes is essential for preimplantation development but dispensable for CG methylation protection. Cell Rep. 2019, 27, 282–293.e4. [Google Scholar] [CrossRef] [Green Version]
- Amouroux, R.; Nashun, B.; Shirane, K.; Nakagawa, S.; Hill, P.W.; D’Souza, Z.; Nakayama, M.; Matsuda, M.; Turp, A.; Ndjetehe, E.; et al. De novo DNA methylation drives 5hmC accumulation in mouse zygotes. Nat. Cell Biol. 2016, 18, 225–233. [Google Scholar] [CrossRef] [Green Version]
- Richard Albert, J.; Au Yeung, W.K.; Toriyama, K.; Kobayashi, H.; Hirasawa, R.; Brind’Amour, J.; Bogutz, A.; Sasaki, H.; Lorincz, M. Maternal DNMT3A-dependent de novo methylation of the paternal genome inhibits gene expression in the early embryo. Nat. Commun. 2020, 11, 5417. [Google Scholar] [CrossRef]
- Gu, T.; Lin, X.; Cullen, S.M.; Luo, M.; Jeong, M.; Estecio, M.; Shen, J.; Hardikar, S.; Sun, D.; Su, J.; et al. DNMT3A and TET1 cooperate to regulate promoter epigenetic landscapes in mouse embryonic stem cells. Genome Biol. 2018, 19, 88. [Google Scholar] [CrossRef] [Green Version]
- Adam, S.; Bracker, J.; Klingel, V.; Osteresch, B.; Radde, N.E.; Brockmeyer, J.; Bashtrykov, P.; Jeltsch, A. Flanking sequences influence the activity of TET1 and TET2 methylcytosine dioxygenases and affect genomic 5hmC patterns. Commun. Biol. 2022, 5, 92. [Google Scholar] [CrossRef]
- Wu, H.; Zhang, Y. Tet1 and 5-hydroxymethylation: A genome-wide view in mouse embryonic stem cells. Cell Cycle 2011, 10, 2428–2436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanna, C.W.; Kelsey, G. The specification of imprints in mammals. Heredity 2014, 113, 176–183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamaguchi, S.; Hong, K.; Liu, R.; Shen, L.; Inoue, A.; Diep, D.; Zhang, K.; Zhang, Y. Tet1 controls meiosis by regulating meiotic gene expression. Nature 2012, 492, 443–447. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Yue, X.; Pastor, W.A.; Lin, L.; Georges, R.; Chavez, L.; Evans, S.M.; Rao, A. Tet proteins influence the balance between neuroectodermal and mesodermal fate choice by inhibiting Wnt signaling. Proc. Natl. Acad. Sci. USA 2016, 113, E8267–E8276. [Google Scholar] [CrossRef] [Green Version]
- Dai, H.Q.; Wang, B.A.; Yang, L.; Chen, J.J.; Zhu, G.C.; Sun, M.L.; Ge, H.; Wang, R.; Chapman, D.L.; Tang, F.; et al. TET-mediated DNA demethylation controls gastrulation by regulating Lefty-Nodal signalling. Nature 2016, 538, 528–532. [Google Scholar] [CrossRef] [PubMed]
- Arand, J.; Chiang, H.R.; Martin, D.; Snyder, M.P.; Sage, J.; Reijo Pera, R.A.; Wossidlo, M. Tet enzymes are essential for early embryogenesis and completion of embryonic genome activation. EMBO Rep. 2022, 23, e53968. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.A. Functions of DNA methylation: Islands, start sites, gene bodies and beyond. Nat. Rev. Genet. 2012, 13, 484–492. [Google Scholar] [CrossRef] [PubMed]
- Percharde, M.; Lin, C.J.; Yin, Y.; Guan, J.; Peixoto, G.A.; Bulut-Karslioglu, A.; Biechele, S.; Huang, B.; Shen, X.; Ramalho-Santos, M. A LINE1-nucleolin partnership regulates early development and ESC Identity. Cell 2018, 174, 391–405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kigami, D.; Minami, N.; Takayama, H.; Imai, H. MuERV-L is one of the earliest transcribed genes in mouse one-cell embryos. Biol. Reprod. 2003, 68, 651–654. [Google Scholar] [CrossRef] [Green Version]
- Han, L.; Ren, C.; Li, L.; Li, X.; Ge, J.; Wang, H.; Miao, Y.L.; Guo, X.; Moley, K.H.; Shu, W.; et al. Embryonic defects induced by maternal obesity in mice derive from Stella insufficiency in oocytes. Nat. Genet. 2018, 50, 432–442. [Google Scholar] [CrossRef]
- Tsukada, Y.; Akiyama, T.; Nakayama, K.I. Maternal TET3 is dispensable for embryonic development but is required for neonatal growth. Sci. Rep. 2015, 5, 15876. [Google Scholar] [CrossRef] [Green Version]
- Inoue, A.; Shen, L.; Matoba, S.; Zhang, Y. Haploinsufficiency, but not defective paternal 5mC oxidation, accounts for the developmental defects of maternal Tet3 knockouts. Cell Rep. 2015, 10, 463–470. [Google Scholar] [CrossRef] [Green Version]
- Jankowska, A.M.; Szpurka, H.; Tiu, R.V.; Makishima, H.; Afable, M.; Huh, J.; O’Keefe, C.L.; Ganetzky, R.; McDevitt, M.A.; Maciejewski, J.P. Loss of heterozygosity 4q24 and TET2 mutations associated with myelodysplastic/myeloproliferative neoplasms. Blood 2009, 113, 6403–6410. [Google Scholar] [CrossRef] [Green Version]
- Macfarlan, T.S.; Gifford, W.D.; Driscoll, S.; Lettieri, K.; Rowe, H.M.; Bonanomi, D.; Firth, A.; Singer, O.; Trono, D.; Pfaff, S.L. Embryonic stem cell potency fluctuates with endogenous retrovirus activity. Nature 2012, 487, 57–63. [Google Scholar] [CrossRef] [Green Version]
- Smallwood, S.A.; Lee, H.J.; Angermueller, C.; Krueger, F.; Saadeh, H.; Peat, J.; Andrews, S.R.; Stegle, O.; Reik, W.; Kelsey, G. Single-cell genome-wide bisulfite sequencing for assessing epigenetic heterogeneity. Nat. Methods 2014, 11, 817–820. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhou, B.; Pache, L.; Chang, M.; Khodabakhshi, A.H.; Tanaseichuk, O.; Benner, C.; Chanda, S.K. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun. 2019, 10, 1523. [Google Scholar] [CrossRef]
- Zhang, M.; Xie, S.; Dong, X.; Zhao, X.; Zeng, B.; Chen, J.; Li, H.; Yang, W.; Zhao, H.; Wang, G.; et al. Genome-wide high resolution parental-specific DNA and histone methylation maps uncover patterns of imprinting regulation in maize. Genome Res. 2014, 24, 167–176. [Google Scholar] [CrossRef] [Green Version]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Qi, Q.; Wang, Q.; Liu, K.; Bian, J.; Yu, Z.; Hou, J. Early Expression of Tet1 and Tet2 in Mouse Zygotes Altered DNA Methylation Status and Affected Embryonic Development. Int. J. Mol. Sci. 2022, 23, 8495. https://doi.org/10.3390/ijms23158495
Qi Q, Wang Q, Liu K, Bian J, Yu Z, Hou J. Early Expression of Tet1 and Tet2 in Mouse Zygotes Altered DNA Methylation Status and Affected Embryonic Development. International Journal of Molecular Sciences. 2022; 23(15):8495. https://doi.org/10.3390/ijms23158495
Chicago/Turabian StyleQi, Qi, Qianqian Wang, Kailing Liu, Jiangyue Bian, Zhixuan Yu, and Jian Hou. 2022. "Early Expression of Tet1 and Tet2 in Mouse Zygotes Altered DNA Methylation Status and Affected Embryonic Development" International Journal of Molecular Sciences 23, no. 15: 8495. https://doi.org/10.3390/ijms23158495
APA StyleQi, Q., Wang, Q., Liu, K., Bian, J., Yu, Z., & Hou, J. (2022). Early Expression of Tet1 and Tet2 in Mouse Zygotes Altered DNA Methylation Status and Affected Embryonic Development. International Journal of Molecular Sciences, 23(15), 8495. https://doi.org/10.3390/ijms23158495